Genetic Diversity of Black Salamanders (Aneides flavipunctatus) across Watersheds in the Klamath Mountains


Abstract
:1. Introduction

2. Methods
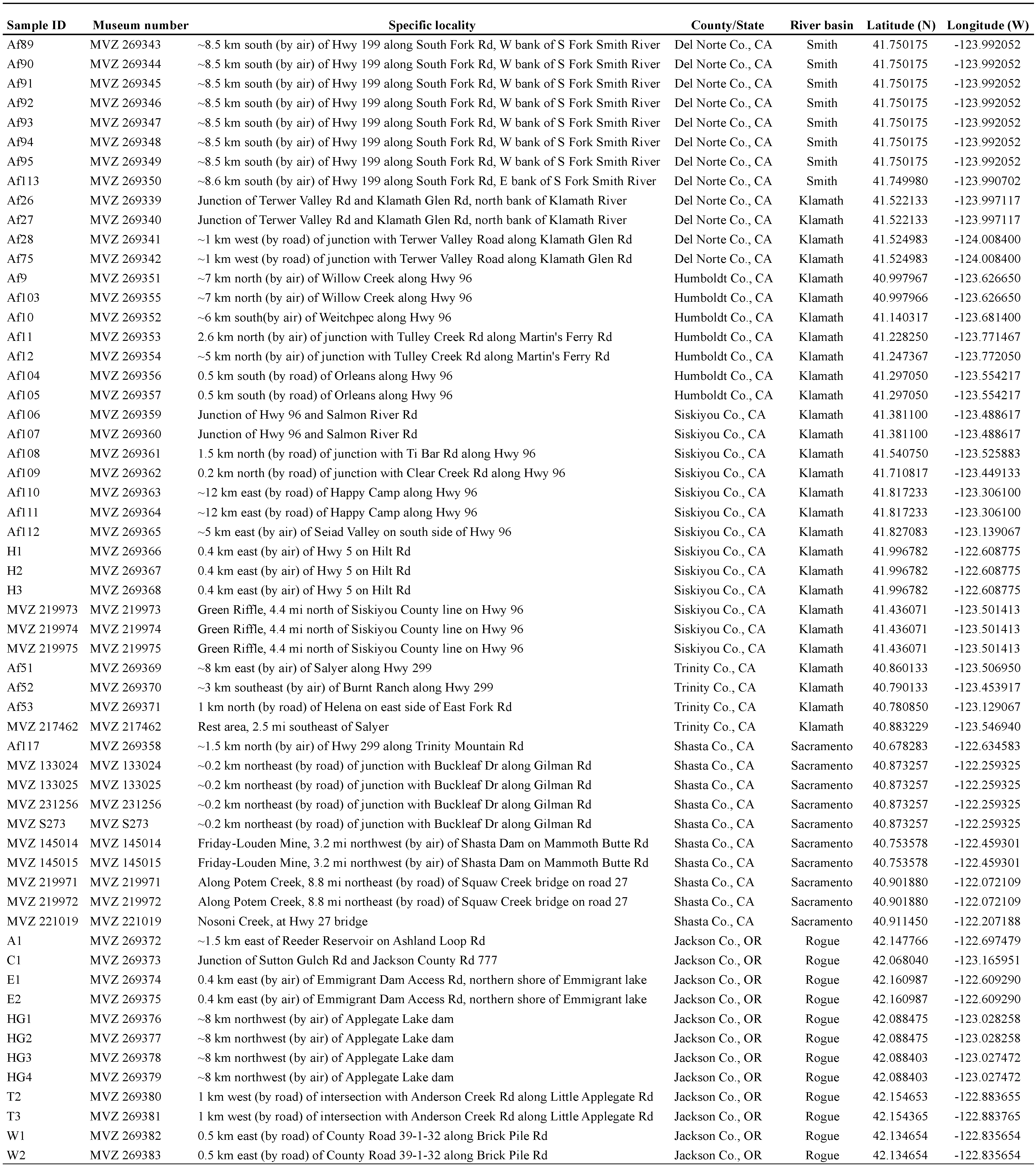
2.1. Sampling and Geographic Distribution
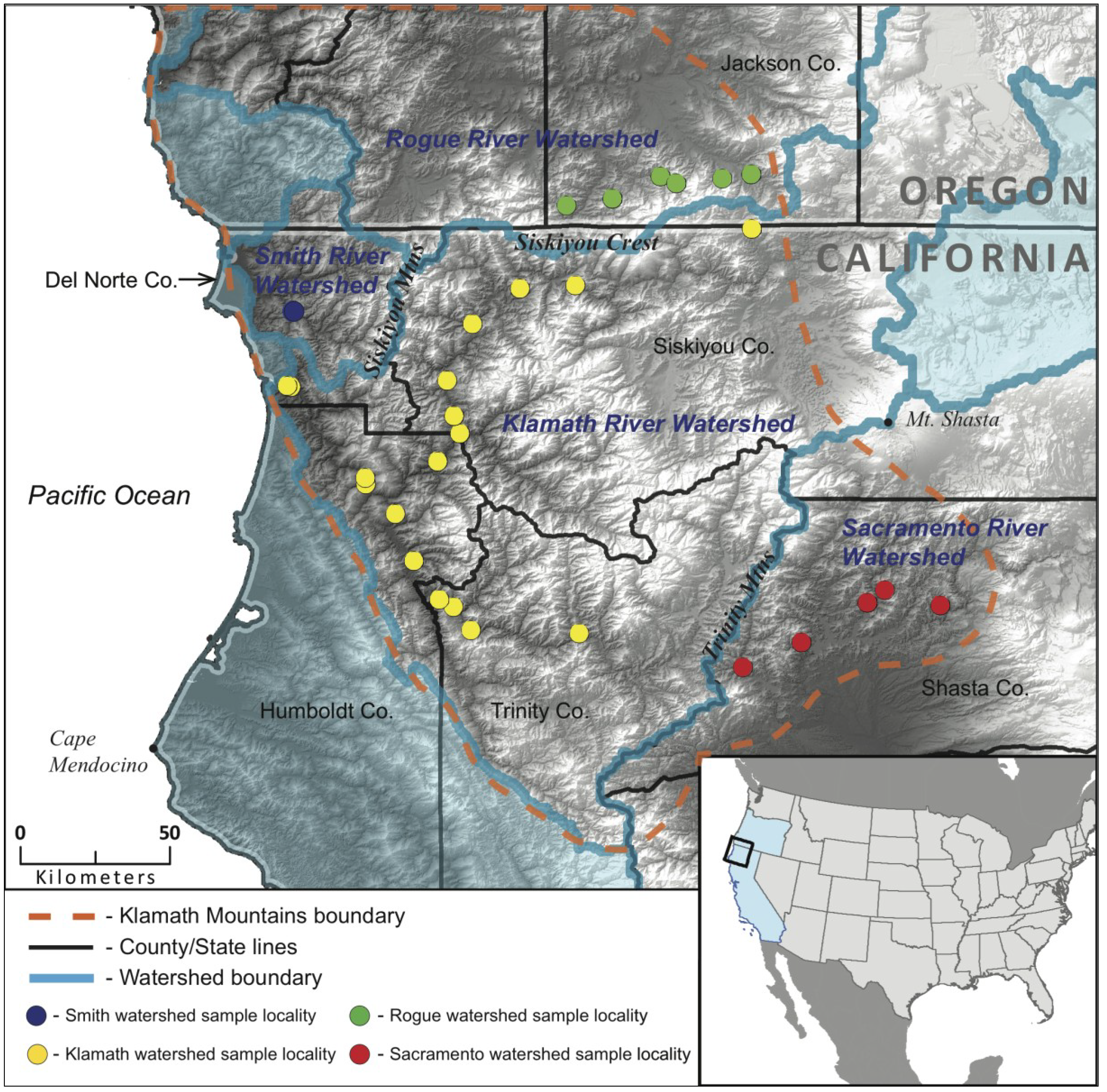
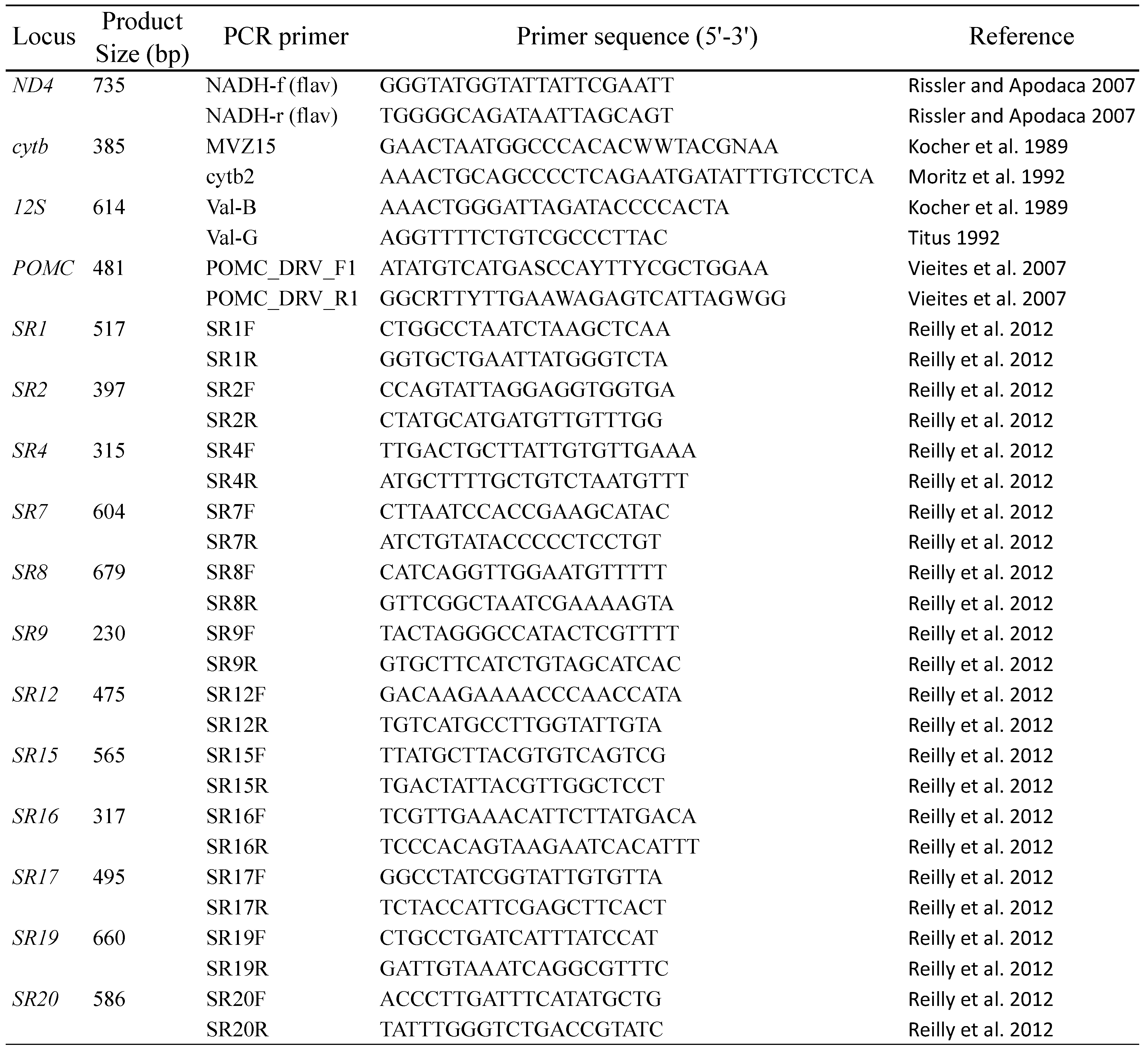
2.2. Molecular Markers
2.3. Data Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Watershed | # sequences | nucleotide diversity | # haplotypes | haplotype diversity | # polymorphic sites | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|
| Rogue | 12 | 0.00064 | 5 | 0.667 | 5 | −1.29119; p > 0.10 | −1.715 |
| Klamath | 28 | 0.01097 | 11 | 0.823 | 28 | −0.15901; p > 0.10 | 0.72 |
| Sacramento | 10 | 0.00908 | 7 | 0.867 | 26 | −0.82555; p > 0.10 | 0.24 |
| Smith | 8 | 0.00046 | 4 | 0.643 | 3 | −1.44751; p > 0.10 | −1.832 |
| Rogue | Klamath | Sacramento | Smith | |
|---|---|---|---|---|
| Rogue | - | 0 | 28 | 18 |
| Klamath | 0.008 | - | 20 | 0 |
| Sacramento | 0.042 | 0.047 | - | 25 |
| Smith | 0.012 | 0.013 | 0.037 | - |
| Locus | No. of bp | No. Sequences | h | Hd | Variable sites (not including gaps) | Parsimony informative sites | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|---|
| SR1 | 477 | 74 | 2 | 0.03 | 1 | 0 | −1.06 | −1.97 |
| SR2 | 334 | 20 | 6 | 0.84 | 16 | 14 | −0.78 | 1.46 |
| SR4 | 236 | 24 | 4 | 0.47 | 6 | 4 | −0.2 | 1.22 |
| SR7 | 564 | 32 | 9 | 0.82 | 14 | 10 | −0.46 | −0.55 |
| SR8 | 639 | 72 | 8 | 0.69 | 7 | 6 | 0.52 | −0.39 |
| SR9 | 188 | 32 | 6 | 0.70 | 6 | 4 | 0.02 | −0.37 |
| SR12 | 434 | 26 | 5 | 0.73 | 12 | 12 | 1.45 | 4.35 |
| SR15 | 525 | 30 | 8 | 0.83 | 10 | 9 | 1.37 | 0.72 |
| SR16 | 273 | 36 | 6 | 0.66 | 7 | 6 | −0.58 | −0.54 |
| SR17 | 455 | 74 | 25 | 0.75 | 24 | 17 | −0.91 | −11.23 |
| SR19 | 604 | 76 | 6 | 0.25 | 8 | 5 | −1.36 | −1.49 |
| SR20 | 577 | 68 | 9 | 0.49 | 8 | 5 | −1.24 | −4.29 |
| POMC | 481 | 98 | 4 | 0.69 | 3 | 3 | 1.46 | 1.62 |
| Watershed | Statistic | POMC | SR1 | SR2 | SR4 | SR7 | SR8 | SR9 | SR12 | SR15 | SR16 | SR17 | SR19 | SR20 | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rogue | Tajima’s D | −0.248 | N/A | N/A | N/A | N/A | −0.954 | N/A | N/A | −0.612 | N/A | 2.002 | N/A | 0.895 | 0.217 |
| Fu’s Fs | 0.23 | N/A | N/A | N/A | N/A | 1.523 | N/A | N/A | 0.172 | N/A | 5.36 | N/A | 1.116 | 1.68 | |
| Nucleotide Diversity | 0.00047 | 0 | N/A | N/A | 0 | 0.00089 | 0 | 0 | 0.00095 | 0 | 0.0045 | 0 | 0.00073 | 0.00069 | |
| Klamath | Tajima’s D | 1.885 | −1.129 | −0.1 | −1.162 | −0.038 | 0.353 | 0.313 | −0.173 | 1.421 | 0.065 | −0.358 | −1.073 | −0.915 | −0.07 |
| Fu’s Fs | 1.846 | −1.407 | 4.515 | −0.7 | 0.252 | −0.278 | −0.175 | −0.674 | 1.716 | 0.324 | −1.56 | 0.312 | −1.776 | 0.184 | |
| Nucleotide Diversity | 0.00191 | 0.00012 | 0.01168 | 0.00055 | 0.00479 | 0.0034 | 0.00965 | 0.00342 | 0.00944 | 0.00443 | 0.00793 | 0.00036 | 0.0015 | 0.00455 | |
| Sacramento | Tajima’s D | N/A | N/A | −0.304 | −1.233 | −0.933 | −1.132 | 0.85 | N/A | 1.754 | 0.839 | 0.159 | −1.28 | 0.932 | −0.035 |
| Fu’s Fs | N/A | N/A | 0.33 | 1.609 | −0.003 | 0.952 | 0.625 | N/A | 3.833 | 0.825 | −2.16 | −0.197 | 0.139 | 0.595 | |
| Nucleotide Diversity | 0 | 0 | 0.00321 | 0.00439 | 0.00062 | 0.00112 | 0.00284 | 0 | 0.00898 | 0.00666 | 0.00549 | 0.00278 | 0.00226 | 0.00295 | |
| Smith | Tajima’s D | 0.156 | N/A | N/A | N/A | −0.754 | −1.233 | −0.71 | N/A | 1.181 | N/A | 0.505 | N/A | −0.05 | −0.129 |
| Fu’s Fs | 0.551 | N/A | N/A | N/A | −0.288 | 1.609 | 1.099 | N/A | 3.146 | N/A | −4.414 | N/A | −0.427 | 0.182 | |
| Nucleotide Diversity | 0.00068 | 0 | N/A | 0 | 0.00266 | 0.00169 | 0.00585 | 0 | 0.00738 | 0 | 0.01029 | 0 | 0.00151 | 0.00251 |
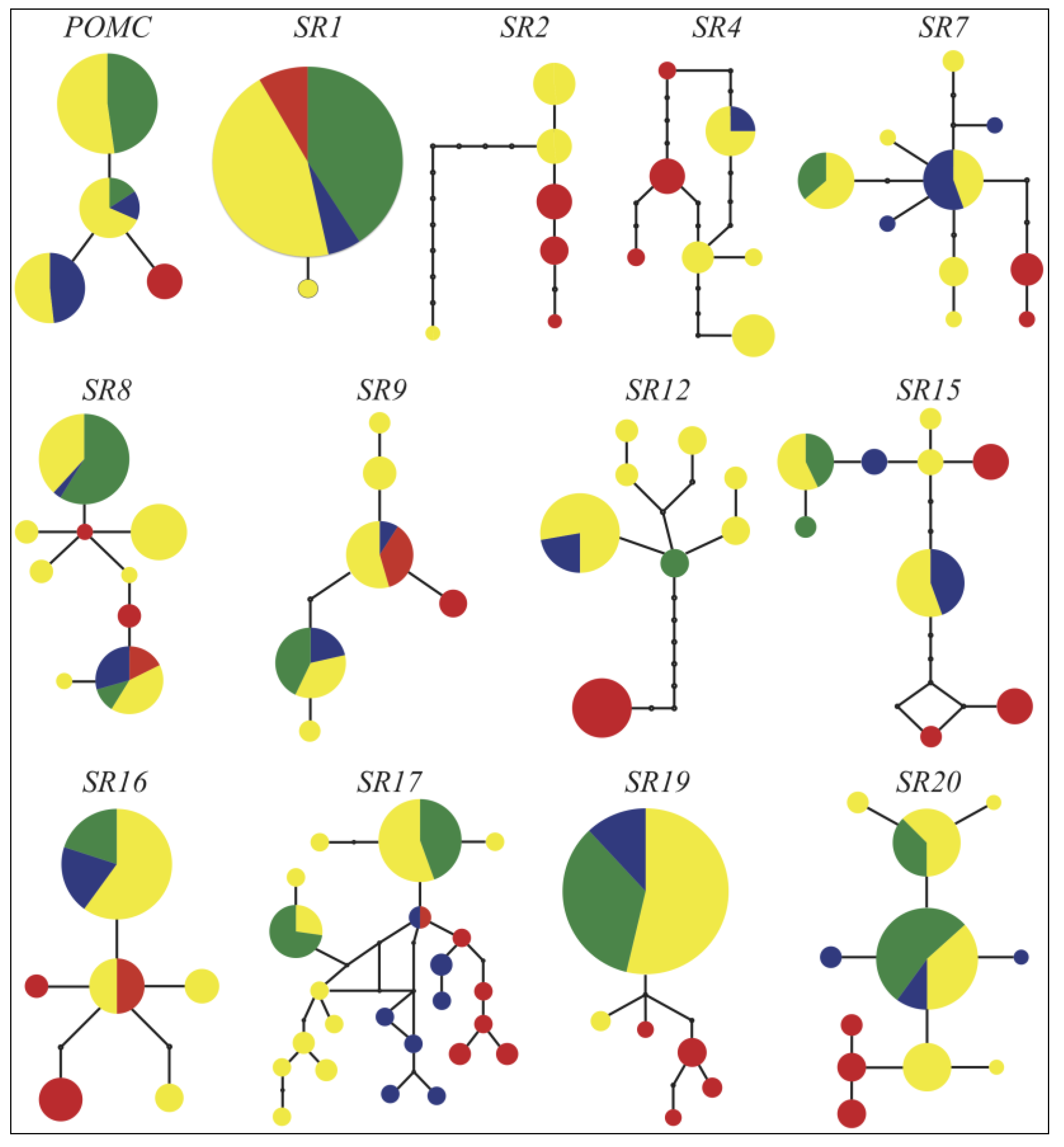
2.4. Mitochondrial Relationships
2.5. Population Structure
2.6. Historical Demography
3. Results and Discussion
3.1. Data Characteristics
3.2. Phylogenetic Analyses
3.3. Population Structure
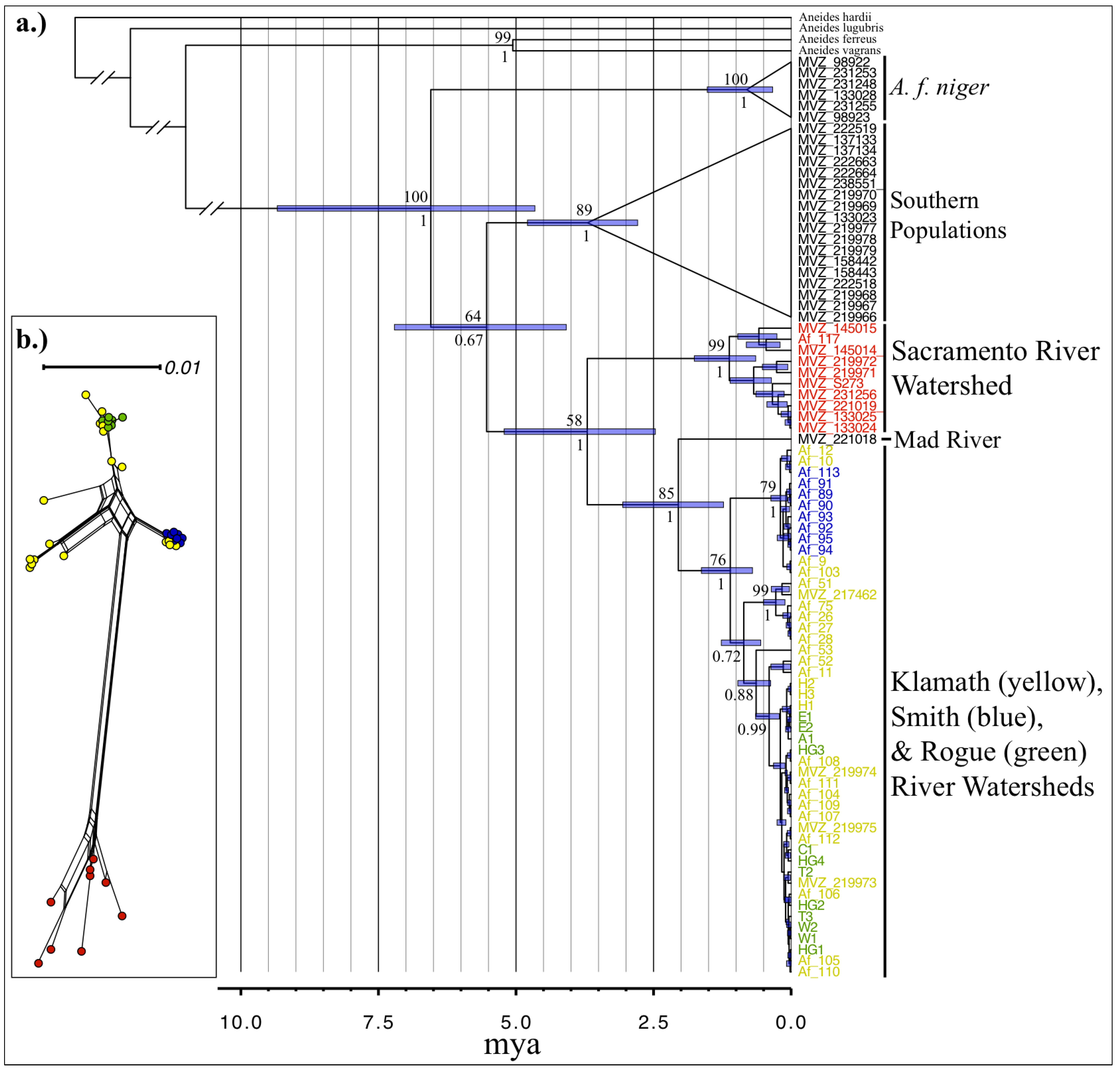
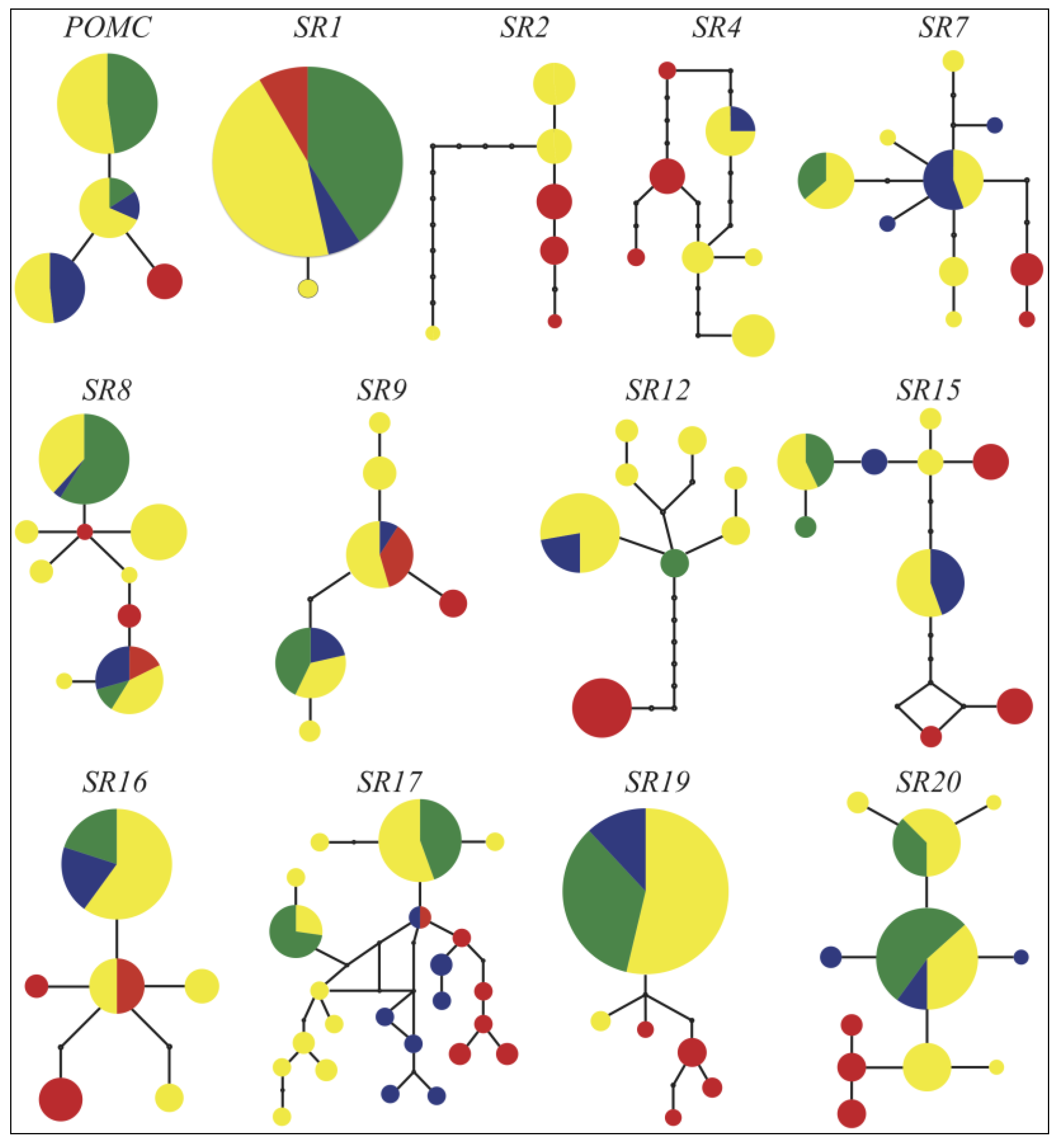
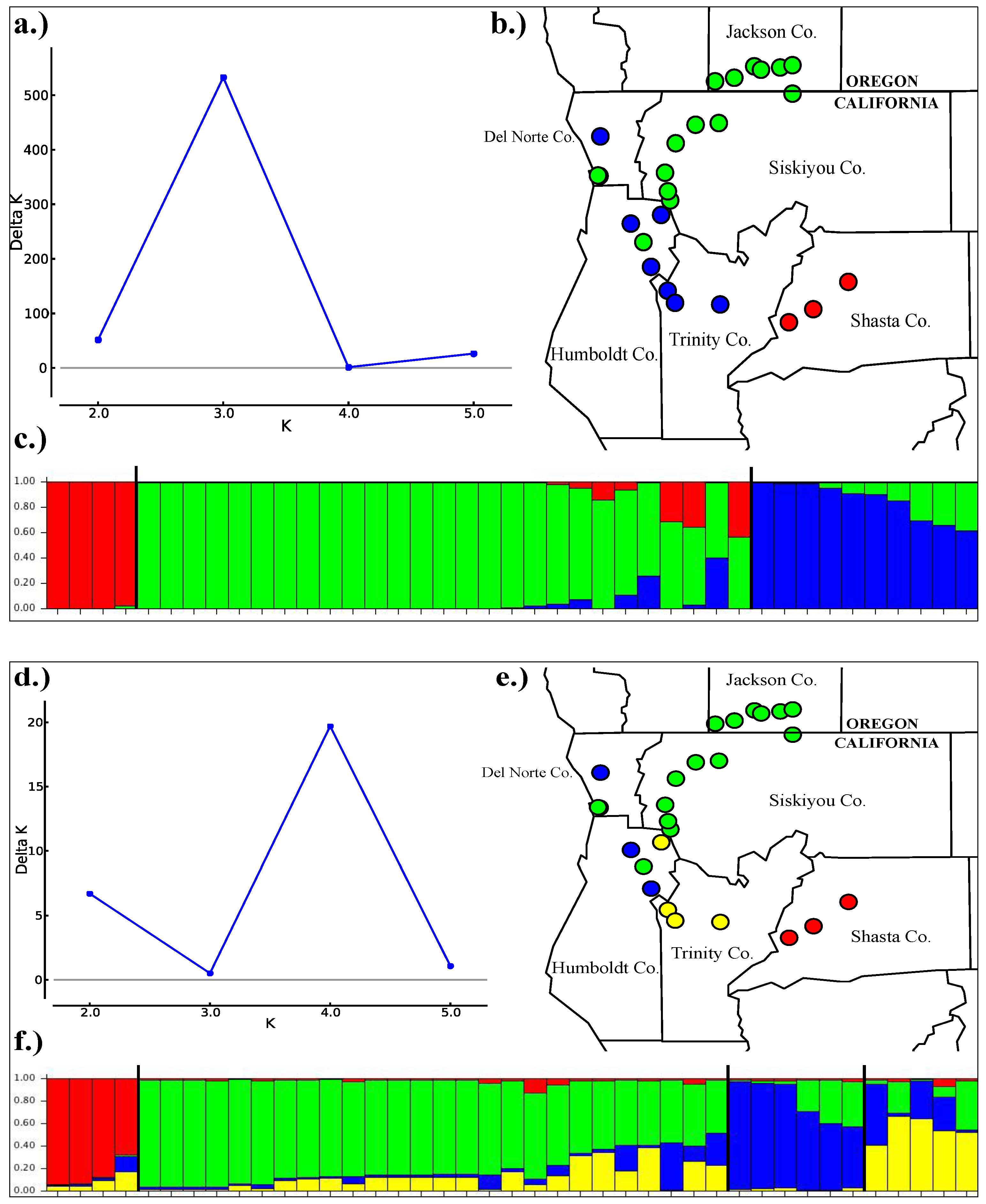
3.4. Historical Demography
| Population Divergence Time (years) | Effective Population Size (individuals) | Population Migration Rate (2Nm) | |||||
|---|---|---|---|---|---|---|---|
| Population Comparion | Value | t0 | Shasta (q0) | Klamath (q1) | Ancestor (q2) | m0>1 | m1>0 |
| Sacramento vs Klamath | HiPt | 766,948 | 34,055 | 82,572 | 125,492 | 0.116 | 0.002 |
| 95%Lo | 599,003 | 19,127 | 54,582 | 20,993 | 0.006 | 0.001 | |
| 95%Hi | 10,899,624 | 68,577 | 125,492 | 898,971 | 0.531 | 0.346 | |
3.5. Refugia in the Klamath Mountains
3.6. Should the Sacramento Watershed Population be Considered a Distinct Species?
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Manel, S.; Schwartz, M.K.; Luikart, G.; Taberlet, P. Landscape genetics: Combining landscape ecology and population genetics. Trends Ecol. Evol. 2003, 18, 189–197. [Google Scholar] [CrossRef]
- Storfer, A.; Murphy, M.A.; Spear, S.F.; Holderegger, R.; Waits, L.P. Landscape genetics: Where are we now? Mol. Ecol. 2010, 19, 3496–3514. [Google Scholar] [CrossRef]
- Orr, W.N.; Orr, E.L. Geology of the Pacific Northwest; Waveland Press, Inc.: Long Grove, IL, USA, 2002. [Google Scholar]
- Agee, J.K. Steward’s Fork: A Sustainable Future for the Klamath Mountains; University of California Press: Berkeley, CA, USA, 2007. [Google Scholar]
- Sawyer, J.O. Northwest California: A Natural History; University of California Press: Berkeley, CA, USA, 2006. [Google Scholar]
- Harden, D.R. California Geology; Prentice Hall: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Oregon Climate Service (OCS). Available online: http://www.ocs.orst.edu (accessed on 15 April 2013).
- Noss, R.F. The Redwood Forest: History, Ecology, and Conservation of the Coast Redwoods; Island Press: Washington, DC, USA, 2000. [Google Scholar]
- AmphibiaWeb: Information on Amphibian Biology and Conservation. Berkeley, CA, USA, 2013. Available online: http://amphibiaweb.org/ (accessed on 20 June 2013).
- Rissler, L.J.; Apodaca, J.J. Adding more ecology into species delimitation: ecological niche models and phylogeography help define cryptic species in the Black Salamander (Aneides flavipunctatus). Syst. Biol. 2007, 56, 924–942. [Google Scholar] [CrossRef]
- Reilly, S.B.; Marks, S.B.; Jennings, W.B. Defining evolutionary boundaries across parapatric ecomorphs of Black Salamanders (Aneides flavipunctatus) with conservation implications. Mol. Ecol. 2012, 21, 5745–5761. [Google Scholar] [CrossRef]
- Lynch, J.F. Patterns of ontogenetic and geographic variation in the black salamander, Aneides flavipunctatus. Smithson. Contrib. Zool. 1981, 324, 1–53. [Google Scholar] [CrossRef]
- Larson, A. Paedomorphosis in relation to rates of morphological and molecular evolution in the salamander Aneides flavipunctatus. Evolution 1980, 34, 1–17. [Google Scholar] [CrossRef]
- Lowe, C.H. Speciation and Ecology in Salamanders of the Genus Aneides. Ph.D. Thesis, University of California, Los Angeles, CA, USA, 1950. [Google Scholar]
- Nauman, R.S.; Olson, D.H. Surveys for terrestrial amphibians in Shasta County, California, with notes on the distribution of Shasta salamanders (Hydromantes shastae). Northwest. Nat. 2004, 85, 35–38. [Google Scholar] [CrossRef]
- Mahoney, M. Molecular systematics and phylogeography of the Plethodon elongatus species group: Combining phylogenetic and population genetic methods to investigate species history. Mol. Ecol. 2004, 13, 149–166. [Google Scholar] [CrossRef]
- Mead, L.S.; Clayton, D.R.; Nauman, R.S.; Olson, D.H.; Pfrender, M.E. Characterization of newly discovered populations of Plethodon from Siskiyou County, California. Herpetologica 2005, 61, 158–177. [Google Scholar] [CrossRef]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Stephens, M.; Scheet, P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data inputation. Am. J. Hum. Genet. 2005, 76, 449–462. [Google Scholar] [CrossRef]
- Rozas, J.; Sanchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Zwickl, D.J. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. Thesis, The University of Texas, Austin, TX, USA, 2006. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Tan, A.M.; Wake, D.B. MtDNA phylogeography of the California newt, Taricha torosa (Caudata, Salamandridae). Mol. Phylogenet. Evol. 1995, 4, 383–394. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer v1.5.0. 2009. Available online: http://beast.bio.ed.ac.uk/ (accessed on 10 Aug 2011).
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1660. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure: Extensions to linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Hey, J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 2010, 27, 905–920. [Google Scholar] [CrossRef]
- Woerner, A.E.; Cox, M.P.; Hammer, M.F. Recombination-filtered genomic datasets by information maximization. Bioinformatics 2007, 23, 1851–1853. [Google Scholar] [CrossRef]
- Hey, J. On the number of New World founders: A population genetic portrait of the peopling of the Americas. Plos Biol. 2005, 3, 965–975. [Google Scholar]
- Kumar, S.; Subramanian, S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 803–808. [Google Scholar] [CrossRef]
- Anderson, P.K. Ecology and evolution in island populations of salamanders in the San Francisco Bay region. Ecol. Monogr. 1960, 30, 359–386. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Luthi, D.; Le Floch, M.; Bereiter, B.; Blunier, T.; Barnola, J.; Siegenthaler, U.; Raynaud, D.; Jouzel, J.; Fischer, H.; Kawamura, K.; et al. High-resolution carbon dioxide concentration record 650,000–800,000 years before present. Nature 2008, 453, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Shafer, A.B.A.; Cullingham, C.I.; Cote, S.D.; Coltman, D.W. Of glaciers and refugia: A decade of study sheds new light on the phylogeography of northwestern North America. Mol. Ecol. 2010, 19, 4589–4621. [Google Scholar] [CrossRef]
- Reilly, S.B.; Gottscho, A.D.; Garwood, J.M.; Jennings, W.B. Phylogenetic analysis of common garter snake (Thamnophis sirtalis) stomach contents detects cryptic range of a secretive salamander (Ensatina eschscholtzii oregonensis). Herpetol. Conserv. Biol. 2010, 5, 395–402. [Google Scholar]
- Goebel, A.M.; Ranker, T.A.; Corn, P.S.; Olmstead, R.G. Mitochondrial DNA evolution in the Anaxyrus boreas species group. Mol. Phylogenet. Evol. 2009, 50, 209–225. [Google Scholar] [CrossRef]
- Nielson, M.; Lohman, K.; Sullivan, J. Phylogeography of the tailed frog (Ascaphus truei): Implications for biogeography of the Pacific Northwest. Evolution 2001, 55, 147–160. [Google Scholar]
- Nielson, M.; Lohman, K.; Daugherty, C.H.; Allendorf, F.W.; Knudsen, K.L.; Sullivan, J. Allozyme and mitochondrial DNA variation in the tailed frog (Anura: Ascaphus): The influence of geography and gene flow. Herpetologica 2006, 62, 235–258. [Google Scholar] [CrossRef]
- Thompson, M.D.; Russell, A.P. Glacial Retreat and Its fluence on Migration of Mitochondrial Genes in the Long-Toed Salamander (Ambystoma macrodactylum) in Western North America. In Migration of Organisms; Elewa, A.M.T., Ed.; Springer: Berlin Heidelberg, Germany, 2005; pp. 205–246. [Google Scholar]
- Steele, C.A.; Storfer, A. Coalescent-based hypothesis testing supports multiple Pleistocene refugia in the Pacific Northwest for the Pacific giant salamander (Dicamptodon tenebrosus). Mol. Ecol. 2006, 15, 2477–2487. [Google Scholar] [CrossRef]
- Kuchta, S.R.; Tan, A. Isolation by distance and post-glacial range expansion in the rough-skinned newt, Taricha granulosa. Mol. Ecol. 2005, 14, 225–244. [Google Scholar] [CrossRef]
- Kuchta, S.R.; Parks, D.S.; Mueller, R.L.; Wake, D.B. Closing the ring: Historical biogeography of the salamander ring species Ensatina eschscholtzii. J. Biogeogr. 2009, 36, 982–995. [Google Scholar] [CrossRef]
- Eckert, A.J.; Tearse, B.R.; Hall, B.D. A phylogeographical analysis of the range disjunction for foxtail pine (Pinus balfouriana, Pinaceae): The role of Pleistocene glaciation. Mol. Ecol. 2008, 17, 1983–1997. [Google Scholar] [CrossRef]
- Kiefer, C.; Dobes, C.; Sharbel, T.F.; Koch, M.A. Phylogeographic structure of the chloroplast DNA gene pool in North American Boechera- A genus and continent-wide perspective. Mol. Phylogenet. Evol. 2009, 52, 303–311. [Google Scholar] [CrossRef]
- Wilke, T.; Duncan, N. Phylogeographic patterns in the American Pacific Northwest: Lessons from the arionid slug Prophysaon coeruleum. Mol. Ecol. 2004, 13, 2303–2315. [Google Scholar] [CrossRef]
- Harrison, S.P. Plant and Animal Endemism in California; University of California Press: Berkeley, CA, USA, 2013. [Google Scholar]
- Ricketts, T.H.; Dinerstein, E.; Olson, D.; Loucks, C.; Eichbaum, W. Terrestrial Ecoregions of North America: A Conservation Assessment; Island Press: Washington, DC, USA, 1999. [Google Scholar]
- Stein, B.A.; Kutner, L.S.; Adams, J.S. Precious Heritage: The Status of Biodiversity in the United States; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Conservation International. Biodiversity Hotspots: California Floristic Provence. Available online: www.biodiversityhotspots.org/xp/Hotspots/california_floristic/ (accessed on 2 May 2013).
- California Department of Fish and Wildlife (CDFW), Atlas of the Biodiversity of California; California Department of Fish and Wildlife: Sacramento, CA, USA, 2003.
- Edwards, S.V.; Beerli, P. Perspective: Gene divergence, population divergence, and the variance in coalescent time in phylogeographic studies. Evolution 2000, 54, 1839–1854. [Google Scholar]
Appendix

© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reilly, S.B.; Mulks, M.F.; Reilly, J.M.; Jennings, W.B.; Wake, D.B. Genetic Diversity of Black Salamanders (Aneides flavipunctatus) across Watersheds in the Klamath Mountains. Diversity 2013, 5, 657-679. https://doi.org/10.3390/d5030657
Reilly SB, Mulks MF, Reilly JM, Jennings WB, Wake DB. Genetic Diversity of Black Salamanders (Aneides flavipunctatus) across Watersheds in the Klamath Mountains. Diversity. 2013; 5(3):657-679. https://doi.org/10.3390/d5030657
Chicago/Turabian StyleReilly, Sean B., Mitchell F. Mulks, Jason M. Reilly, W. Bryan Jennings, and David B. Wake. 2013. "Genetic Diversity of Black Salamanders (Aneides flavipunctatus) across Watersheds in the Klamath Mountains" Diversity 5, no. 3: 657-679. https://doi.org/10.3390/d5030657
APA StyleReilly, S. B., Mulks, M. F., Reilly, J. M., Jennings, W. B., & Wake, D. B. (2013). Genetic Diversity of Black Salamanders (Aneides flavipunctatus) across Watersheds in the Klamath Mountains. Diversity, 5(3), 657-679. https://doi.org/10.3390/d5030657