Band Gap Tuning in 2D Layered Materials by Angular Rotation

 ,
,
Abstract
:
1. Introduction
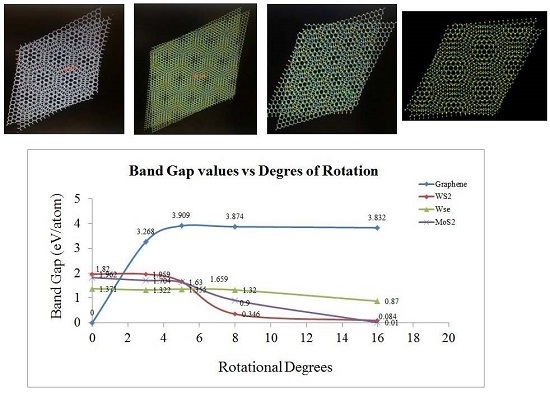
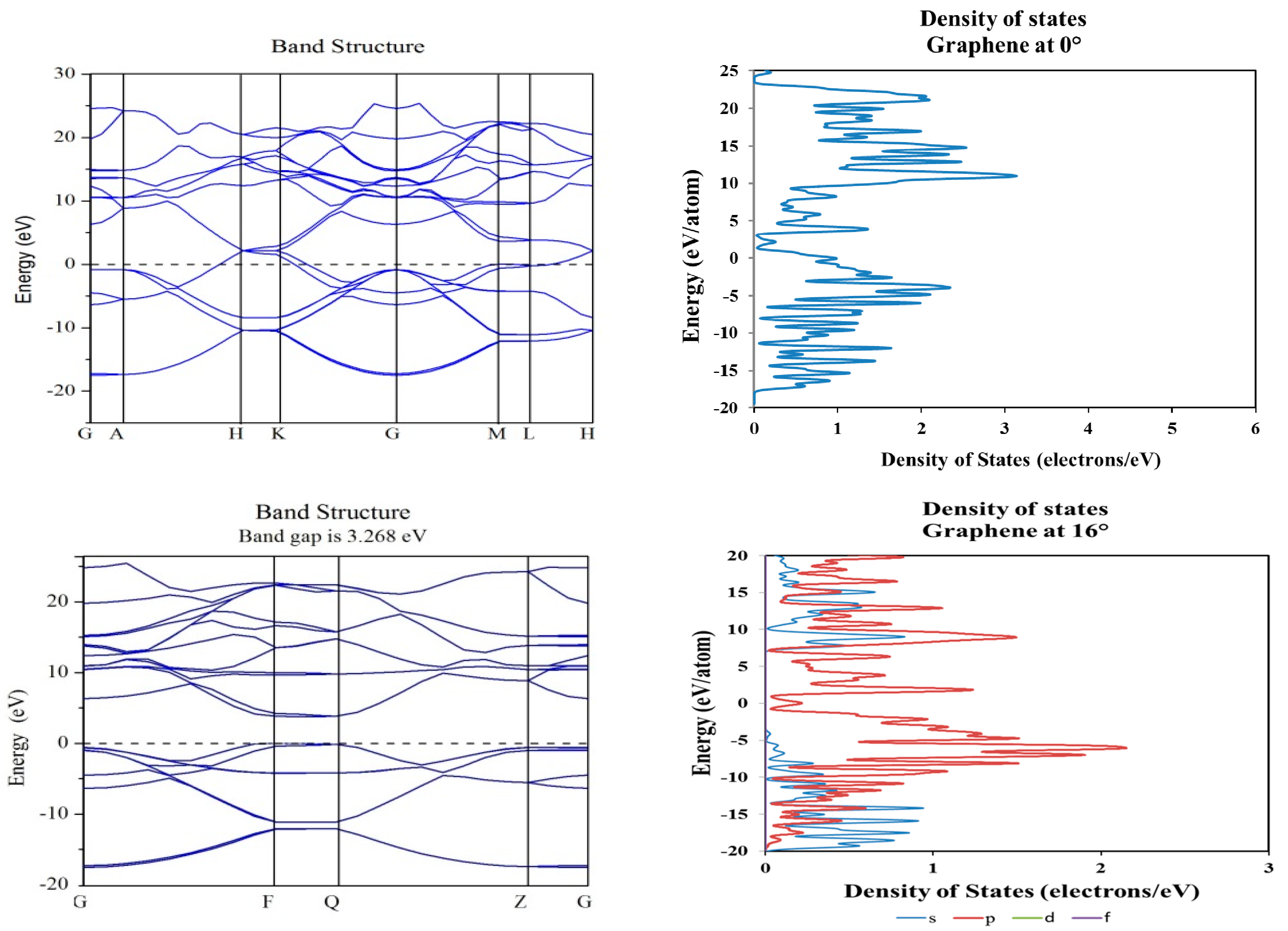
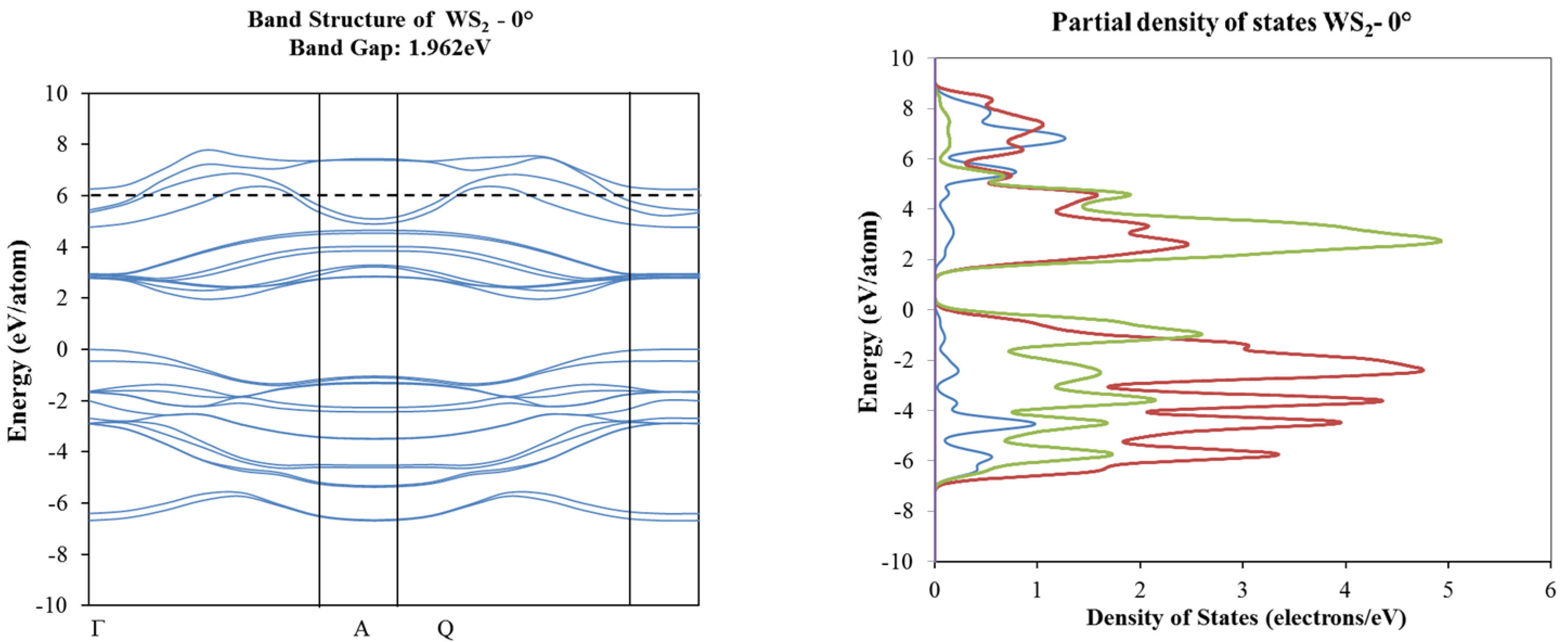
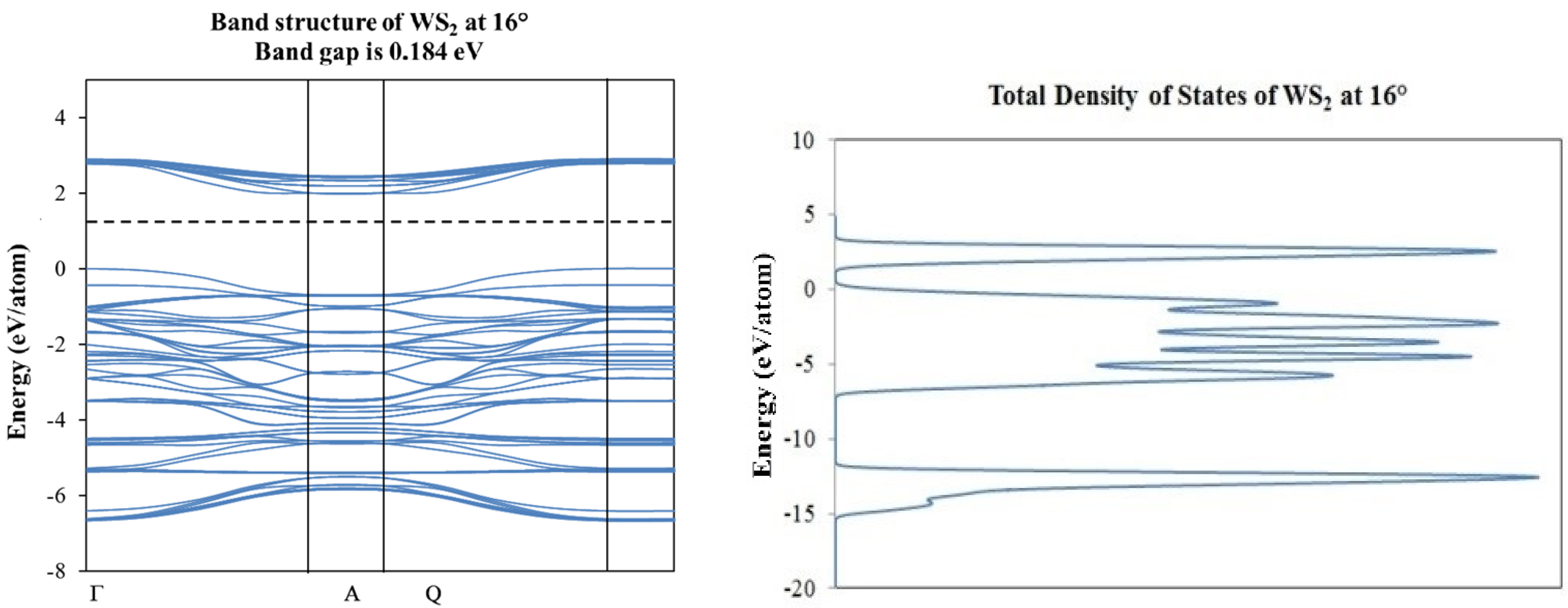
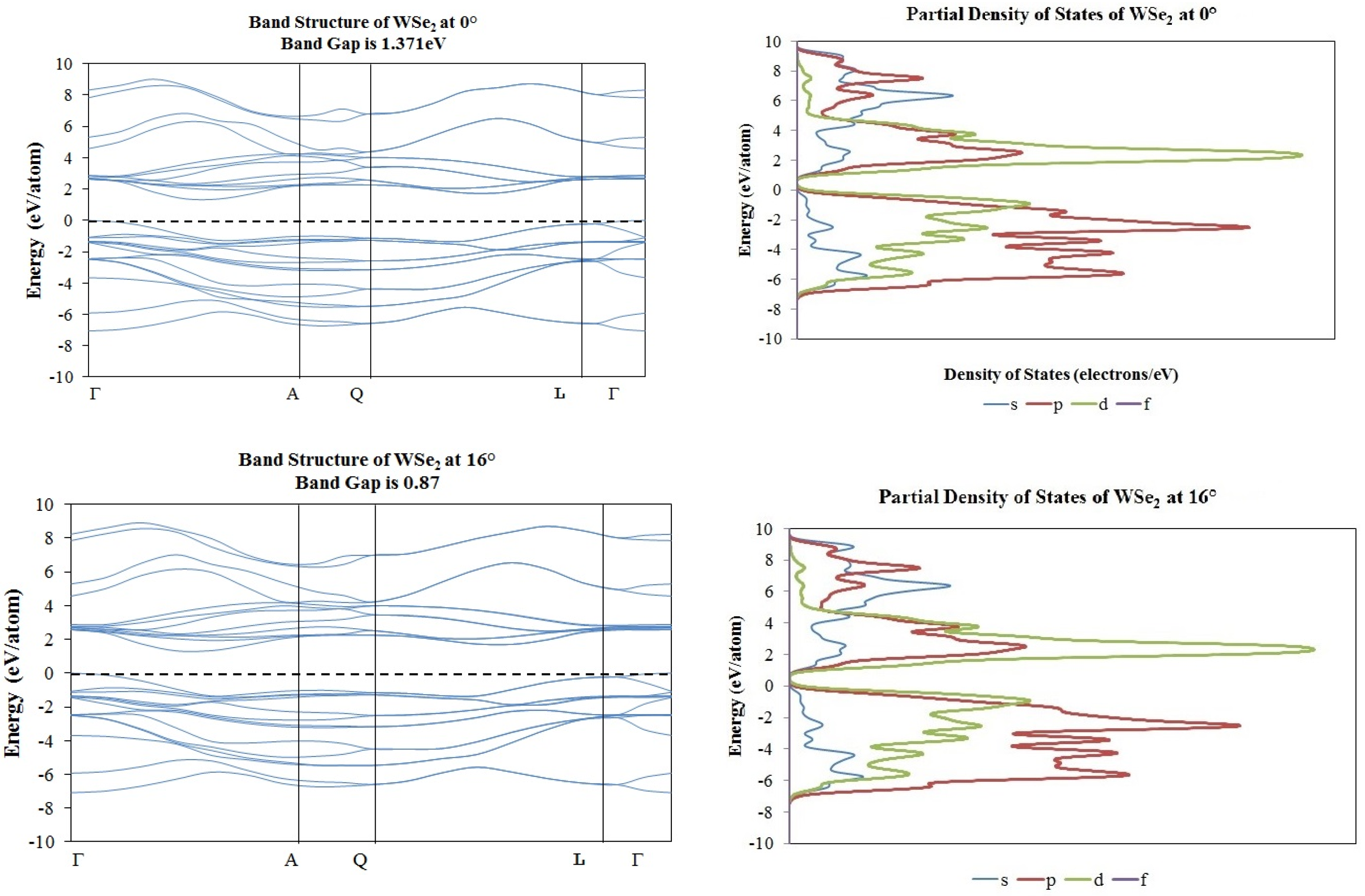
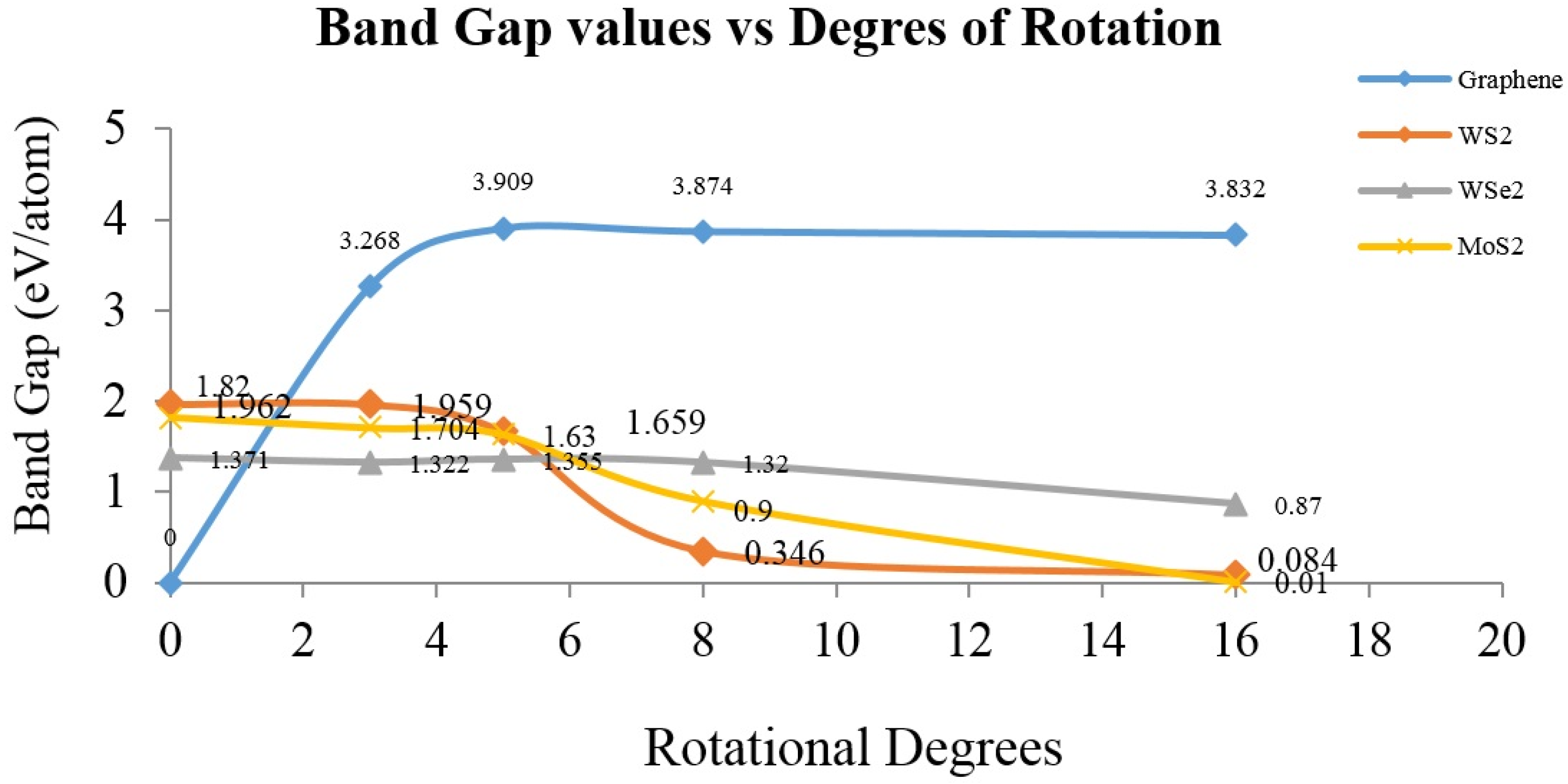
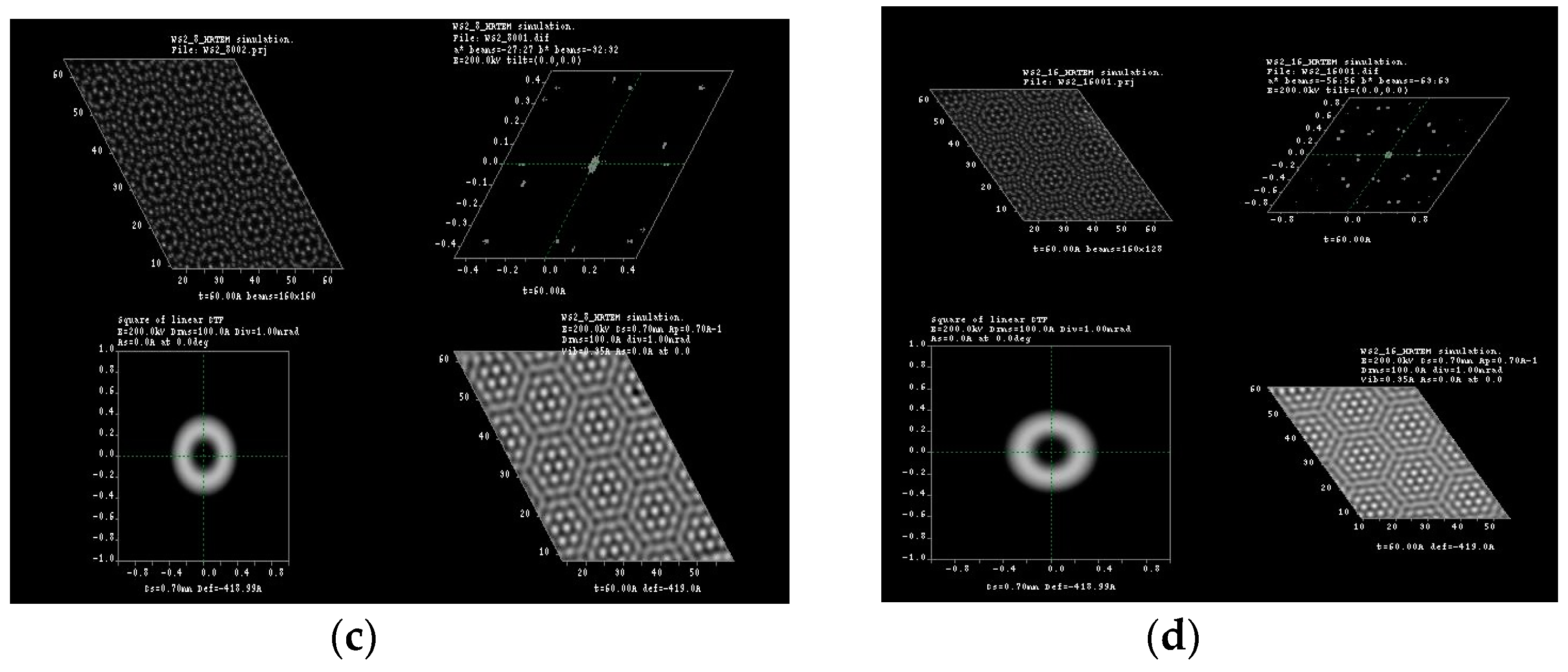
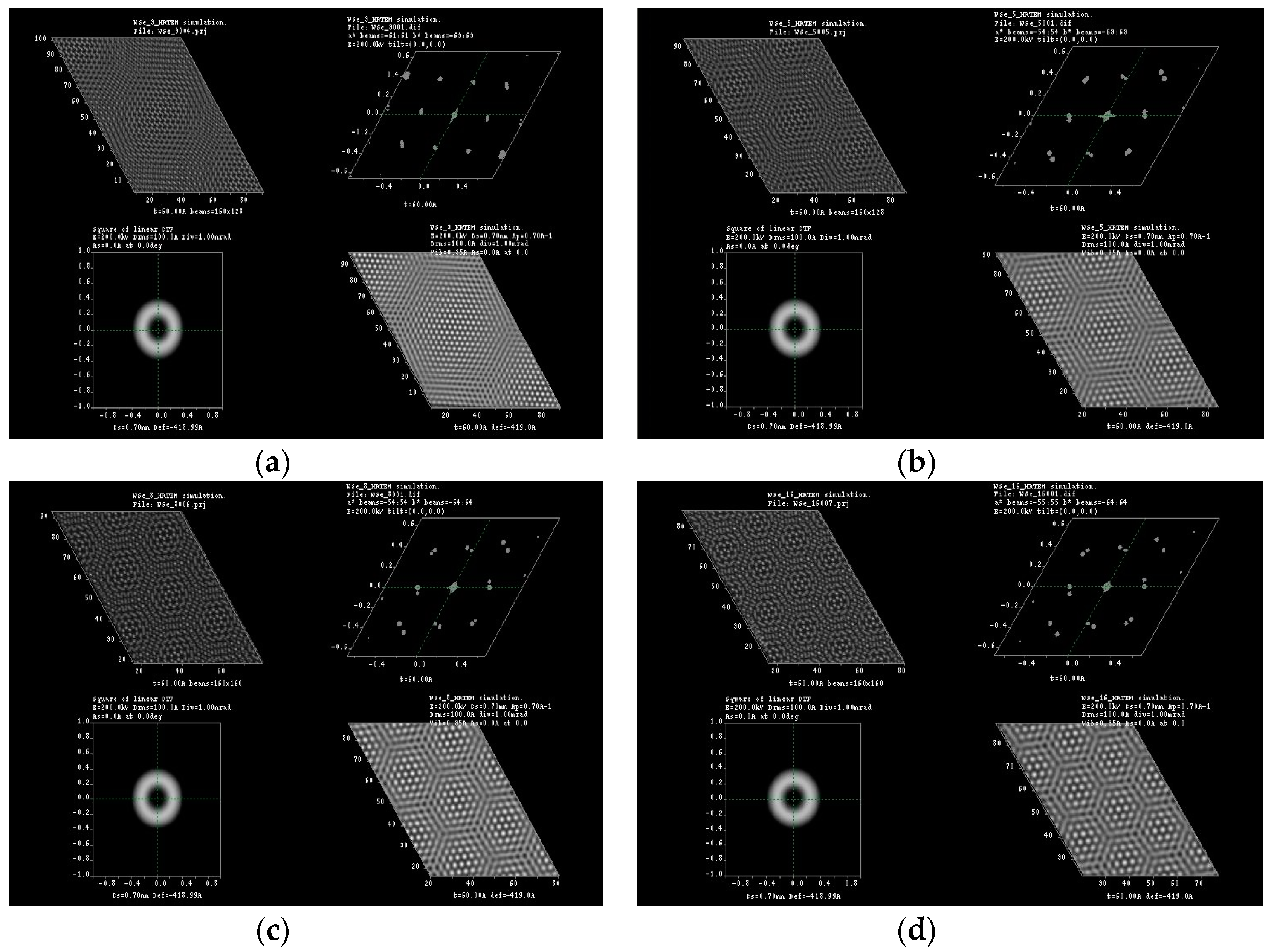
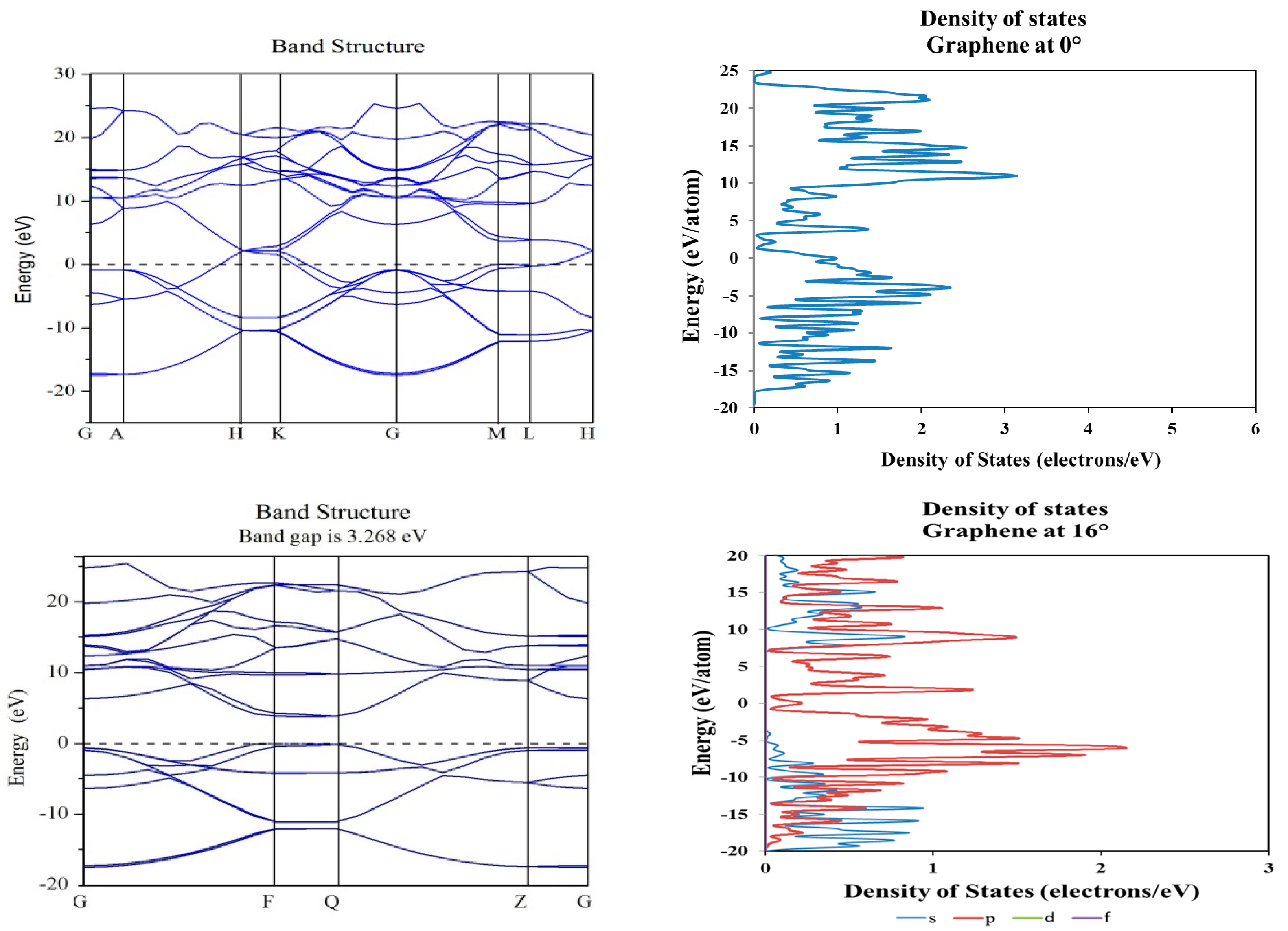
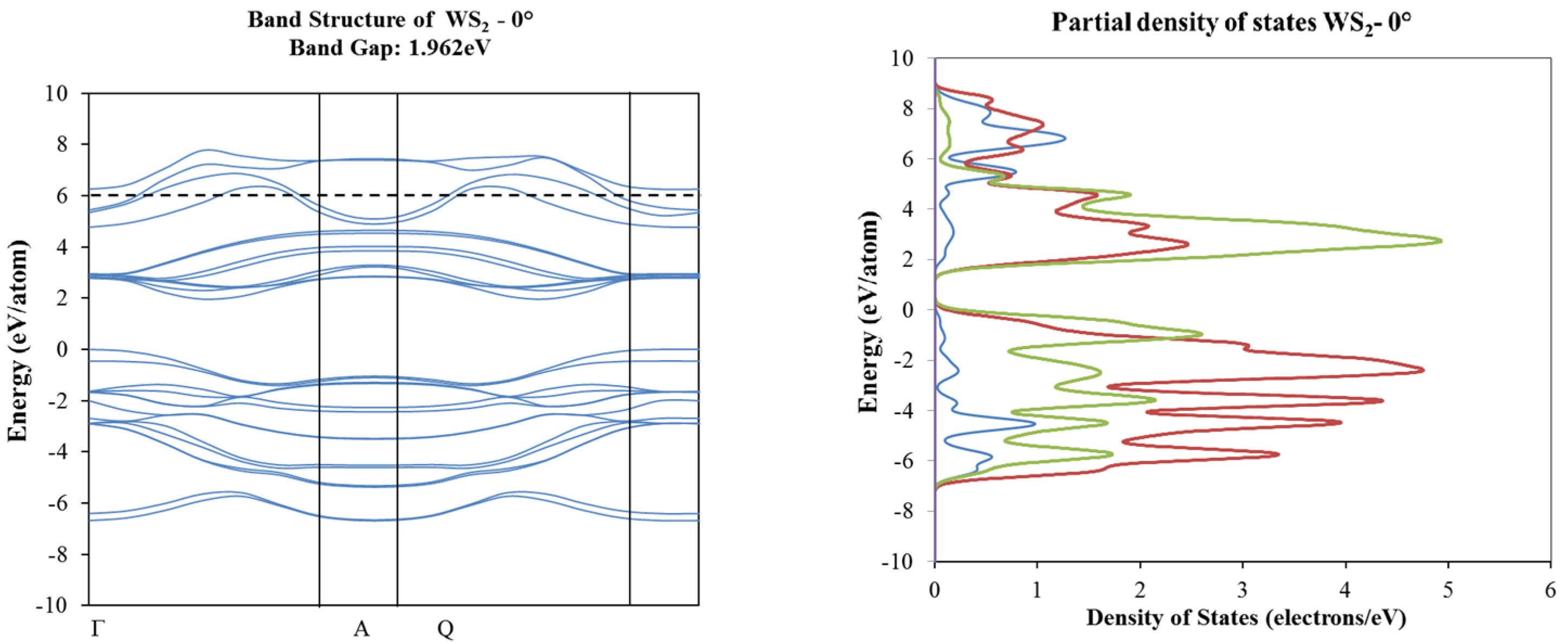
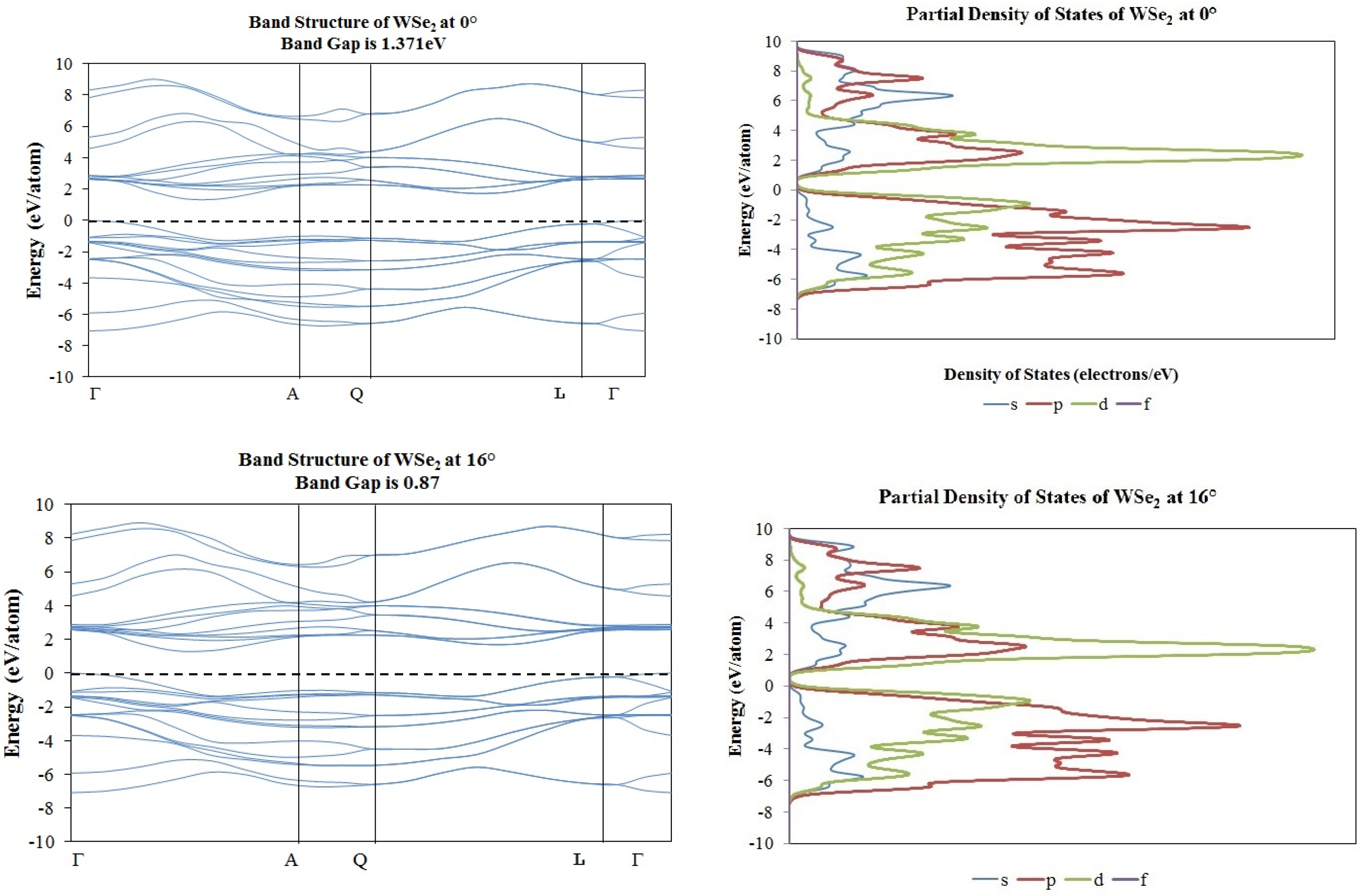
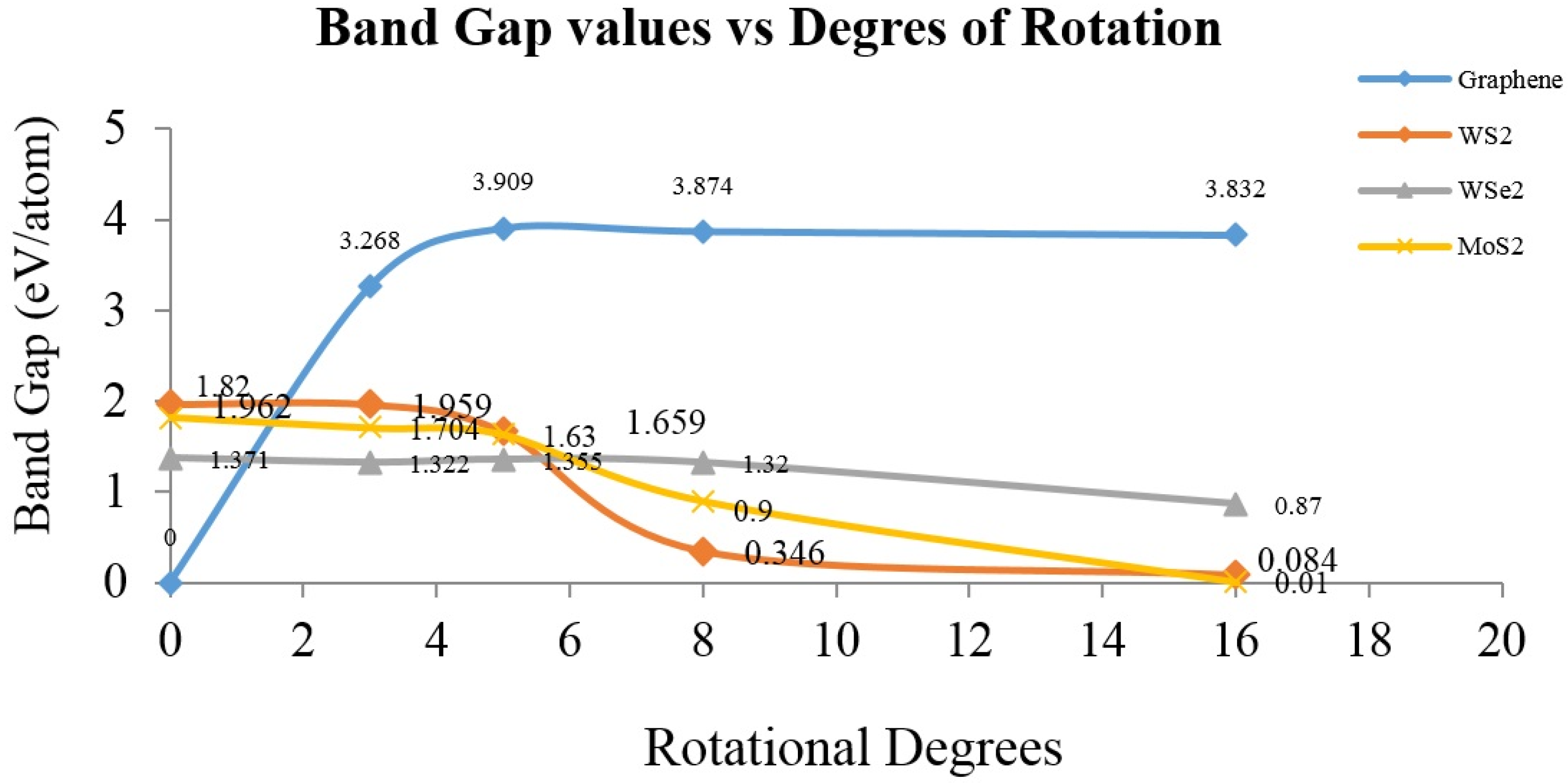
2. Results and Discussion
3. Computational Details
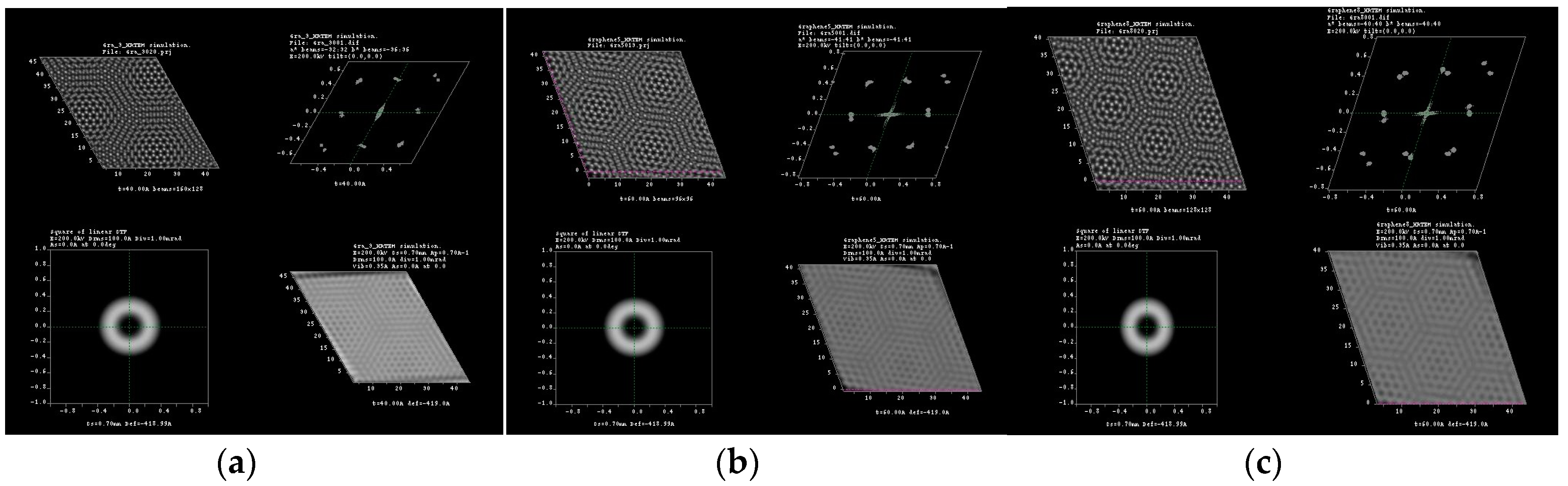
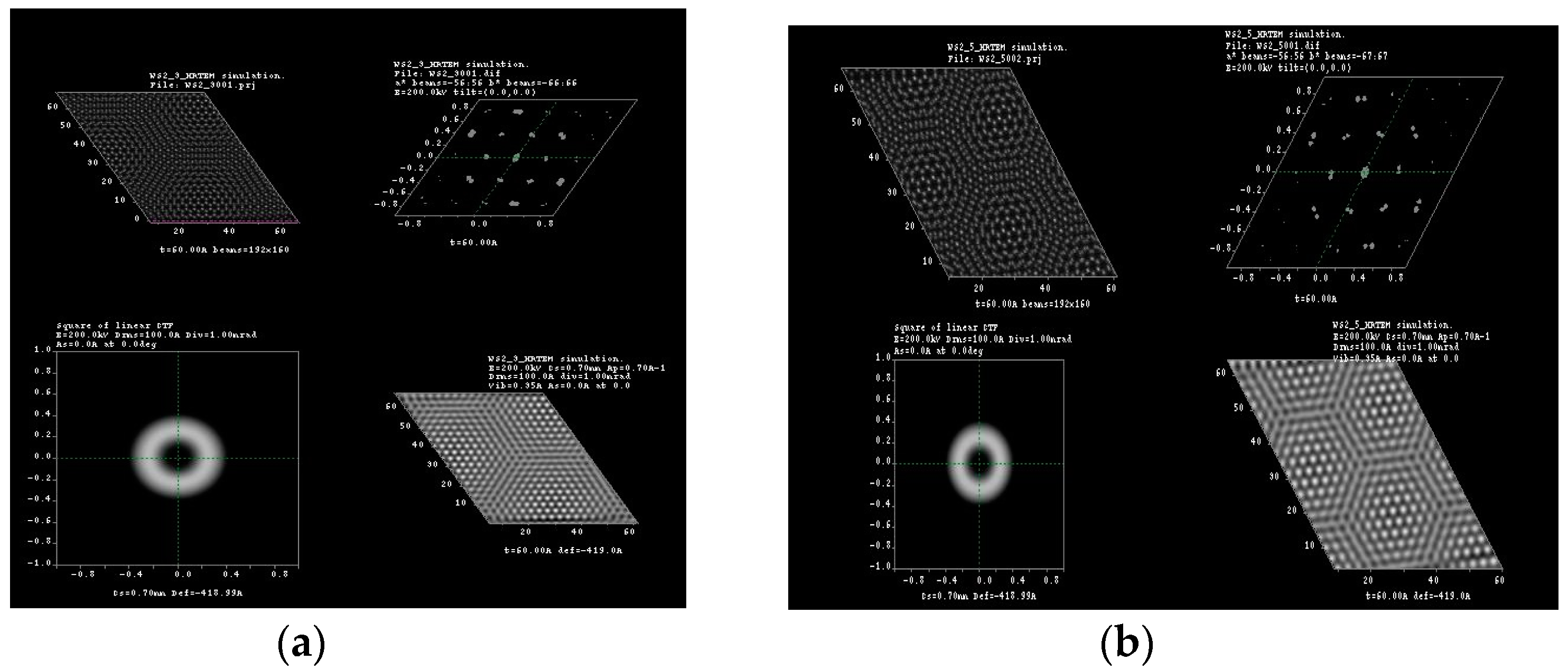
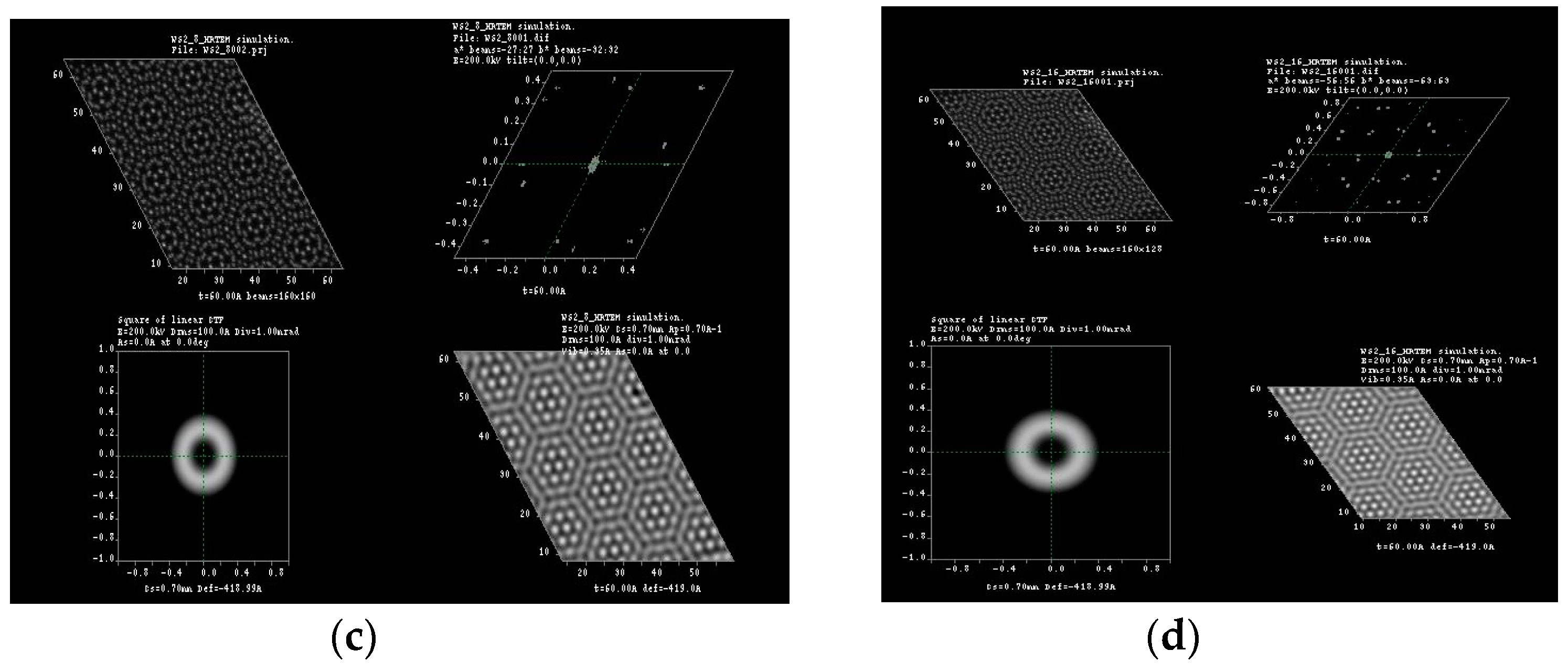
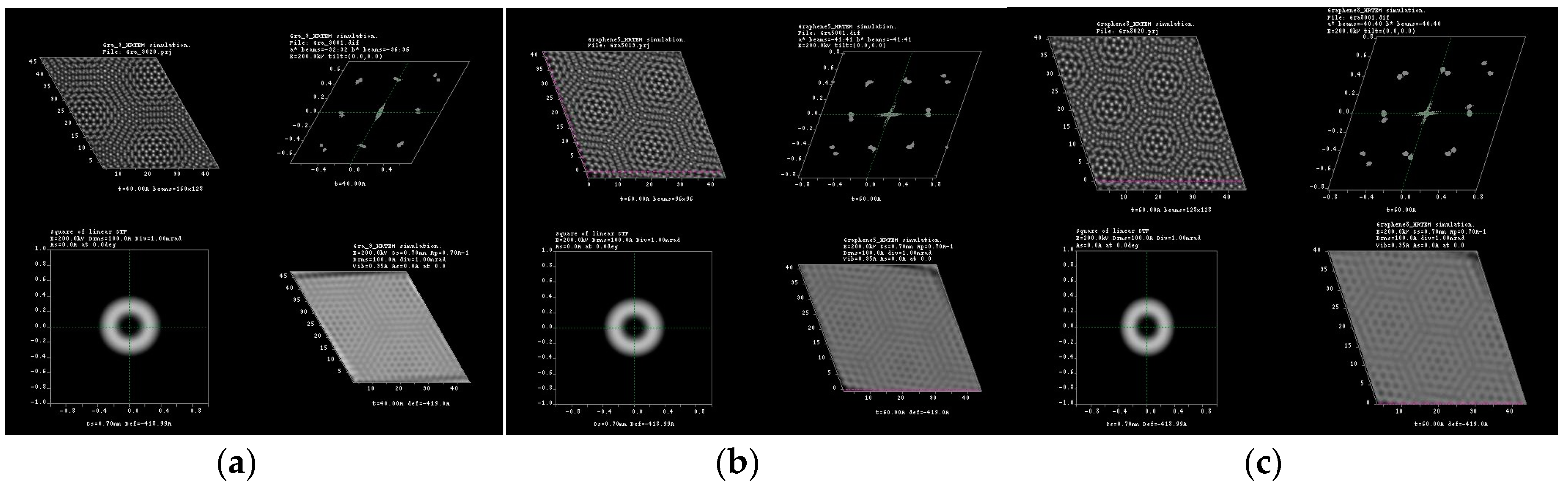
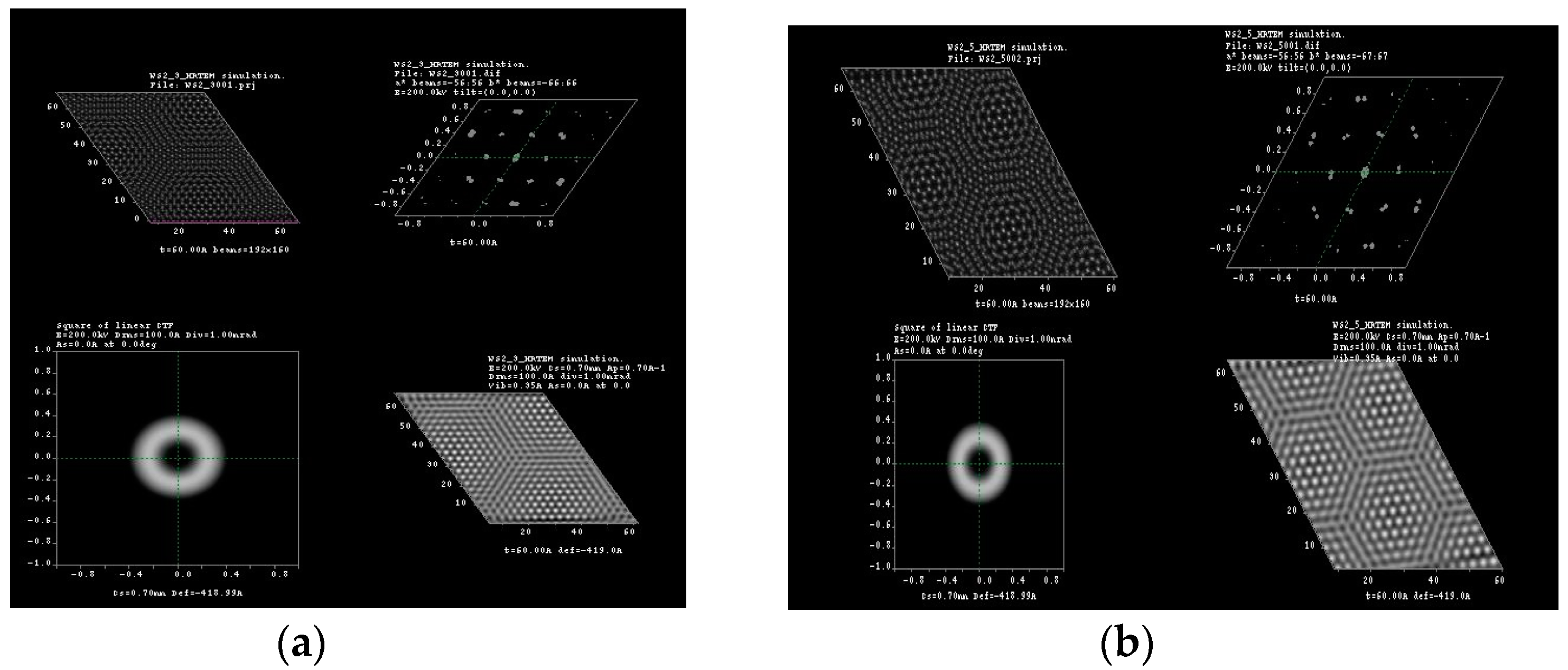
3.1. Multislice HRTEM Simulations
3.2. Density Functional Theory Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weisser, O.; Landa, S. Sulfide Catalysts, Their Properties and Applications; Pergamon Press: New York, NY, USA, 1973. [Google Scholar]
- Perea-López, N.; Lin, Z.; Pradhan, N.R.; Iñiguez-Rábago, A.; Elías, A.L.; McCreary, A.; Lou, J.; Ajayan, P.M.; Terrones, H.; Balicas, L.; et al. CVD-grown monolayered MoS2 as an effective photosensor operating at low-voltage. 2D Mater. 2014, 1, 011004. [Google Scholar] [CrossRef]
- Chianelli, R.R.; Ruppert, A.F.; Jose, M.; Va, A. HREM studies of layered transition metal sulfide catalytic materials. Catal. Today 1995, 23, 269–281. [Google Scholar] [CrossRef]
- Vazquez, A. High Resolution electron microscopy of MoS2:Ni, MoS2:Co and MoS2:Fe layered crystals. Mater. Lett. 1998, 35, 22–27. [Google Scholar] [CrossRef]
- Kobayashi, K. Moiré patterns in scanning tunneling microscopy images of layered materials. J. Vac. Sci. Technol. B 1996, 14, 175–178. [Google Scholar] [CrossRef]
- Jellinek, F.; Brauer, G.; Müller, H. Molybdenum and Niobium Sulphides. Nature 1960, 185, 376–377. [Google Scholar] [CrossRef]
- Reyes-Gasga, J.; Tehuacanero, S.; Yacamán, M.J. Moiré patterns in high resolution electron microscopy images of MoS2. Microsc. Res. Tech. 1998, 40, 2–9. [Google Scholar] [CrossRef]
- Jasinski, J.B.; Dumpala, S.; Sumanasekera, G.U.; Sunkara, M.K.; Ouseph, P.J. Observation and interpretation of adjacent Moire patterns of different shapes in bilayer graphene. Appl. Phys. Lett. 2011, 99, 073104. [Google Scholar] [CrossRef]
- Ramos, M.A.; Chianelli, R.; Enriquez-Carrejo, J.L.; Gonzalez, G.A.; Berhault, G. Angular depdence of metallic states in 2H-MoS2 slabs. Comput. Mater. Sci. 2014, 84, 18–22. [Google Scholar] [CrossRef]
- Bollman, W. Crysrtal Defects And Crystalline Interfaces; Springer: Berlin, Germany, 1970. [Google Scholar]
- Remskar, M.; Mrzel, A.; Skraba, Z.; Jesih, A.; Ceh, M.; Demšar, J.; Stadelmann, P.; Lévy, F.; Mihailovic, D. Self-Assembly of Subnanometer-Diameter Single-Wall MoS2 Nanotubes. Science 2001, 292, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Gómez Rodríguez, A.; Romeu Casajuana, D. Reciprocity relations for Bollmann's O-lattice. Rev. Mex. Fís. 2007, 53, 139–143. [Google Scholar]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, T.T.; Girit, C.; Hao, Z.; Martin, M.C.; Zettl, A.; Crommie, M.F.; Shen, R.Y.; Wang, F. Direct observation of a widely tunable bandgap in bilayer graphene. Nat. Lett. 2009, 459, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Rodríguez, A.; Beltrán-del-Río, L.M.; Herrera-Becerra, R. SimulaTEM: Multislice simulations for general objects. Ultramicroscopy 2010, 110, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Almlöf, J. General methods for geometry and wave function optimization. J. Phys. Chem. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rotation Angle | Graphene | MoS2 | WSe2 | WS2 |
|---|---|---|---|---|
| 3° | 12.300 Å | 25.328 Å | 22.974 Å | 18.948 Å |
| 5° | 9.840 Å | 15.830 Å | 19.692 Å | 15.796 Å |
| 8° | 8.639 Å | 15.736 Å | 14.899 Å | 9.950 Å |
| 16° | 7.380 Å | 12.725 Å | 9.846 Å | 9.474 Å |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polanco-Gonzalez, J.; Carranco-Rodríguez, J.A.; Enríquez-Carrejo, J.L.; Mani-Gonzalez, P.G.; Domínguez-Esquivel, J.M.; Ramos, M. Band Gap Tuning in 2D Layered Materials by Angular Rotation. Materials 2017, 10, 147. https://doi.org/10.3390/ma10020147
Polanco-Gonzalez J, Carranco-Rodríguez JA, Enríquez-Carrejo JL, Mani-Gonzalez PG, Domínguez-Esquivel JM, Ramos M. Band Gap Tuning in 2D Layered Materials by Angular Rotation. Materials. 2017; 10(2):147. https://doi.org/10.3390/ma10020147
Chicago/Turabian StylePolanco-Gonzalez, Javier, Jesús Alfredo Carranco-Rodríguez, José L. Enríquez-Carrejo, Pierre G. Mani-Gonzalez, José Manuel Domínguez-Esquivel, and Manuel Ramos. 2017. "Band Gap Tuning in 2D Layered Materials by Angular Rotation" Materials 10, no. 2: 147. https://doi.org/10.3390/ma10020147