Statistical Evaluation of Monophyly in the ‘Broad-Nosed Weevils’ through Molecular Phylogenetic Analysis Combining Mitochondrial Genome and Single-Locus Sequences (Curculionidae: Entiminae, Cyclominae, and Hyperinae)


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. ‘Backbone’ Phylogeny
2.2. Public Database Sequences
2.3. Multiple Sequence Alignment and Dataset Concatenation
2.4. Monophyly Constraints
2.5. Phylogenetic Analyses
2.6. Statistical Hypothesis Testing
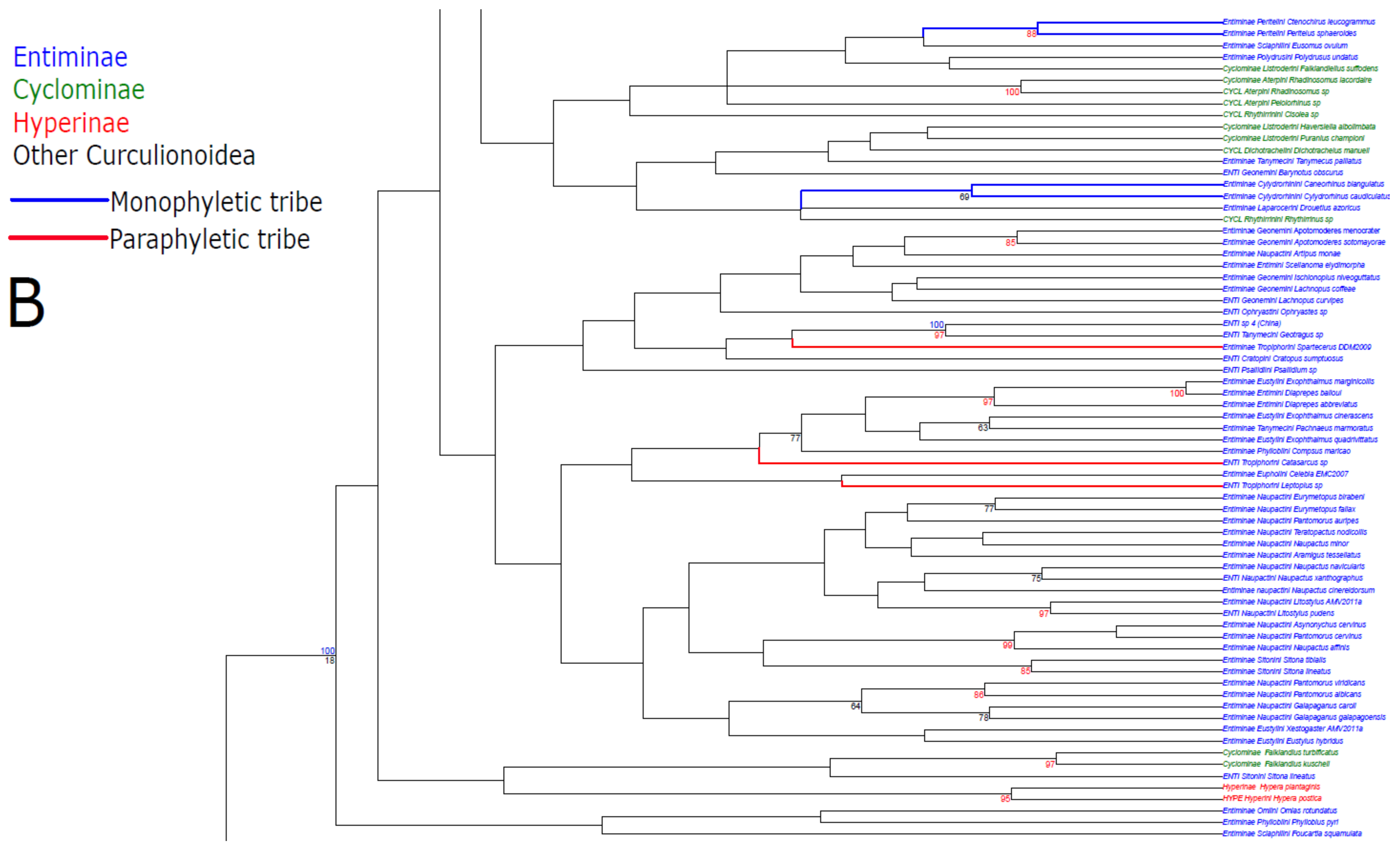
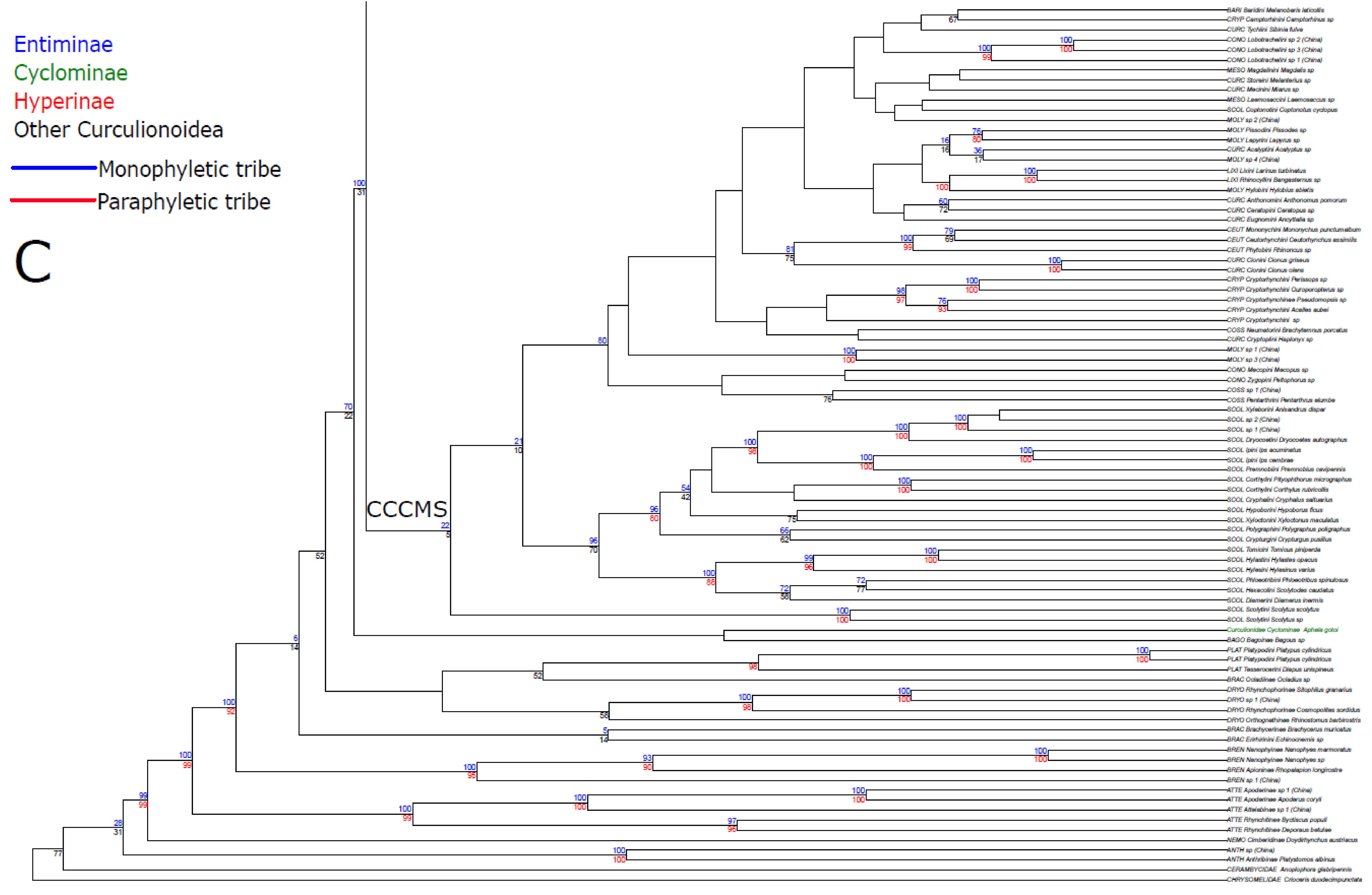
3. Results
3.1. Public Database Sequences
3.2. Phylogenetic Analyses
3.3. Statistical Hypothesis Testing
4. Discussion
4.1. Unconstrained Analysis
4.2. Constraint Analyses and Statistical Tests of Monophyly
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harrison, C.J.; Langdale, J.A. A step by step guide to phylogeny reconstruction. Plant J. 2006, 45, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Wiley, E.O.; Lieberman, B.S. Phylogenetics: Theory and Practice of Phylogenetic Systematics, 2nd ed.; Wiley-Blackwell: Hoboken, NY, USA, 2011; pp. 1–432. ISBN 978-0-470-90596-8. [Google Scholar]
- Franz, N.M.; Engel, M.S. Can higher-level phylogenies of weevils explain their evolutionary success? A critical review. Syst. Entoml. 2010, 35, 597–606. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum-likelihood estimate of the evolutionary tree topologies from DNA-sequence data, and the branching order in Hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.A. Testing tree topologies. In The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing, 2nd ed.; Lemey, P., Salemi, M., Vandamme, A.M., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 381–404. ISBN 9780521730716. [Google Scholar]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Jordal, B.H.; Smith, S.M.; Cognato, A.I. Classification of weevils as a data-driven science: Leaving opinion behind. Zookeys 2014, 439, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gillett, C.P.D.T.; Crampton-Platt, A.; Timmermans, M.J.; Jordal, B.H.; Emerson, B.C.; Vogler, A.P. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 2014, 31, 2223–2237. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Vera, G.; Caldara, R.; Tosevski, I.; Emerson, B.C. Molecular phylogenetic analysis of archival tissue reveals the origin of a disjunct southern African-Palaearctic weevil radiation. J. Biogeogr. 2013, 40, 1348–1359. [Google Scholar] [CrossRef]
- Lacordaire, T. Histoire Naturelle des Insectes. Genera des Coléoptères, ou Exposé Méthodique et Critique de Tous Les Genres Proposés Jusqu’ici Dans cet Ordre D’insectes; pp Tome sixième contenant la famille des curculionides; Librairie Encyclopédique de Roret: Paris, France, 1863; pp. 1–637. [Google Scholar]
- Thompson, R.T. Observations on the morphology and classification of weevils (Coleoptera, Curculionoidea) with a key to major groups. J. Nat. Hist. 1992, 26, 835–891. [Google Scholar] [CrossRef]
- Velazquez De Castro, A.J.; Alonso-Zarazaga, M.A.; Outerelo, R. Systematics of Sitonini (Coleoptera: Curculionidae: Entiminae), with a hypothesis on the evolution of feeding habits. Syst. Entomol. 2007, 32, 312–331. [Google Scholar] [CrossRef]
- Oberprieler, R.G.; Marvaldi, A.E.; Anderson, R.S. Weevils, weevils, weevils everywhere. Zootaxa 2007, 1668, 491–520. [Google Scholar] [CrossRef]
- Kuschel, G. A phylogenetic classification of Curculionoidea to families and subfamilies. Mem. Entomol. Soc. Wash. 1995, 14, 5–33. [Google Scholar]
- Marvaldi, A.E. Higher level phylogeny of Curculionidae (Coleoptera: Curculionoidea) based mainly on larval characters, with special reference to broad-nosed weevils. Cladistics 1997, 13, 285–312. [Google Scholar] [CrossRef]
- Bouchard, P.; Bousquet, Y.; Davies, A.E.; Alonso-Zarazaga, M.A.; Lawrence, J.F.; Lyal, C.H.C.; Newton, A.F.; Reid, C.A.M.; Schmitt, M.; Ślipiński, S.A.; et al. Family-group names in Coleoptera (Insecta). Zookeys 2011, 88, 1–972. [Google Scholar] [CrossRef] [PubMed]
- Hundsdoerfer, A.K.; Rheinheimer, J.; Wink, M. Towards the phylogeny of the Curculionoidea (Coleoptera): Reconstructions from mitochondrial and nuclear ribosomal DNA sequences. Zool. Anz. 2009, 248, 9–31. [Google Scholar] [CrossRef]
- McKenna, D.D.; Sequeira, A.S.; Marvaldi, A.E.; Farrell, B.D. Temporal lags and overlap in the diversification of weevils and flowering plants. Proc. Natl. Acad. Sci. USA 2009, 106, 7083–7088. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.; Timmermans, M.J.T.N.; Vogler, A.P. Mitogenome sequences stabilize the phylogenetics of weevils (Curculionoidea) and establish the monophyly of larval ectophagy. Mol. Phylogenet. Evol. 2013, 67, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Gunter, N.L.; Oberprieler, R.G.; Cameron, S.L. Molecular phylogenetics of Australian weevils (Coleoptera: Curculionoidea): Exploring relationships in a hyperdiverse lineage through comparison of independent analyses. Austral Entomol. 2015, 55, 217–233. [Google Scholar] [CrossRef]
- Shin, S.; Clarke, D.J.; Lemmon, A.R.; Aitken, A.L.; Haddad, S.; Farrell, B.D.; Marvaldi, A.E.; Oberprieler, R.G.; McKenna, D.D. Phylogenomic data yield new and robust insights into the phylogeny and evolution of weevils. Mol. Biol. Evol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Zarazaga, M.A.; Lyal, C.H.C. A World Catalogue of Families and Genera of Curculionoidea (Insecta: Coleoptera) (Excepting Scolytidae and Platypodidae); Entomopraxis: Barcelona, Spain, 1999; pp. 1–315. ISBN 84-605-9994-9. [Google Scholar]
- Hunt, T.; Bergsten, J.; Levkanicova, Z.; Papadopoulou, A.; John, O.S.; Wild, R.; Hammond, P.M.; Ahrens, D.; Balke, M.; Caterino, M.S.; et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science 2007, 318, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.; Vogler, A.P. A protocol for large-scale rRNA sequence analysis: Towards a detailed phylogeny of Coleoptera. Mol. Phylogenet. Evol. 2008, 47, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Bocak, L.; Barton, C.; Crampton-Platt, A.; Chesters, D.; Ahrens, D.; Vogler, A.P.V. Building the Coleoptera tree-of-life for >8000 species: Composition of public DNA data and fit with Linnaean classification. Syst. Entomol. 2013, 39, 97–110. [Google Scholar] [CrossRef]
- Oberprieler, R.G. A reclassification of the weevil subfamily Cyclominae (Coleoptera: Curculionidae). Zootaxa 2010, 2515, 1–35. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, H.; Fink, T. New and interesting Peritelini of the Western Mediterranean fauna. XX. A novel Meira (Jacquelin du Val, 1852) species from the Ligurian Alps. Zootaxa 2013, 3716, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, H.; Germann, C.; Braunert, C. New or interesting Peritelini of the West-Mediterranean fauna. XXIV. Two new Simmeiropsis Pierotti & Bello, 2013 from Portugal (Coleoptera, Curculionidae, Entiminae). Zootaxa 2013, 3734, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, H.; Bello, C.; Alonso-Zarazaga, M.A. Contribution to the systematic rearrangement of the Palaearctic Peritelini. VI. A synthesis of the Spanish Peritelini (Coleoptera: Curculionidae: Entiminae). Zootaxa 2010, 2376, 1–96. [Google Scholar]
- Marvaldi, A.E. Larvae of South American Entimini (Coleoptera: Curculionidae), and phylogenetic implications of certain characters. Rev. Chil. Entomol. 1998, 25, 21–44. [Google Scholar]
- Lachowska, D.; Rozek, M.; Holecova, M. Cytotaxonomy and karyology of the tribe Otiorhynchini (Coleoptera: Curculionidae). Eur. J. Entomol. 2008, 105, 175–184. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Jones, A.G.; Hammond, P.M.; Vogler, A.P. DNA taxonomy and phylogeography of beetles of the Falkland Islands (Islas Malvinas). Mol. Phylogenet. Evol. 2009, 53, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Botero-Castro, F.; Tilak, M.K.; Justy, F.; Catzeflis, F.; Delsuc, F.; Douzery, E.J.P. Next-generation sequencing and phylogenetic signal of complete mitochondrial genomes for resolving the evolutionary history of leaf-nosed bats (Phyllostomidae). Mol. Phylogenet. Evol. 2013, 69, 728–739. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Constrained Taxon | Generic Diversity (No. Genera) * | No. of Genera in Constraint | No. of Terminals in Constraint |
|---|---|---|---|
| Broad-nosed weevils | 1585 | 89 | 142 |
| Entiminae | 1370 | 74 | 121 |
| Cyclominae | 180 | 14 | 18 |
| Hyperinae | 35 | 1 | 3 |
| Brachyderini | 24 | 2 | 6 |
| Celeuthetini | 75 | 8 | 8 |
| Cyphicerini | 120 | 1 | 2 |
| Elytrurini | 6 | 2 | 3 |
| Eustylini | 17 | 6 | 9 |
| Geonemini | 39 | 5 | 7 |
| Laparocerini | 9 | 3 | 9 |
| Naupactini | 65 | 9 | 19 |
| Otiorhynchini | 27 | 1 | 6 |
| Polydrusini | 14 | 3 | 6 |
| Rhyncogonini | 3 | 1 | 3 |
| Sciaphilini | 46 | 4 | 4 |
| Sitonini | 8 | 1 | 4 |
| Tanymecini | 42 | 5 | 6 |
| Trachyphloeini | 23 | 1 | 2 |
| Tropiphorini | 115 | 6 | 9 |
| UNCONSTRAINED | 147 | 229 |
| Rank (By Likelihood) | Taxon Constrained in ML Tree | ∆Log Likelihood to Best Tree | AU Test p-Value | KH Test p-Value | SH Test p-Value |
|---|---|---|---|---|---|
| 1 | Sitonini | −4.1 | 0.621 | 0.526 | 0.971 |
| 2 | UNCONSTRAINED | 4.1 | 0.605 | 0.474 | 0.948 |
| 3 | Hyperinae | 8.7 | 0.527 | 0.396 | 0.968 |
| 4 | Laparocerini | 11.7 | 0.573 | 0.430 | 0.961 |
| 5 | Rhyncogonini | 18.0 | 0.513 | 0.409 | 0.921 |
| 6 | Broad-nosed weevils | 21.4 | 0.442 | 0.378 | 0.913 |
| 7 | Polydrusini | 23.7 | 0.431 | 0.357 | 0.942 |
| 8 | Cyphicerini | 24.1 | 0.425 | 0.362 | 0.865 |
| 9 | Geonemini | 26.7 | 0.411 | 0.355 | 0.873 |
| 10 | Elytrurini | 29.2 | 0.395 | 0.340 | 0.876 |
| 11 | Celeuthetini | 55.6 | 0.202 | 0.213 | 0.719 |
| 12 | Naupactini | 56.4 | 0.206 | 0.185 | 0.726 |
| 13 | Cyclominae | 70.6 | 0.132 | 0.174 | 0.627 |
| 14 | Eustylini | 72.6 | 0.176 | 0.125 | 0.619 |
| 15 | Sciaphilini | 78.0 | 0.119 | 0.153 | 0.573 |
| 16 | Entiminae | 88.0 | 0.080 | 0.100 | 0.505 |
| 17 | Trachyphloeini | 94.3 | 0.083 | 0.059 | 0.463 |
| 18 | Tanymecini | 99.3 | 0.054 | 0.059 | 0.426 |
| 19 | Otiorhynchini | 204.2 | 2 × 10−4 | 0.006 | 0.048 |
| 20 | Brachyderini | 241.0 | 6 × 10−51 | 3 × 10−5 | 0.007 |
| 21 | Tropiphorini | 483.0 | 0.001 | 0 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillett, C.P.D.T.; Lyal, C.H.; Vogler, A.P.; Emerson, B.C. Statistical Evaluation of Monophyly in the ‘Broad-Nosed Weevils’ through Molecular Phylogenetic Analysis Combining Mitochondrial Genome and Single-Locus Sequences (Curculionidae: Entiminae, Cyclominae, and Hyperinae). Diversity 2018, 10, 21. https://doi.org/10.3390/d10020021
Gillett CPDT, Lyal CH, Vogler AP, Emerson BC. Statistical Evaluation of Monophyly in the ‘Broad-Nosed Weevils’ through Molecular Phylogenetic Analysis Combining Mitochondrial Genome and Single-Locus Sequences (Curculionidae: Entiminae, Cyclominae, and Hyperinae). Diversity. 2018; 10(2):21. https://doi.org/10.3390/d10020021
Chicago/Turabian StyleGillett, Conrad P.D.T., Christopher H. Lyal, Alfried P. Vogler, and Brent C. Emerson. 2018. "Statistical Evaluation of Monophyly in the ‘Broad-Nosed Weevils’ through Molecular Phylogenetic Analysis Combining Mitochondrial Genome and Single-Locus Sequences (Curculionidae: Entiminae, Cyclominae, and Hyperinae)" Diversity 10, no. 2: 21. https://doi.org/10.3390/d10020021
APA StyleGillett, C. P. D. T., Lyal, C. H., Vogler, A. P., & Emerson, B. C. (2018). Statistical Evaluation of Monophyly in the ‘Broad-Nosed Weevils’ through Molecular Phylogenetic Analysis Combining Mitochondrial Genome and Single-Locus Sequences (Curculionidae: Entiminae, Cyclominae, and Hyperinae). Diversity, 10(2), 21. https://doi.org/10.3390/d10020021