
Retinoic Acid Upregulates METTL14 Expression and the m6A Modification Level to Inhibit the Proliferation of Embryonic Palate Mesenchymal Cells in Cleft Palate Mice


Abstract
:1. Introduction
2. Results
2.1. Morphological Observation of Two Fetal Mouse Models with Cleft Palate
2.2. atRA Upregulated the m6A Level and the Expression of METTL14 in the Embryonic Palatal Mesenchyme
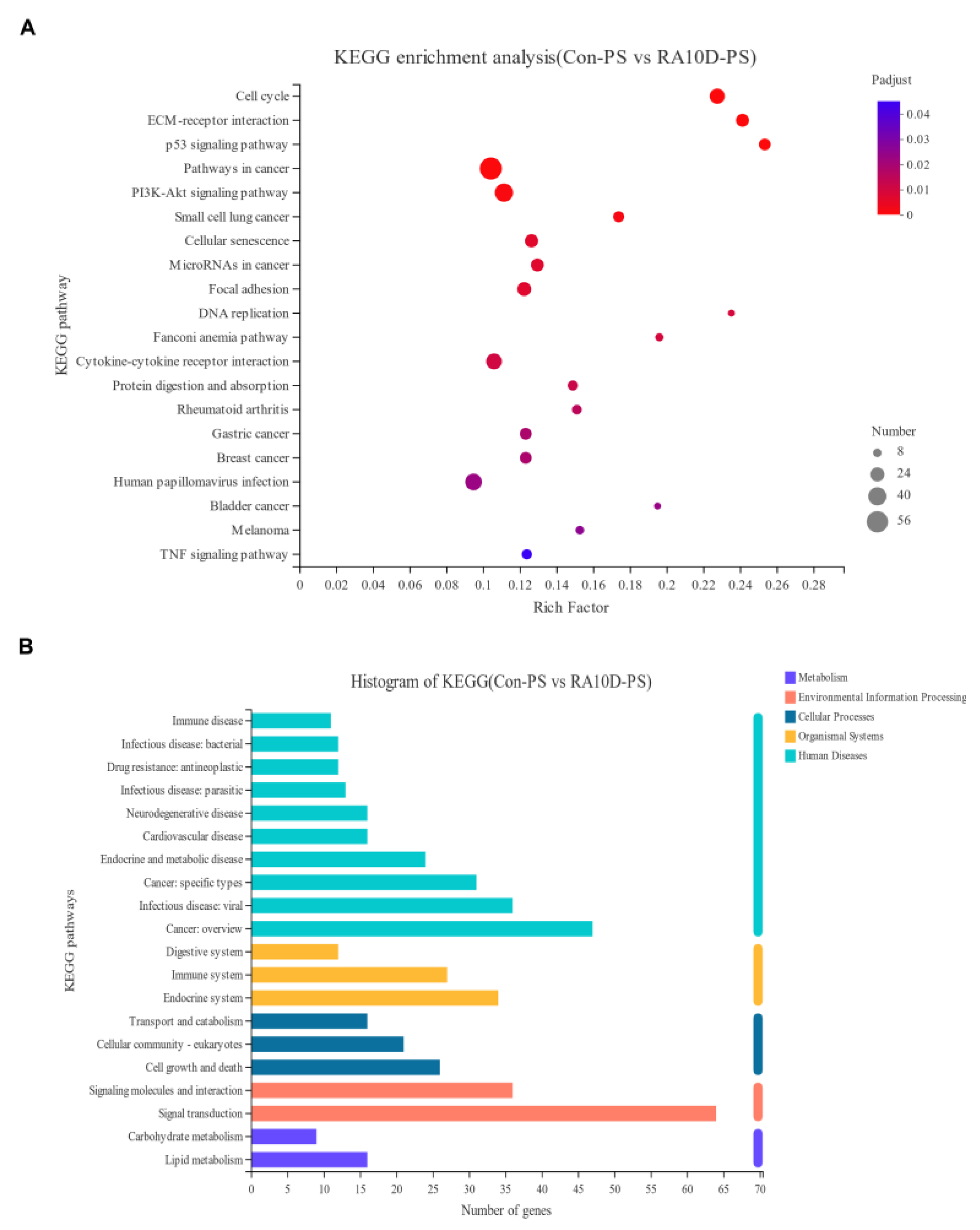
2.3. Environmental Factors Affect the Cell Cycle and p53 Signaling Pathway, Leading to Cleft Palate
2.4. Inhibition of Mesenchymal Cell Proliferation in Cleft Palate Mice May Be Related to the Abnormal Expression of METTL14
2.5. Knockdown of METTL14 or Inhibition of m6A Methylation Modification Can Partially Rescue the Decline in Cell Proliferation Induced by atRA
3. Discussion
4. Materials and Methods
- Mice
- Collection of palatal shelves and isolation and culture of primary MEPM cells
- Histological staining
- RNA extraction and Quantitative reverse transcription–PCR (qRT-PCR)
- RNA-seq
- Protein extraction and Western blot
- Dot blot
- Immunofluorescence identification of primary MEPM cells
- Flow cytometry for cell purity, stemness, and cell cycle detection
- Preparation of special culture medium
- Cell Proliferation Assay
- RNA interference
- Statistical analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Stanier, P.; Moore, G.E. Genetics of cleft lip and palate: Syndromic genes contribute to the incidence of non-syndromic clefts. Hum. Mol. Genet. 2004, 13, 73R–81R. [Google Scholar] [CrossRef] [PubMed]
- Mossey, P.A.; Little, J.; Munger, R.G.; Dixon, M.J.; Shaw, W.C. Cleft lip and palate. Lancet 2009, 374, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lan, Y.; Jiang, R. Molecular and Cellular Mechanisms of Palate Development. J. Dent. Res. 2017, 96, 1184–1191. [Google Scholar] [CrossRef]
- Barkhordarian, A.; Sison, J.; Cayabyab, R.; Mahanian, N.; Chiappelli, F. Epigenetic regulation of osteogenesis: Human embryonic palatal mesenchymal cells. Bioinformation 2011, 5, 278–281. [Google Scholar] [CrossRef]
- Seelan, R.S.; Mukhopadhyay, P.; Pisano, M.M.; Greene, R.M. Developmental epigenetics of the murine secondary palate. ILAR J. 2012, 53, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef]
- Grosswendt, S.; Kretzmer, H.; Smith, Z.D.; Kumar, A.S.; Hetzel, S.; Wittler, L.; Klages, S.; Timmermann, B.; Mukherji, S.; Meissner, A. Epigenetic regulator function through mouse gastrulation. Nature 2020, 584, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Greene, R.M.; Pisano, M.M. Cigarette smoke induces proteasomal-mediated degradation of DNA methyltransferases and methyl CpG-/CpG domain-binding proteins in embryonic orofacial cells. Reprod. Toxicol. 2015, 58, 140–148. [Google Scholar] [CrossRef]
- Kuriyama, M.; Udagawa, A.; Yoshimoto, S.; Ichinose, M.; Sato, K.; Yamazaki, K.; Matsuno, Y.; Shiota, K.; Mori, C. DNA methylation changes during cleft palate formation induced by retinoic acid in mice. Cleft Palate Craniofac. J. 2008, 45, 545–551. [Google Scholar] [CrossRef]
- Krauss, R.S.; Hong, M. Gene-Environment Interactions and the Etiology of Birth Defects. Curr. Top. Dev. Biol. 2016, 116, 569–580. [Google Scholar]
- Juriloff, D.M.; Harris, M.J. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 2008, 82, 63–77. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Casella, G.; Tsitsipatis, D.; Abdelmohsen, K.; Gorospe, M. mRNA methylation in cell senescence. Wiley Interdiscip Rev. RNA 2019, 10, e1547. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef]
- Ma, L.; Zhou, X.; Yao, S.; Zhang, X.; Mao, J.; Vona, B.; Fan, L.; Lou, S.; Li, D.; Wang, L.; et al. METTL3-dependent m(6)A modification of PSEN1 mRNA regulates craniofacial development through the Wnt/beta-catenin signaling pathway. Cell Death Dis. 2024, 15, 229. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Lei, H.; He, X.; Liu, Y.; Wang, A.; Ren, Z.; Liu, X.; Yan, G.; Wang, W.; Wang, Y.; et al. METTL14 Regulates Osteogenesis of Bone Marrow Mesenchymal Stem Cells via Inducing Autophagy Through m6A/IGF2BPs/Beclin-1 Signal Axis. Stem Cells Transl. Med. 2022, 11, 987–1001. [Google Scholar] [CrossRef]
- Tian, C.; Huang, Y.; Li, Q.; Feng, Z.; Xu, Q. Mettl3 Regulates Osteogenic Differentiation and Alternative Splicing of Vegfa in Bone Marrow Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 551. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y. Downregulation of METTL14 improves postmenopausal osteoporosis via IGF2BP1 dependent posttranscriptional silencing of SMAD1. Cell Death Dis. 2022, 13, 919. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, L.; Wang, M.; Xiong, Q.; Guo, Y.; Liang, Y.; Li, J.; Sheng, R.; Deng, P.; Wang, Y.; et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 2018, 9, 4772. [Google Scholar] [CrossRef] [PubMed]
- Radlanski, R.J.; Renz, H. Genes, forces, and forms: Mechanical aspects of prenatal craniofacial development. Dev. Dyn. 2006, 235, 1219–1229. [Google Scholar] [CrossRef]
- Ozekin, Y.H.; O’Rourke, R.; Bates, E.A. Single cell sequencing of the mouse anterior palate reveals mesenchymal heterogeneity. Dev. Dyn. 2023, 252, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Suzuki, A.; Iwaya, C.; Pei, G.; Chen, X.; Yoshioka, H.; Yu, M.; Simon, L.M.; Iwata, J.; Zhao, Z. Single-cell multiomics decodes regulatory programs for mouse secondary palate development. Nat. Commun. 2024, 15, 821. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.H.; Klezovitch, O.; Fernandez, T.E.; Delrow, J.; Vasioukhin, V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 2006, 311, 1609–1612. [Google Scholar] [CrossRef]
- Kappil, M.; Lambertini, L.; Chen, J. Environmental Influences on Genomic Imprinting. Curr. Environ. Health Rep. 2015, 2, 155–162. [Google Scholar] [CrossRef]
- Marazita, M.L. Gene×environment associations in orofacial clefting. Curr. Top. Dev. Biol. 2023, 152, 169–192. [Google Scholar] [PubMed]
- Garland, M.A.; Sun, B.; Zhang, S.; Reynolds, K.; Ji, Y.; Zhou, C.J. Role of epigenetics and miRNAs in orofacial clefts. Birth Defects Res. 2020, 112, 1635–1659. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, H.; Zhan, Z.; Zhang, X.; Zhang, M.; You, J.; Ma, J. Unveiling dysregulated lncRNAs and networks in non-syndromic cleft lip with or without cleft palate pathogenesis. Sci. Rep. 2024, 14, 1047. [Google Scholar] [CrossRef]
- Ma, L.; Shi, B.; Zheng, Q. Cell Polarity and PAR Complex Likely to Be Involved in Dexamethasone-Induced Cleft Palate. J. Craniofac. Surg. 2018, 29, 260–263. [Google Scholar] [CrossRef]
- Zhao, S.F.; Chai, M.Z.; Wu, M.; He, Y.H.; Meng, T.; Shi, B. Effect of vitamin B12 on cleft palate induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin and dexamethasone in mice. J. Zhejiang Univ. Sci. B 2014, 15, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Liu, X.; Cui, L.; Liu, X.; Chen, Y.; He, Z.; Ji, M.; Gao, Z.; Li, N.; Wan, Z.; et al. Oct4 plays a role in 2, 3, 7, 8—Tetrachlorobenzo-p-dioxin (TCDD) inducing cleft palate and inhibiting mesenchymal proliferation. Toxicology 2020, 438, 152444. [Google Scholar] [CrossRef] [PubMed]
- Abbott, B.D.; Harris, M.W.; Birnbaum, L.S. Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 1989, 40, 533–553. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; McCaffery, P.J.; Drager, U.C.; De Luca, L.M. Retinoids in embryonal development. Physiol. Rev. 2000, 80, 1021–1054. [Google Scholar] [CrossRef] [PubMed]
- Clagett-Dame, M.; DeLuca, H.F. The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr. 2002, 22, 347–381. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.H. New agents for the treatment of acute myelogenous leukemia: Focus on topotecan and retinoids. Leukemia 1998, 12 (Suppl. S1), S13–S15. [Google Scholar] [PubMed]
- Mi, J.Q.; Li, J.M.; Shen, Z.X.; Chen, S.J.; Chen, Z. How to manage acute promyelocytic leukemia. Leukemia 2012, 26, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Chandola, U.; Das, R.; Panda, B. Role of the N6-methyladenosine RNA mark in gene regulation and its implications on development and disease. Brief Funct Genom. 2015, 14, 169–179. [Google Scholar] [CrossRef]
- Chen, T.; Hao, Y.J.; Zhang, Y.; Li, M.M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L.; et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 2015, 16, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Cao, S.; Huang, Q.; Xia, L.; Deng, M.; Yang, M.; Jia, G.; Liu, X.; Shi, J.; Wang, W.; et al. The RNA N(6)-methyladenosine modification landscape of human fetal tissues. Nat. Cell Biol. 2019, 21, 651–661. [Google Scholar] [CrossRef]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadee, C.; et al. The SMAD2/3 interactome reveals that TGFbeta controls m(6)A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Jiang, D.; Wang, Y.; Wang, X. N (6)-Methyladenosine (m(6)A) Methylation in mRNA with A Dynamic and Reversible Epigenetic Modification. Mol. Biotechnol. 2016, 58, 450–459. [Google Scholar] [CrossRef]
- Wang, M.K.; Gao, C.C.; Yang, Y.G. Emerging Roles of RNA Methylation in Development. Acc. Chem. Res. 2023, 56, 3417–3427. [Google Scholar] [CrossRef]
- Chen, B.; Liu, S.; Zhang, W.; Xiong, T.; Zhou, M.; Hu, X.; Mao, H.; Liu, S. Profiling Analysis of N6-Methyladenosine mRNA Methylation Reveals Differential m6A Patterns during the Embryonic Skeletal Muscle Development of Ducks. Animals 2022, 12, 2593. [Google Scholar] [CrossRef] [PubMed]
- van Gool, J.D.; Hirche, H.; Lax, H.; De Schaepdrijver, L. Folic acid and primary prevention of neural tube defects: A review. Reprod. Toxicol. 2018, 80, 73–84. [Google Scholar] [CrossRef]
- Stephen, L.J.; Harden, C.; Tomson, T.; Brodie, M.J. Management of epilepsy in women. Lancet Neurol. 2019, 18, 481–491. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, J.; Ren, F.; Ji, C.; Aniagu, S.; Chen, T. PM2.5-induced extensive DNA methylation changes in the heart of zebrafish embryos and the protective effect of folic acid. Environ. Pollut. 2019, 255 Pt 3, 113331. [Google Scholar] [CrossRef]
- Buker, S.M.; Gurard-Levin, Z.A.; Wheeler, B.D.; Scholle, M.D.; Case, A.W.; Hirsch, J.L.; Ribich, S.; Copeland, R.A.; Boriack-Sjodin, P.A. A Mass Spectrometric Assay of METTL3/METTL14 Methyltransferase Activity. SLAS Discov. 2020, 25, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Kennedy, S.; Hajian, T.; Gibson, E.; Seitova, A.; Xu, C.; Arrowsmith, C.H.; Vedadi, M. A Radioactivity-Based Assay for Screening Human m6A-RNA Methyltransferase, METTL3-METTL14 Complex, and Demethylase ALKBH5. J. Biomol. Screen. 2016, 21, 290–297. [Google Scholar] [CrossRef]
- He, Y.; Wang, W.; Xu, X.; Yang, B.; Yu, X.; Wu, Y.; Wang, J. Mettl3 inhibits the apoptosis and autophagy of chondrocytes in inflammation through mediating Bcl2 stability via Ythdf1-mediated m(6)A modification. Bone 2022, 154, 116182. [Google Scholar] [CrossRef]
- Zhou, Y.; Zeng, P.; Li, Y.H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xu, H.; Wu, Y.; Wang, J.; Yuan, Q. In silico genome-wide identification of m6A-associated SNPs as potential functional variants for periodontitis. J. Cell. Physiol. 2020, 235, 900–908. [Google Scholar] [CrossRef]
- Zhu, R.; Tian, D.; Zhao, Y.; Zhang, C.; Liu, X. Genome-Wide Detection of m(6)A-Associated Genetic Polymorphisms Associated with Ischemic Stroke. J. Mol. Neurosci. 2021, 71, 2107–2115. [Google Scholar] [CrossRef]
- Thieme, F.; Ludwig, K.U. The Role of Noncoding Genetic Variation in Isolated Orofacial Clefts. J. Dent. Res. 2017, 96, 1238–1247. [Google Scholar] [CrossRef]
- Liu, H.; Leslie, E.J.; Carlson, J.C.; Beaty, T.H.; Marazita, M.L.; Lidral, A.C.; Cornell, R.A. Identification of common non-coding variants at 1p22 that are functional for non-syndromic orofacial clefting. Nat. Commun. 2017, 8, 14759. [Google Scholar] [CrossRef]
- Yu, Y.; Zuo, X.; He, M.; Gao, J.; Fu, Y.; Qin, C.; Meng, L.; Wang, W.; Song, Y.; Cheng, Y.; et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 2017, 8, 14364. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Nie, P.; Peng, D.; He, Z.; Liu, M.; Xie, Y.; Miao, Y.; Zuo, Z.; Ren, J. m6AVar: A database of functional variants involved in m6A modification. Nucleic Acids Res. 2018, 46, D139–D145. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar]
- Wang, F.; Zhu, Y.; Cai, H.; Liang, J.; Wang, W.; Liao, Y.; Zhang, Y.; Wang, C.; Hou, J. N6-Methyladenosine Methyltransferase METTL14-Mediated Autophagy in Malignant Development of Oral Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 738406. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total Number of Embryonic Mice | Number of Stillbirths/ Absorptions | Number of Embryonic Mice with Cleft Palate | Survival Rate (%) | Incidence of Cleft Palate (%) |
|---|---|---|---|---|---|
| RA10D & | 136 | 25 | 108 | 81.6% | 97.3% |
| RA12D | 116 | 8 | 92 | 93.1% | 85.2% |
| Control * | 105 | 0 | 0 | 100.0% | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Zhang, Y.; Jiang, Y.; Cai, H.; Liang, J.; Li, H.; Wang, C.; Hou, J. Retinoic Acid Upregulates METTL14 Expression and the m6A Modification Level to Inhibit the Proliferation of Embryonic Palate Mesenchymal Cells in Cleft Palate Mice. Int. J. Mol. Sci. 2024, 25, 4538. https://doi.org/10.3390/ijms25084538
Zhu Y, Zhang Y, Jiang Y, Cai H, Liang J, Li H, Wang C, Hou J. Retinoic Acid Upregulates METTL14 Expression and the m6A Modification Level to Inhibit the Proliferation of Embryonic Palate Mesenchymal Cells in Cleft Palate Mice. International Journal of Molecular Sciences. 2024; 25(8):4538. https://doi.org/10.3390/ijms25084538
Chicago/Turabian StyleZhu, Yue, Yadong Zhang, Yaoqi Jiang, Hongshi Cai, Jianfeng Liang, Hongyu Li, Cheng Wang, and Jinsong Hou. 2024. "Retinoic Acid Upregulates METTL14 Expression and the m6A Modification Level to Inhibit the Proliferation of Embryonic Palate Mesenchymal Cells in Cleft Palate Mice" International Journal of Molecular Sciences 25, no. 8: 4538. https://doi.org/10.3390/ijms25084538