
Comparative Analysis of Gut Bacterial Community Composition in Two Tropical Economic Sea Cucumbers under Different Seasons of Artificial Environment
 ,
,
Abstract
:1. Introduction
2. Results
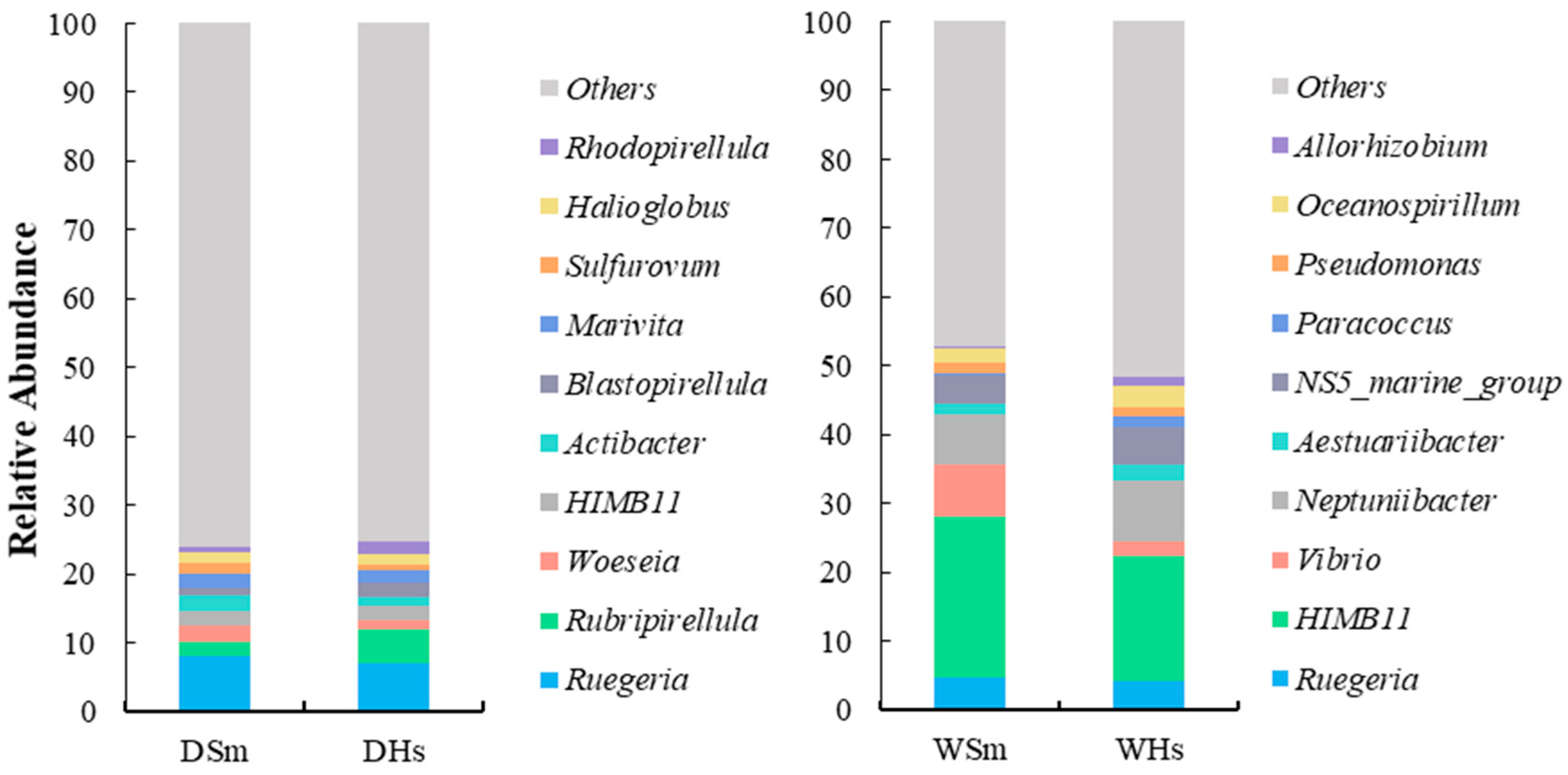
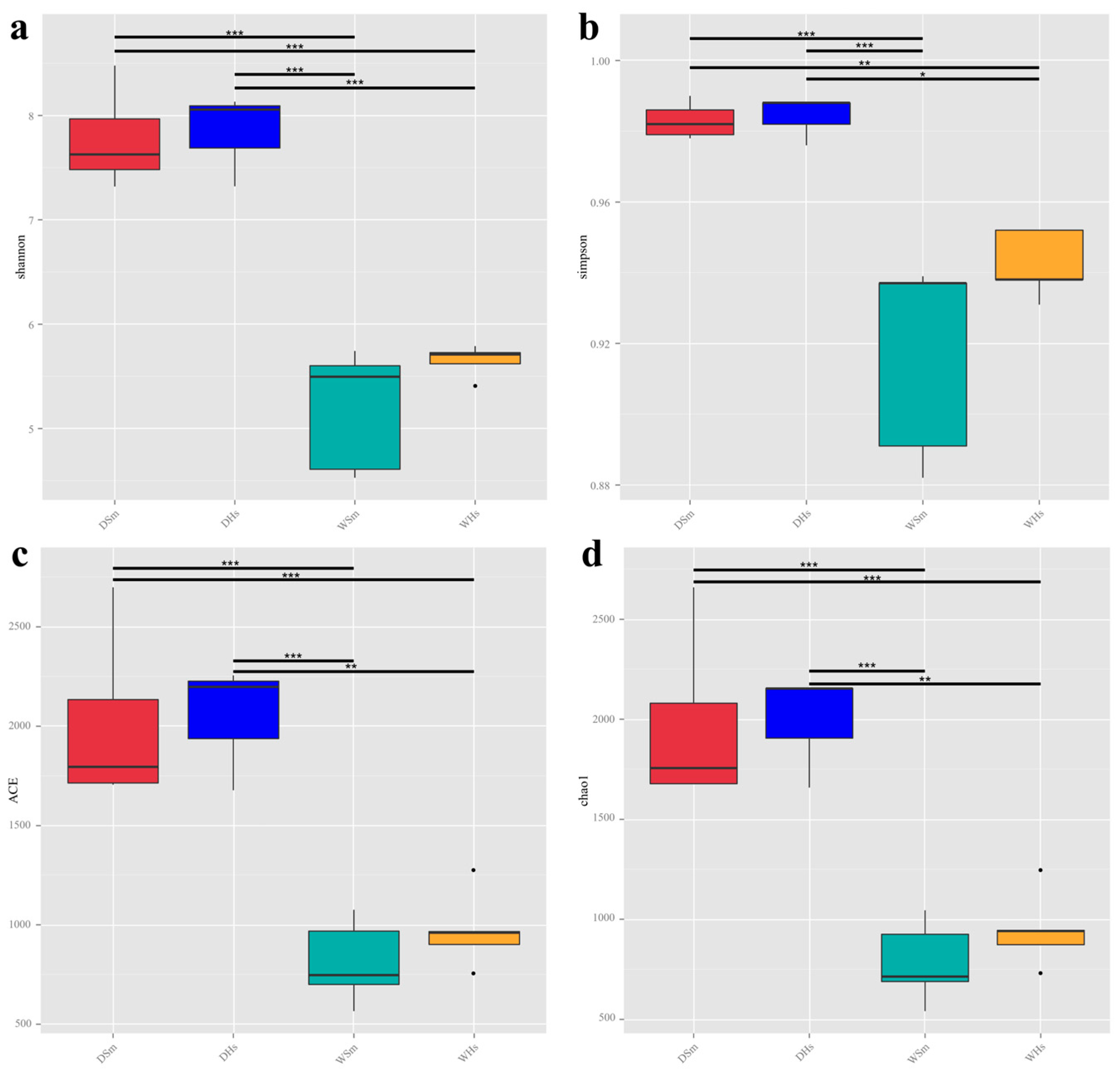
2.1. Composition and Diversity of Gut Bacteria
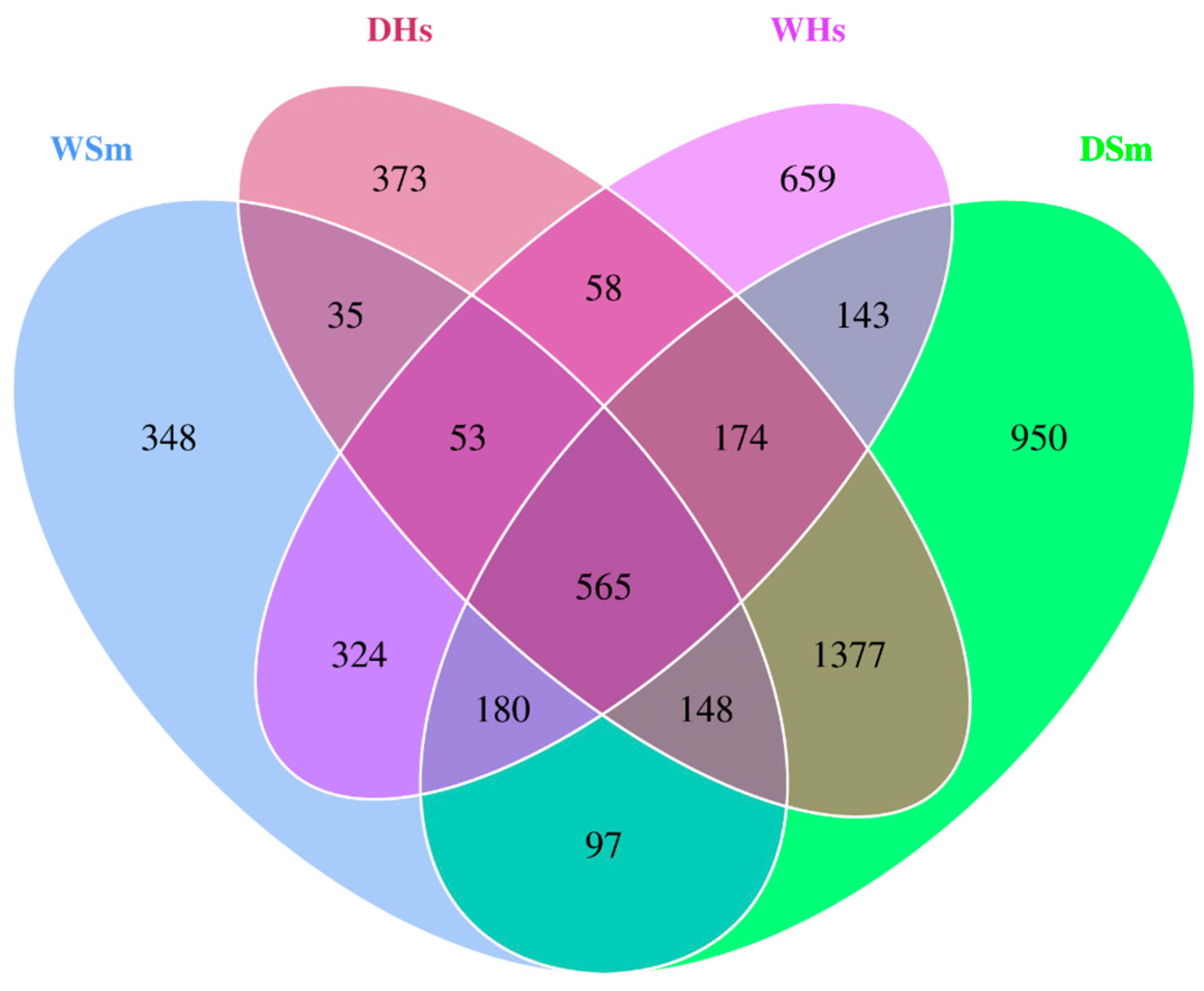
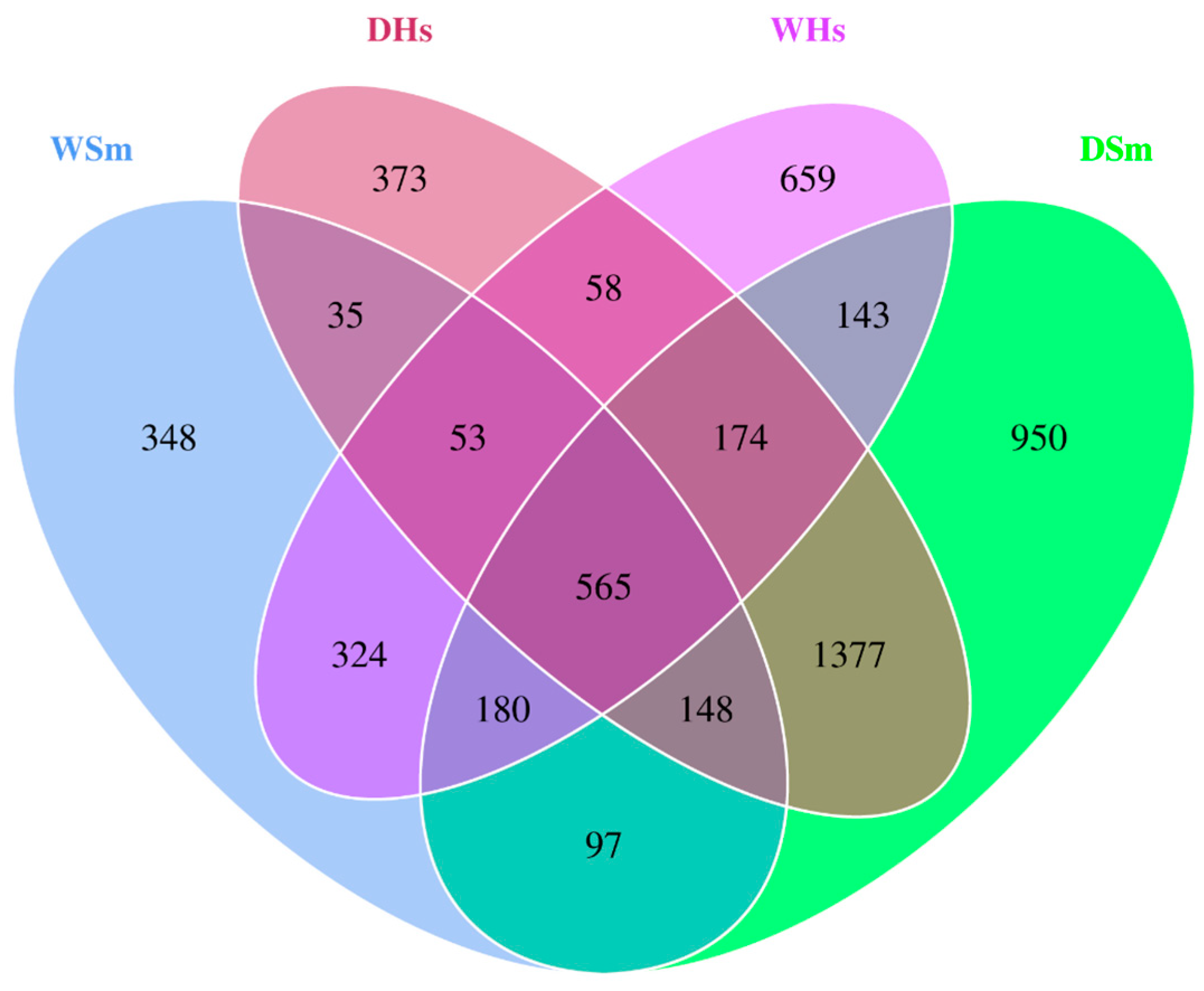
2.2. Analysis of Core Bacterial Community Shared by All Samples
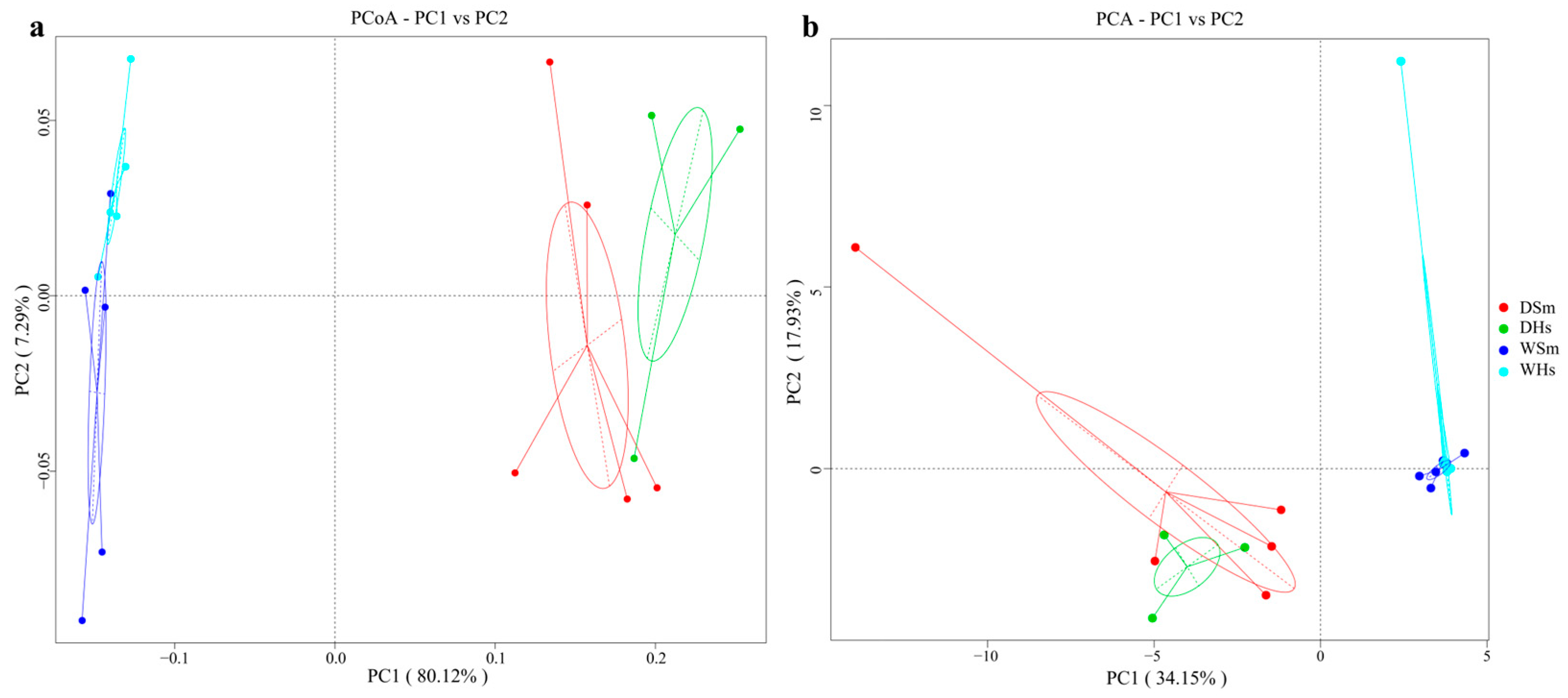
2.3. Analysis of the Gut Bacterial Distinctions of Sea Cucumbers in Different Seasons
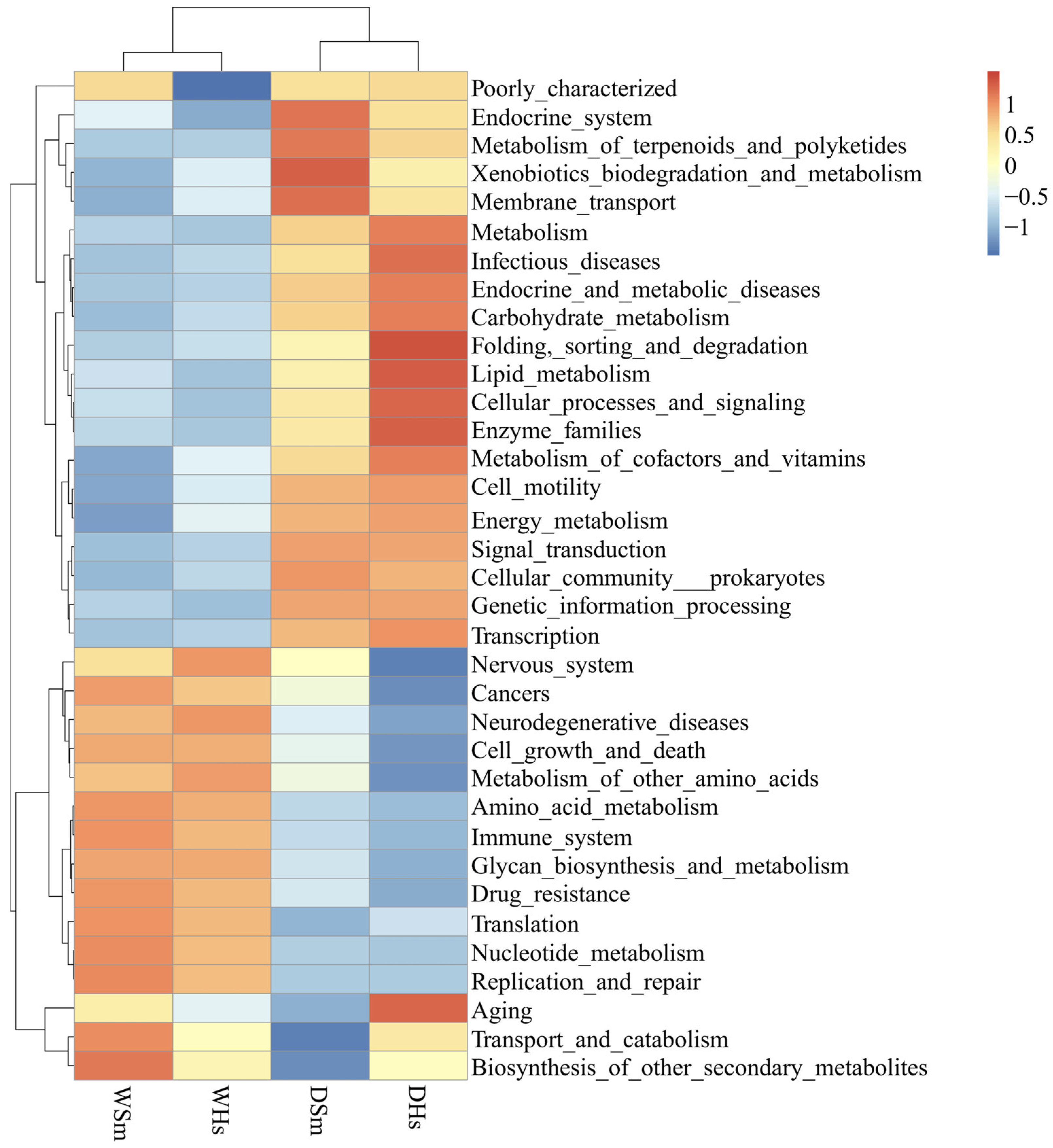
2.4. Gene Function Prediction of Gut Bacterial Community
3. Discussion
3.1. Dominant Gut Bacterial Community
3.2. Comparison of Gut Bacterial Communities among Different Seasons
3.3. The Functional Prediction of Bacteria in the Gut of Sea Cucumber
4. Materials and Methods
4.1. Sample Collection
4.2. PCR Amplification and High-Throughput Sequencing
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liao, Y. Fauna Sinica: Echinoderma: Holothuroidea, 1st ed.; Science Press: Beijing, China, 1997; pp. 99–156. (In Chinese) [Google Scholar]
- Purcell, S.W.; Lovatelli, A.; Vasconcellos, M.; Ye, Y. Managing Sea Cucumber Fisheries with An Ecosystem Approach, 1st ed.; FAO: Rome, Italy, 2010; p. 15. [Google Scholar]
- Purcell, S.W.; Samyn, Y.; Conand, C. Commercially Important Sea Cucumbers of the World. In FAO Species Catalogue for Fishery Purposes No.6; Angelis, N.D., Lovatelli, A., Eds.; FAO: Rome, Italy, 2012; pp. 6+80–107. [Google Scholar]
- WoRMS. Holothuroidea. 2024. Available online: https://www.marinespecies.org/aphia.php?p=taxdetails&id=123083 (accessed on 3 January 2024).
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Zhang, X.; Nakahara, T.; Miyazaki, M.; Nogi, Y.; Kudo, T. Diversity and function of aerobic culturable bacteria in the intestine of the sea cucumber Holothuria leucospilota. J. Gen. Appl. Microbiol. 2012, 58, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tomomi, N.; Shinji, M.; Hideaki, N.; Tetsushi, I.; Toshiaki, K. Physiological characterization of aerobic culturable bacteria in the intestine of the sea cucumber Apostichopus japonicus. J. Gen. Appl. Microbiol. 2013, 59, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Liang, Z. Research status of utilization value of intestinal flora of sea cucumber. Food Res. Dev. 2021, 42, 184–188, (In Chinese with English abstract). [Google Scholar]
- Gerlach, S.A. Food-chain relationships in subtidal silty sand marine sediments and the role of meiofauna in stimulating bacterial productivity. Oecologia 1978, 33, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sun, H.; Xu, Q.; Tian, J.; Wang, Q. PCR-DGGE analysis of bacterial community composition in the gut contents of Apostichopus japonicus. J. Fish. Sci. China 2010, 17, 671–680. [Google Scholar]
- Moriarty, D. Feeding of Holothuria atra and Stichopus chloronotus on bacteria, organic carbon and organic nitrogen in sediments of the Great Barrier Reef. Mar. Freshw. Res. 1982, 33, 255–263. [Google Scholar] [CrossRef]
- Fenchel, M.T. Detritus food chains of aquatic ecosystems: The role of bacteria. In Advances in Microbial Ecology; Springer: Berlin/Heidelberg, Germany, 1977; pp. 1–58. [Google Scholar]
- Deming, J.W.; Colwell, R.R. Barophilic bacteria associated with digestive tracts of abyssal holothurians. Appl. Environ. Microbiol. 1982, 44, 1222. [Google Scholar] [CrossRef] [PubMed]
- Amaro, T.; Witte, H.; Herndl, G.J.; Cunha, M.R.; Billett, D.S.M. Deep-sea bacterial communities in sediments and guts of deposit-feeding holothurians in Portuguese canyons (NE Atlantic). Deep-Sea Res. Part I 2009, 56, 1834–1843. [Google Scholar] [CrossRef]
- Zhou, H.; Ma, H.; Zhang, W.; Xu, W.; Mai, K. Effects of potential probiotics on growth performance and immune response of the juvenile sea cucumber (Apostichopus japonicus). J. Fish. China 2010, 34, 775–783, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Byrne, M.; Rowe, F.; Uthicke, S. Molecular taxonomy, phylogeny and evolution in the family Stichopodidae (Aspidochirotida: Holothuroidea) based on COI and 16S mitochondrial DNA. Mol Phylogenet. Evol. 2010, 56, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Yaghmour, F.; Whittington-Jones, B. First record of Holothuria (Metriatyla) scabra Jaeger, 1833 (Echinodermata: Holothuroidea) from the coastal waters of the United Arab Emirates. PeerJ 2018, 6, e5555. [Google Scholar] [CrossRef] [PubMed]
- Asha, P.S.; Diwakar, K. Effect of stocking density on the hatching rate, larval and early juvenile rearing of edible sea cucumber Holothuria scabra (Jaeger, 1883). Indian J. Geo-Mar. Sci. 2013, 42, 191–195. [Google Scholar]
- Jia, Z.; Yu, G.; Qin, C.; Huang, J.; Bai, D. Research status of tropical sea cucumber breeding and culture. Guangdong Agric. Sci. 2013, 18, 120–122, (In Chinese with English abstract). [Google Scholar]
- Chen, Y.F.; Hu, C.Q.; Ren, C.H. Sole and binary fresh waste from shrimp (Litopenaeus vannamei) forculture of sea cucumber (Stichopus monotuberculatus). South China Fish. Sci. 2014, 10, 1–8. [Google Scholar]
- Feng, Y.; Weng, W.; Fang, Z. Large-scale breeding techniques of rough sea cucumber Holothuria scabra. Fish. Sci. 2021, 40, 750–756, (In Chinese with English abstract). [Google Scholar]
- Cheng, C.; Wu, F.; Ren, C.; Jiang, X.; Zhang, X.; Li, X. Aquaculture of the tropical sea cucumber, Stichopus monotuberculatus: Induced spawning, detailed records of gonadal and embryonic development, and improvements in larval breeding by digestive enzyme supply in diet. Aquaculture 2021, 540, 736690. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Dougal, K.; Fuente, G.D.L.; Harris, P.A.; Girdwood, S.E.; Pinioche, E.; Newbold, J. Identification of a core bacterial community within the large intestine of the horse. PLoS ONE 2013, 8, e77660. [Google Scholar] [CrossRef]
- Hu, F.; Chang, J.; Tong, Q.; Yu, J.; Niu, H. High-throughput sequencing analysis of intestinal flora diversity of two freshwater snails (Radix auricularia and Planorbella trivolvis). Chin. J. Biotechnol. 2020, 36, 2622–2634, (In Chinese with English abstract). [Google Scholar]
- Tzuc, J.; Escalante, D.; Herrera, R.; Cortés, G.; Ortiz, M. Microbiota from Litopenaeus vannamei: Digestive Tract Microbial Community of Pacific White Shrimp (Litopenaeus vannamei); Springer Plus: Berlin/Heidelberg, Germany, 2014; Volume 3, p. 280. [Google Scholar]
- Rungrassamee, W.; Klanchui, A.; Maibunkaew, S.; Chaiyapechara, S.; Karoonuthaisiri, N. Characterization of intestinal bacteria in wild and domesticated adult black tiger shrimp (Penaeus monodon). PLoS ONE 2014, 9, e91853. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Pan, L.; Huang, F.; Song, M.; Tian, C.; Zhang, M. Metagenomic insights into the structure and function of intestinal microbiota of the farmed Pacific white shrimp (Litopenaeus vannamei). Aquaculture 2019, 499, 109–118. [Google Scholar] [CrossRef]
- Zeng, C.; Lin, M.; Li, Z.; Ma, Y.; Wang, S. The structural and functional characteristics of the gut microbiota of Marsupenaeus japonicus as revealed by 16S rRNA gene amplicon sequencing. Microbiol. China 2020, 47, 1857–1866, (In Chinese with English abstract). [Google Scholar]
- Wang, Y.; Zhang, Y.; Jia, C.; Xu, Q.; Rong, Y.; Xu, Z.; Wang, Y.; Gao, F. Comparative analysis of gut microbial community structure of three tropical sea cucumber species. Diversity 2023, 15, 855. [Google Scholar] [CrossRef]
- Sha, Y.; Liu, M.; Wang, B.; Jiang, K.; Sun, G.; Wang, L. Gut bacterial diversity of farmed sea cucumbers Apostichopus japonicus with different growth rates. Microbiology 2016, 85, 109–115. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, X.; Chen, M.; Li, W.; Zhang, P. Comparison of intestinal microbiota and activities of digestive and immune-related enzymes of sea cucumber Apostichopus japonicus in two habitats. J. Oceanol. Limnol. 2018, 36, 990–1001. [Google Scholar] [CrossRef]
- Ward-Rainey, N.; Rainey, F.A.; Stackebrandt, E. A study of the bacterial flora associated with Holothuria atra. J. Exp. Mar. Biol. Ecol. 1996, 203, 11–26. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Y.; Wu, P.; Chen, M.; He, L.; Xu, Q.; Wang, A. Bacterial community composition in gut content and ambient sediment of two tropical wild sea cucumbers (Holothuria atra and H. leucospilota). J. Oceanol. Limnol. 2022, 40, 360–372. [Google Scholar] [CrossRef]
- Miura, N.; Motone, K.; Takagi, T.; Aburaya, S.; Watanabe, S.; Aoki, W.; Ueda, M. Ruegeria sp. strains isolated from the reef-building coral Galaxea fascicularis inhibit growth of the temperature-dependent pathogen Vibrio coralliilyticus. Mar. Biotechnol. 2019, 21, 1–8. [Google Scholar] [CrossRef]
- Kitamura, R.; Miura, N.; Ito, M.; Takagi, T.; Yamashiro, H.; Nishikawa, Y.; Nishimura, Y.; Kobayashi, K.; Kataoka, M. Specific detection of coral-associated Ruegeria, a potential probiotic bacterium, in corals and subtropical seawater. Mar. Biotechnol. 2021, 23, 576–589. [Google Scholar] [CrossRef]
- Dittnann, K.K. Interaction between Fish Probiotic Roseobacters and the Natural Microbiota in Aquaculture Settings. Ph.D. Thesis, Technical University of Denmark, Copenhagen, Denmark, 2019. [Google Scholar]
- Durham, B.; Grote, J.; Whittaker, K. Draft genome sequence of marine alphaproteobacterial strain HIMB11, the first cultivated representative of a unique lineage within the Roseobacter clade possessing an unusually small genome. BioMed. Cent. 2014, 9, 632–645. [Google Scholar] [CrossRef]
- Plotieau, T.; Lavitra, T.; Lavitra, C.; Eeckhaut, I. Bacterial diversity of the sediments transiting through the gut of Holothuria scabra (Holothuroidea; Echinodermata). Mar. Biol. 2013, 160, 3087–3101. [Google Scholar] [CrossRef]
- Gao, F.; Li, F.; Tan, J.; Yan, J.; Sun, H. Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by 16S rRNA gene pyrosequencing. PLoS ONE 2014, 9, e100092. [Google Scholar] [CrossRef]
- Gao, F.; Tan, J.; Sun, H.L.; Yan, J.P. Bacterial diversity of gut content in sea cucumber (Apostichopus japonicus) and its habitat surface sediment. J. Ocean Univ. China 2014, 13, 303–310. [Google Scholar] [CrossRef]
- Gao, F.; Xu, Q.; Yang, H. Seasonal variations of food sources in Apostichopus japonicus indicated by fatty acid biomarkers analysis. J. Fish. China 2010, 34, 760–767, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Feng, J.; Zhang, L.; Tang, X.; Xia, X.; Hu, W.; Zhou, P. Season and geography induced variation in sea cucumber (Stichopus japonicus) nutritional composition and gut microbiota. J. Food Compos. Anal. 2021, 101, 103838. [Google Scholar] [CrossRef]
- Tiralerdpanich, P.; Nasaree, S.; Pinyakong, O.; Sonthiphand, P. Variation of the mangrove sediment microbiomes and their phenanthrene biodegradation rates during the dry and wet seasons. Environ. Pollut. 2021, 289, 11749. [Google Scholar] [CrossRef]
- Wang, L.; Liang, Z.; Guo, Z.; Cong, W.; Song, M.; Wang, Y.; Jiang, Z. Response mechanism of microbial community to seasonal hypoxia in marine ranching. Sci. Total Environ. 2022, 811, 152387. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, Y.; Xu, Q.; Sun, C.; Wang, Y.; Gao, F. Comparative analysis of in situ eukaryotic food sources in three tropical sea cucumber species by metabarcoding. Animals 2022, 12, 2303. [Google Scholar] [CrossRef]
- Xu, N.; Wang, W.; Xu, K.; Xu, Y.; Ji, D.; Chen, C.; Xie, C. Cultivation of different seaweed species and seasonal changes cause divergence of the microbial community in coastal seawaters. Front. Microbiol. 2024, 13, 988743. [Google Scholar] [CrossRef]
- Thornton, D. Dissolved organic matter (DOM) release by phytoplankton in the contemporary and future ocean. Eur. J. Phycol. 2014, 49, 20–46. [Google Scholar] [CrossRef]
- Li, B.; Rong, X.; Liao, M.; Zhang, Z.; Wang, Y.; Wang, L.; Liu, Z.; Xue, T. Annual changes of total heterotrophic bacteria and Vibrios in the intestine of Apostichopus japonicas and its culture pond. Mar. Sci. 2012, 36, 63–67. [Google Scholar]
- Zhang, C.; Liang, W.; Zhang, W.; Li, C. Characterization of a metalloprotease involved in Vibrio splendidus infection in the sea cucumber, Apostichopus japonicus. Microb. Pathog. 2016, 101, 96–103. [Google Scholar] [CrossRef]
- Ndi, O.; Barton, M. Resistance determinants of Pseudomonas species from aquaculture in Australia. J. Aquac. Res. Dev. 2012, 3, 1000119. [Google Scholar] [CrossRef]
- Kang, S.; Asaf, S.; Khan, A.; Lubna; Khan, A.; Mun, B.; Khan, M.; Gul, H.; Lee, I. Complete Genome Sequence of Pseudomonas psychrotolerans CS51, a Plant Growth-Promoting Bacterium, Under Heavy Metal Stress Conditions. Microorganisms 2020, 8, 382. [Google Scholar] [CrossRef]
- De Schrijver, R.; Ollevier, F. Protein digestion in juvenile turbot (Scophthalmus maximus) and effects of dietary administration of Vibrio proteolyticus. Aquaculture 2000, 186, 107–116. [Google Scholar] [CrossRef]
- Verschuere, L.; Heang, H.; Criel, G.; Sorgeloos, P.; Verstraete, W. Selected bacterial strains protect Artemia spp. from the pathogenic effects of Vibrio proteolyticus CW8T2. Appl. Environ. Microbiol. 2000, 66, 1139–1146. [Google Scholar] [CrossRef]
- Pankaj, G.; Divya, B.; Kumar, M. Isolation of malathion degrading Pseudomonas xanthomarina with plant growth promoting activity. Res. J. Chem. Environ. 2013, 17, 59–66. [Google Scholar]
- Crovadore, J.; Cochard, B.; Calmin, G.; Chablais, R.; Schulz, T.; Lefort, F. Whole-genome sequence of Pseudomonas xanthomarina strain UASWS0955, a potential biological agent for agricultural and environmental uses. Genome Announc. 2016, 4, e01136-16. [Google Scholar] [CrossRef]
- Sopeña, F.; Laiz, L.; Morillo, E.; Sanchez-Trujillo, M.; Villaverde, J.; Jurado, V.; Saiz-Jimenez, C. Phenanthrene Biodegradation by Pseudomonas xanthomarina Isolated from an Aged Contaminated Soil. Clean Soil Air Water 2014, 42, 785–790. [Google Scholar] [CrossRef]
- Abdullahi, R.; Lihan, S.; Carlos, B.; Maurice, B.; Michelle, K.; Felecia, C. Detection of oprL gene and antibiotic resistance of Pseudomonas aeruginosa from aquaculture environment. Eur. J. Exp. Biol. 2013, 3, 148–152. [Google Scholar]
- Igbinosa, H.; Beshiru, A.; Igbinosa, E. Antibiotic resistance profile of Pseudomonas aeruginosa isolated from aquaculture and abattoir environments in urban communities. Asian Pac. J. Trop. Dis. 2017, 7, 47–52. [Google Scholar] [CrossRef]
- Gomez-Gil, B.; Thompson, F.; Thompson, C.; Garcia-Gasca, A.; Roque, A.; Swings, J. Vibrio hispanicus sp. nov., isolated from Artemia sp. and sea water in Spain. Int. J. Syst. Evol. Microbiol. 2004, 54, 261–265. [Google Scholar] [CrossRef]
- Ray, A.; Kinch, L.; Santos, M.; Grishin, N.; Salomon, D. Proteomics Analysis Reveals Previously Uncharacterized Virulence Factors in Vibrio proteolyticus. mBio 2016, 7, 01077-16. [Google Scholar] [CrossRef]
- Gómez-León, J.; Villamil, L.; Lemos, M.; Novoa, B.; Figueras, A. Isolation of Vibrio alginolyticus and Vibrio splendidus from Aquacultured Carpet Shell Clam (Ruditapes decussatus) Larvae Associated with Mass Mortalities. Appl. Environ. Microbiol. 2005, 71, 98–104. [Google Scholar] [CrossRef]
- Lorenz, N.; Reiger, M.; Toro-Nahuelpan, M.; Brachmann, A.; Poettinger, L.; Plener, L.; Lassak, J.; Jung, K. Identification and Initial Characterization of Prophages in Vibrio campbellii. PLoS ONE 2016, 11, e0156010. [Google Scholar] [CrossRef]
- Travis, J.; Catherine, S.; Himanshu, V.; Hannah, D.; Amro, H. CRISPR/Cas9 mutagenesis reveals a role for ABCB1 in gut immune responses to Vibrio diazotrophicus in sea urchin larvae. J. Exp. Biol. 2021, 224, jeb232272. [Google Scholar]
- Dong, Y.; Dong, S. Advances of ecological physiology in sea cucumber, Apostichopus Japonicus Selenka. Period. Ocean Univ. China 2009, 39, 908–912, (In Chinese with English abstract). [Google Scholar]
- Yu, S. Antioxidant Defense Regulatory Function Gene of Apostichopus japonicus and Its Genes Expression and Polymorphism Analysis during Aestivation. Master’s Thesis, Dalian Ocean University, Dalian, China, 2018. (In Chinese with English abstract). [Google Scholar]
- Li, F.; Gao, F.; Tan, J.; Fan, C.; Sun, H.; Yan, J.; Chen, S.; Wang, X. Characterization and identification of enzyme-producing microflora isolated from the gut of sea cucumber Apostichopus japonicus. Chin. J. Oceanol. Limnol. 2016, 34, 153–162. [Google Scholar] [CrossRef]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Bend, W.; Rolf, D.; Peter, M. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Family | Genus | DSm | DHs | WSm | WHs |
|---|---|---|---|---|---|---|
| Proteobacteria | Rhodobacteraceae | Ruegeria | 0.081799 | 0.070116 | 0.048307 | 0.041341 |
| Proteobacteria | Rhodobacteraceae | HIMB11 | 0.020323 | 0.019825 | 0.231674 | 0.181732 |
| Proteobacteria | Rhodobacteraceae | Shimia | 0.007181 | 0.0033 | 0.001552 | 0.000949 |
| Proteobacteria | Oceanospirillaceae | Oceanospirillum | 0.006382 | 0.00764 | 0.021083 | 0.030632 |
| Proteobacteria | Thioglobaceae | SUP05_cluster | 0.005579 | 0.005339 | 0.006931 | 0.009897 |
| Proteobacteria | Vibrionaceae | Vibrio | 0.004714 | 0.004229 | 0.076026 | 0.021355 |
| Proteobacteria | Moraxellaceae | Acinetobacter | 0.003266 | 0.00271 | 0.00438 | 0.006747 |
| Proteobacteria | Nitrincolaceae | Neptuniibacter | 0.002958 | 0.003417 | 0.07232 | 0.088948 |
| Bacteroidota | Flavobacteriaceae | NS5_marine_group | 0.002439 | 0.002103 | 0.039845 | 0.056475 |
| Proteobacteria | Pseudomonadaceae | Pseudomonas | 0.002222 | 0.001355 | 0.015521 | 0.014391 |
| Proteobacteria | Alteromonadaceae | Aestuariibacter | 0.001938 | 0.002278 | 0.017226 | 0.022461 |
| Proteobacteria | Marinomonadaceae | Marinomonas | 0.001637 | 0.003318 | 0.004282 | 0.004573 |
| Cyanobacteria | unidentified_Chloroplast | unidentified_Chloroplast | 0.00156 | 0.001291 | 0.000658 | 0.000838 |
| Proteobacteria | Nitrincolaceae | Corallomonas | 0.001332 | 0.002202 | 0.010918 | 0.021142 |
| Proteobacteria | Halieaceae | OM60(NOR5)_clade | 0.001234 | 0.000654 | 0.003614 | 0.004501 |
| Proteobacteria | Nitrincolaceae | Marinobacterium | 0.00115 | 0.001525 | 0.002413 | 0.003123 |
| Proteobacteria | Litoricolaceae | Litoricola | 0.001009 | 0.00097 | 0.017966 | 0.019806 |
| Proteobacteria | SAR116_clade | unidentified_SAR116_clade | 0.000911 | 0.000847 | 0.009324 | 0.011897 |
| Proteobacteria | Alteromonadaceae | Alteromonas | 0.000803 | 0.000613 | 0.008214 | 0.008191 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, C.; Wang, Y.; Zheng, B.; Wang, Y.; He, L.; Xu, Q.; Gao, F. Comparative Analysis of Gut Bacterial Community Composition in Two Tropical Economic Sea Cucumbers under Different Seasons of Artificial Environment. Int. J. Mol. Sci. 2024, 25, 4573. https://doi.org/10.3390/ijms25084573
Jia C, Wang Y, Zheng B, Wang Y, He L, Xu Q, Gao F. Comparative Analysis of Gut Bacterial Community Composition in Two Tropical Economic Sea Cucumbers under Different Seasons of Artificial Environment. International Journal of Molecular Sciences. 2024; 25(8):4573. https://doi.org/10.3390/ijms25084573
Chicago/Turabian StyleJia, Chenghao, Yuanhang Wang, Bojun Zheng, Yanan Wang, Linwen He, Qiang Xu, and Fei Gao. 2024. "Comparative Analysis of Gut Bacterial Community Composition in Two Tropical Economic Sea Cucumbers under Different Seasons of Artificial Environment" International Journal of Molecular Sciences 25, no. 8: 4573. https://doi.org/10.3390/ijms25084573