
Role of Protein Tyrosine Phosphatase Receptor Type E (PTPRE) in Chemoresistant Retinoblastoma
 , and
, and 
Abstract
:1. Introduction
2. Results
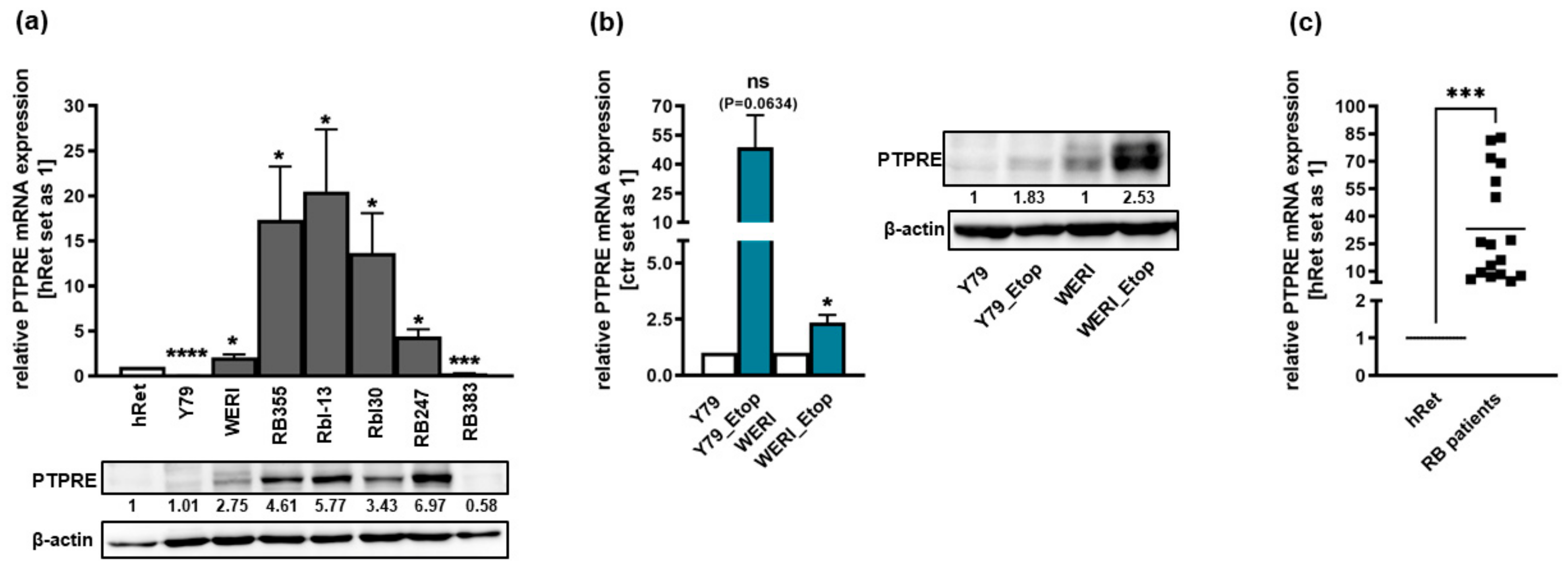
2.1. PTPRE Is Differentially Expressed in Retinoblastoma Cell Lines and Patient Tumors
2.2. PTPRE Expression Is Not Regulated by Promotor Methylation
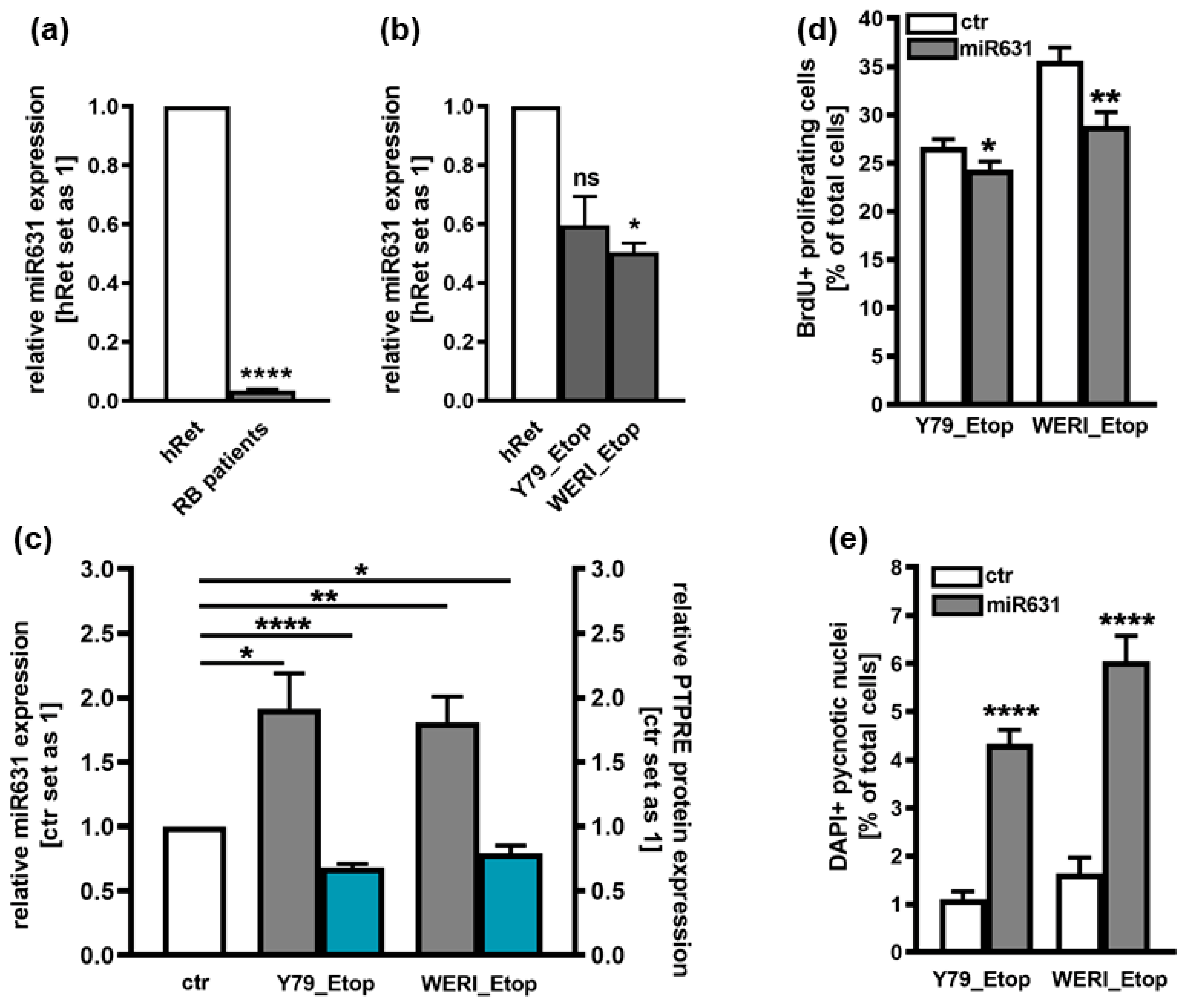
2.3. Involvement of miR631 in the Regulation of PTPRE Expression
2.4. Involvement of FGFb Signaling in the Regulation of PTPRE Expression
2.5. PTPRE Knockdown Influences Cell Viability, Proliferation and Growth of Etoposide-Resistant Y79 and WERI Retinoblastoma Cell Lines
2.6. PTPRE Knockdown Induces Caspase Dependent Apoptosis in Etoposide-Resistant RB Cell Lines
2.7. PTPRE Knockdown Reduces Anchorage-Independent Growth of Etoposide-Resistant RB Cell Lines
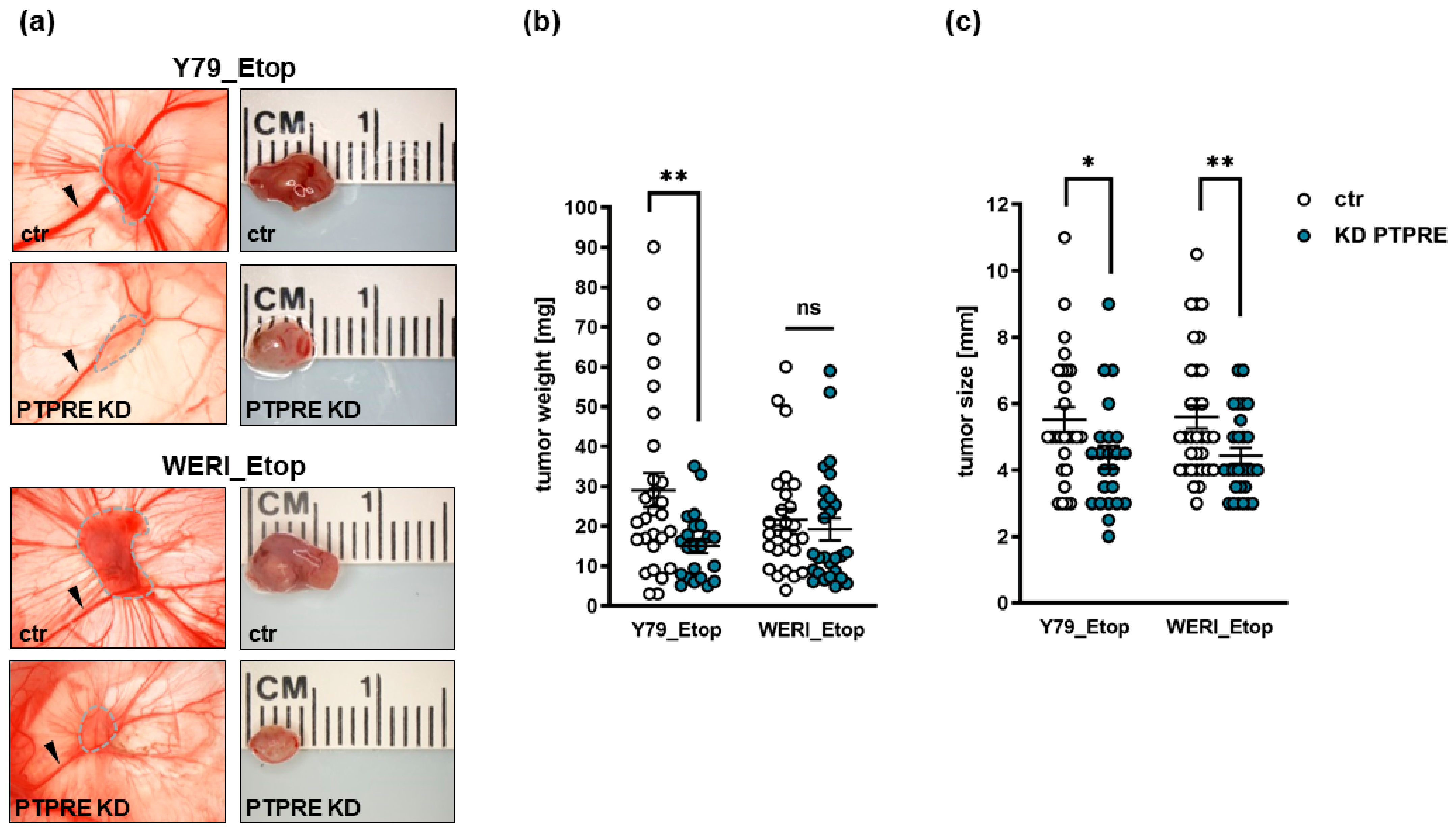
2.8. PTPRE Knockdown Decreases Tumorigenicity and Migration Potential of Etoposide-Resistant RB Cells In Vivo
2.9. Re-Sensitization of Resistant RB Cells towards Etoposide after PTPRE Knockdown
2.10. PTPRE Downstream Signaling
3. Discussion
4. Materials and Methods
4.1. Human Retina and Retinoblastoma Samples
4.2. Cell Lines and Culture
4.3. Lentiviral PTPRE Knockdown
4.4. PTPRE Promotor Analysis
4.5. Plasmids and miR631 Overexpression
4.6. FGF Receptor Inhibitor Studies
4.7. Re-Sensitization Studies
4.8. RNA Extraction and Quantitative Real-Time PCR
4.9. Western Blotting
4.10. Cell Viability Assays
4.11. Growth Kinetic
4.12. Cell Proliferation and Apoptosis Detection
4.13. Caspase Dependent Apoptosis
4.14. Colony Formation and Soft Agarose Assay
4.15. CAM Assays
4.16. Immunohistochemistry
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaewkhaw, R.; Rojanaporn, D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers 2020, 12, 2304. [Google Scholar] [CrossRef] [PubMed]
- Bornfeld, N.; Lohmann, D.; Bechrakis, N.E.; Biewald, E. Retinoblastom. Ophthalmol. Z. Dtsch. Ophthalmol. Ges. 2020, 117, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivelä, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Prog. Retin. Eye Res. 2019, 73, 100764. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.L.; Kogachi, K.; Aziz, H.A.; McGovern, K.; Zolfaghari, E.; Murphree, A.L.; Jubran, R.; Kim, J.W. Risk of metastasis and orbital recurrence in advanced retinoblastoma eyes treated with systemic chemoreduction versus primary enucleation. Pediatr. Blood Cancer 2017, 64, e26270. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Lally, S.E.; Leahey, A.M.; Jabbour, P.M.; Caywood, E.H.; Schwendeman, R.; Shields, J.A. Targeted retinoblastoma management: When to use intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Curr. Opin. Ophthalmol. 2014, 25, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.D.; Schündeln, M.M.; Biewald, E.; Astrahantseff, K.; Wieland, R.; Bornfeld, N.; et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr. Blood Cancer 2017, 64, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Grandis, J.R. Receptor-type protein tyrosine phosphatases in cancer. Chin. J. Cancer 2015, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.J.; Ter Steege, E.; den Hertog, J. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev. Biol. 2017, 428, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Berman-Golan, D.; Granot-Attas, S.; Elson, A. Protein tyrosine phosphatase epsilon and Neu-induced mammary tumorigenesis. Cancer Metastasis Rev. 2008, 27, 193–203. [Google Scholar] [CrossRef]
- Liang, J.; Shi, J.; Wang, N.; Zhao, H.; Sun, J. Tuning the protein phosphorylation by receptor type protein tyrosine phosphatase epsilon (PTPRE) in normal and cancer cells. J. Cancer 2019, 10, 105–111. [Google Scholar] [CrossRef]
- Frankson, R.; Yu, Z.-H.; Bai, Y.; Li, Q.; Zhang, R.-Y.; Zhang, Z.-Y. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [PubMed]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular pathways: Targeting protein tyrosine phosphatases in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Kabir, N.N.; Rönnstrand, L.; Kazi, J.U. Deregulation of protein phosphatase expression in acute myeloid leukemia. Med. Oncol. 2013, 30, 517. [Google Scholar] [CrossRef] [PubMed]
- LACZMANSKA, I.; LACZMANSKI, L.; SASIADEK, M.M. Expression analysis of tyrosine phosphatase genes at different stages of renal cell carcinoma. Anticancer Res. 2020, 40, 5667–5671. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.; Ye, R.; Ha, Y.; Xiong, Y.; Li, R.; Di, X.; Zou, Z.; Sun, Y.; Zhang, Z. Prediction biomarkers associated with lymph node metastasis and prognosis were identified in papillary thyroid carcinoma via integrated bioinformatics analysis. Comb. Chem. High Throughput Screen. 2021, 24, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Goryca, K.; Kunicki, J.; Bonicki, W.; Siedlecki, J.A.; Bujko, M. DNA methylation profiling in nonfunctioning pituitary adenomas. Mol. Cell. Endocrinol. 2018, 473, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liao, Z.; Qi, Y.; Zhang, H.; Su, C.; Liang, H.; Zhang, B.; Chen, X. miR-631 Inhibits intrahepatic metastasis of hepatocellular carcinoma by targeting PTPRE. Front. Oncol. 2020, 10, 565266. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.E.; Elson, A.; Pulido, R. Epidermal growth factor receptor (EGFR)-mediated positive feedback of protein-tyrosine phosphatase epsilon (PTPepsilon) on ERK1/2 and AKT protein pathways is required for survival of human breast cancer cells. J. Biol. Chem. 2012, 287, 3433–3444. [Google Scholar] [CrossRef] [PubMed]
- Gil-Henn, H.; Elson, A. Tyrosine phosphatase-epsilon activates Src and supports the transformed phenotype of Neu-induced mammary tumor cells. J. Biol. Chem. 2003, 278, 15579–15586. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yamada, N.; Shimizu, H.; Shiota, M.; Tamura, M.; Kim-Mitsuyama, S.; Miyazaki, H. Tyrosine phosphatase epsilonM stimulates migration and survival of porcine aortic endothelial cells by activating c-Src. Biochem. Biophys. Res. Commun. 2004, 325, 314–319. [Google Scholar] [CrossRef]
- Peretz, A.; Gil-Henn, H.; Sobko, A.; Shinder, V.; Attali, B.; Elson, A. Hypomyelination and increased activity of voltage-gated K(+) channels in mice lacking protein tyrosine phosphatase epsilon. EMBO J. 2000, 19, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Siffroi-Fernandez, S. Acidic Fibroblast Growth Factor (FGF-1) and FGF receptor 1 signaling in human Y79 retinoblastoma. Arch. Ophthalmol. 2005, 123, 368. [Google Scholar] [CrossRef] [PubMed]
- Wabakken, T.; Hauge, H.; Finne, E.F.; Wiedlocha, A.; Aasheim, H. Expression of human protein tyrosine phosphatase epsilon in leucocytes: A potential ERK pathway-regulating phosphatase. Scand. J. Immunol. 2002, 56, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. Second AKT: The rise of SGK in cancer signalling. Growth Factors 2010, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xu, Z.; Wang, J.; Cigliano, A.; Pilo, M.G.; Ribback, S.; Zhang, S.; Qiao, Y.; Che, L.; Pascale, R.M.; et al. Functional role of SGK3 in PI3K/Pten driven liver tumor development. BMC Cancer 2019, 19, 343. [Google Scholar] [CrossRef] [PubMed]
- Raji, L.; Tetteh, A.; Amin, A.R. Role of c-Src in carcinogenesis and drug resistance. Cancers 2023, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Lu, Z.; Lai, K.; Li, D.; Hao, B.; Xu, C.; Pan, S.; Li, N.; Geng, Q. Identification of an inflammatory response signature associated with prognostic stratification and drug sensitivity in lung adenocarcinoma. Sci. Rep. 2022, 12, 10110. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, C.; Yu, W.; Li, L.; Zhang, Z.; Liu, T.; Zhang, Y.; Fan, G.; Huangfu, H. Receptor Type Protein Tyrosine Phosphatase Epsilon (PTPRE) plays an oncogenic role in thyroid carcinoma by activating the AKT and ERK1/2 signaling pathway. Curr. Cancer Drug Targets 2023, 23, 471–481. [Google Scholar]
- Fu, D.; Liu, B.; Zang, L.E.; Jiang, H. MiR-631/ZAP70: A novel axis in the migration and invasion of prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 469, 345–351. [Google Scholar] [CrossRef]
- Lv, H.; Yu, J.; Zhang, H.; Qian, X.; Wang, Q.; Lu, B.; Sun, Y. MicroRNA-631 deriving from bone marrow mesenchymal stem cell exosomes facilitates the malignant behavior of non-small cell lung cancer via modulating the E2F family of transcription factor 2/phosphatidylinositol 3-kinase/Akt signaling pathway. Bioengineered 2022, 13, 8382–8395. [Google Scholar] [CrossRef] [PubMed]
- Najafi, F.; Kelaye, S.K.; Kazemi, B.; Foruzandeh, Z.; Allahverdizadeh, F.; Vakili, S.; Rad, K.K.; Derakhshani, M.; Solali, S.; Alivand, M.R. The role of miRNA-424 and miR-631 in various cancers: Focusing on drug resistance and sensitivity. Pathol. Res. Pract. 2022, 239, 154130. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, F.; Turcotte, S.; Roy, J.; Bissonnette, E.; Rola-Pleszczynski, M.; Stankova, J. RPTPε promotes M2-polarized macrophage migration through ROCK2 signaling and podosome formation. J. Cell Sci. 2020, 133, jcs234641. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Chen, L.; Zhang, X.; Zhang, H.; Tan, X.; Dong, K.; Lu, X.; Zhu, H.; Liu, Q.; Zhang, Z.; et al. PTPRε Acts as a metastatic promoter in hepatocellular carcinoma by facilitating recruitment of SMAD3 to TGF-β receptor 1. Hepatology 2020, 72, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Freeman, M.J.; Lu, H.; Wang, X.; Forster, C.L.; Sarver, A.L.; Hallstrom, T.C. Retinoblastoma cells activate the AKT pathway and are vulnerable to the PI3K/mTOR inhibitor NVP-BEZ235. Oncotarget 2017, 8, 38084–38098. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. AKT-independent PI3-K signaling in cancer-emerging role for SGK3. Cancer Manag. Res. 2013, 5, 281–292. [Google Scholar] [PubMed]
- Dong, R.; Wang, T.; Dong, W.; Zhu, H.; Liu, Q.; Liang, H.; Chen, X.; Zhang, B.; Zhang, X. Inhibition of PTPRE suppresses tumor progression and improves sorafenib response in hepatocellular carcinoma. Biomed. Pharmacother. 2024, 173, 116366. [Google Scholar] [CrossRef] [PubMed]
- Griegel, S.; Hong, C.; Frötschl, R.; Hülser, D.F.; Greger, V.; Horsthemke, B.; Rajewsky, M.F. Newly established human retinoblastoma cell lines exhibit an “immortalized” but not an invasive phenotype in vitro. Int. J. Cancer 1990, 46, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.W.; Albert, D.M.; Rabson, A.S.; Russell, P.; Craft, J.; Chu, E.W.; Tralka, T.S.; Wilcox, J.L. Characteristics of an established cell line of retinoblastoma. J. Nat. Cancer Inst. 1974, 53, 347–360. [Google Scholar] [CrossRef]
- McFall, R.C.; Sery, T.W.; Makadon, M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res. 1977, 37, 1003–1010. [Google Scholar]
- Busch, M.; Papior, D.; Stephan, H.; Dünker, N. Characterization of etoposide- and cisplatin-chemoresistant retinoblastoma cell lines. Oncol. Rep. 2017, 39, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.; Neveling, K.; Borkens, S.; Schneider, H.; Freund, M.; Grassman, E.; Theiss, S.; Wawer, A.; Burdach, S.; Auerbach, A.D.; et al. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am. J. Hum. Genet. 2010, 87, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Große-Kreul, J.; Busch, M.; Winter, C.; Pikos, S.; Stephan, H.; Dünker, N. Forced Trefoil Factor Family Peptide 3 (TFF3) expression reduces growth, viability, and tumorigenicity of human retinoblastoma cell lines. PLoS ONE 2016, 11, e0163025. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Philippeit, C.; Weise, A.; Dünker, N. Re-characterization of established human retinoblastoma cell lines. Histochem. Cell Biol. 2015, 143, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Weise, A.; Dünker, N. High trefoil factor 1 (TFF1) expression in human retinoblastoma cells correlates with low growth kinetics, increased cyclin-dependent kinase (CDK) inhibitor levels and a selective down-regulation of CDK6. Histochem. Cell Biol. 2013, 139, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, A.; Mellor, R.; Panzarella, G.; Aimes, R.T.; Hooper, J.D.; Marchenko, N.D.; Quigley, J.P. A quantitative analysis of rate-limiting steps in the metastatic cascade using human-specific real-time polymerase chain reaction. Cancer Res. 2002, 62, 7083–7092. [Google Scholar] [PubMed]
- Palmer, T.D.; Lewis, J.; Zijlstra, A. Quantitative analysis of cancer metastasis using an avian embryo model. J. Vis. Exp. JoVE 2011, 51, e2815. [Google Scholar]
- Kim, J.; Yu, W.; Kovalski, K.; Ossowski, L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell 1998, 94, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Dräger, O.; Metz, K.; Busch, M.; Dünker, N. Role of L1CAM in retinoblastoma tumorigenesis: Identification of novel therapeutic targets. Mol. Oncol. 2022, 16, 957–981. [Google Scholar] [CrossRef]
- Van Meenen, D.; Doege, A.; Alefeld, E.; Haase, A.; Beier, M.; Kiefer, T.; Biewald, E.; Metz, K.; Dräger, O.; Busch, M.A.; et al. ADAM10 and ADAM17-novel players in retinoblastoma carcinogenesis. Int. J. Mol. Sci. 2022, 23, 12621. [Google Scholar] [CrossRef]
- Elso, C.M.; Roberts, L.J.; Smyth, G.K.; Thomson, R.J.; Baldwin, T.M.; Foote, S.J.; Handman, E. Leishmaniasis host response loci (lmr1-3) modify disease severity through a Th1/Th2-independent pathway. Genes Immun. 2004, 5, 93–100. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Company | Dilution |
|---|---|---|
| PTPRE (Ma5-25072) | Thermofisher Scientific, Darmstadt, Germany | 1:1000 |
| SRC (#2105) | Cell signaling technology, Danvers, MA, USA | 1:1000 |
| pSRC (#2123) | Cell signaling technology, Danvers, MA, USA | 1:1000 |
| ERK (#9102) | Cell signaling technology, Danvers, MA, USA | 1:1000 |
| pERK (#4370) | Cell signaling technology, Danvers, MA, USA | 1:1000 |
| AKT (#4685) | Cell signaling technology, Danvers, MA, USA | 1:1000 |
| pAKT (#9271) | Cell signaling technology, Danvers, MA, USA | 1:500 |
| SGK3 (Sc-166847) | Santa Cruz Biotechnology, Dallas, TX, USA | 1:500 |
| pSGK3 (#5642) | Cell signaling technology, Danvers, MA, USA | 1:500 |
| FGFb (ab215373) | Abcam, Berlin, Germany | 1:1000 |
| ß-actin (#4967) | Cell signaling technology, Danvers, MA, USA | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohren, L.; Doege, A.; Miroschnikov, N.; Dräger, O.; Busch, M.A.; Dünker, N. Role of Protein Tyrosine Phosphatase Receptor Type E (PTPRE) in Chemoresistant Retinoblastoma. Int. J. Mol. Sci. 2024, 25, 4572. https://doi.org/10.3390/ijms25084572
Mohren L, Doege A, Miroschnikov N, Dräger O, Busch MA, Dünker N. Role of Protein Tyrosine Phosphatase Receptor Type E (PTPRE) in Chemoresistant Retinoblastoma. International Journal of Molecular Sciences. 2024; 25(8):4572. https://doi.org/10.3390/ijms25084572
Chicago/Turabian StyleMohren, Lars, Annika Doege, Natalia Miroschnikov, Oliver Dräger, Maike Anna Busch, and Nicole Dünker. 2024. "Role of Protein Tyrosine Phosphatase Receptor Type E (PTPRE) in Chemoresistant Retinoblastoma" International Journal of Molecular Sciences 25, no. 8: 4572. https://doi.org/10.3390/ijms25084572