Monoclinic 122-Type BaIr2Ge2 with a Channel Framework: A Structural Connection between Clathrate and Layered Compounds


1
Department of Chemistry, Louisiana State University, Baton Rouge, LA 70803, USA
2
Department of Physics, National Cheng Kung University, Tainan 70101, Taiwan
3
Department of Chemistry, Princeton University, Princeton, NJ 08540, USA
*
Author to whom correspondence should be addressed.
Materials 2017, 10(7), 818; https://doi.org/10.3390/ma10070818
Submission received: 26 June 2017
/
Revised: 8 July 2017
/
Accepted: 10 July 2017
/
Published: 18 July 2017
(This article belongs to the Special Issue Metal-Insulator Transition)
Abstract
:A new 122-type phase, monoclinic BaIr2Ge2 is successfully synthesized by arc melting; X-ray diffraction and scanning electron microscopy are used to purify the phase and determine its crystal structure. BaIr2Ge2 adopts a clathrate-like channel framework structure of the monoclinic BaRh2Si2-type, with space group P21/c. Structural comparisons of clathrate, ThCr2Si2, CaBe2Ge2, and BaRh2Si2 structure types indicate that BaIr2Ge2 can be considered as an intermediate between clathrate and layered compounds. Magnetic measurements show it to be diamagnetic and non-superconducting down to 1.8 K. Different from many layered or clathrate compounds, monoclinic BaIr2Ge2 displays a metallic resistivity. Electronic structure calculations performed for BaIr2Ge2 support its observed structural stability and physical properties.
1. Introduction
Quasi-two-dimensional (2D) layered intermetallic compounds attract broad interest in condensed matter physics and solid state chemistry for their various physical and structural properties. Some of the most well-known examples are Fe-based superconducting families, such as FeSe1−x [1,2], LaFeAsO1−xFx [3], and Ba1−xKxFe2As2 [4]. In addition to the high temperature superconductors, layered compounds host other strong quantum thermal and spin fluctuations, for example, charge-density-waves (CDWs) [5] and spin-density-waves (SDWs) [6]. Structurally, many two-dimensional layered intermetallics, especially of the 122-type, can be traced back to the parent structure, body-centered tetragonal BaAl4 (space group I4/mmm) [7,8]. In BaAl4, the Al atoms, on two independent crystallographic (4d and 4e) sites, form Al@Al4 tetrahedral layers separated by Ba atoms. Derived from BaAl4, two major ternary intermetallic families are the ThCr2Si2 and CaBe2Ge2-types, with 4d and 4e sites occupied by transition metals (T) and metalloids (M) [9,10]. Identical by symmetry through the body centering, the ThCr2Si2 structure contains two equivalent T2M2 layers per cell. Not only are high Tc Fe-based superconductors known in this structure, but strongly correlated electron behavior and magnetic ordering transitions tuned by chemical and physical pressure are also observed [11]. In contrast to ThCr2Si2, the primitive tetragonal CaBe2Ge2 structure consists of alternating T2M2 and M2T2 layers. Currently, no high Tc superconductors have been reported in the CaBe2Ge2 structure; but many exotic properties are found for materials in this structure, such as multiple bands leading to the coexistence of charge density waves and superconductivity in SrPt2As2 [12]. By removing the inversion center from the BaAl4 structure, non-centrosymmetric CeCoGe3 and CePt3Si form, for example [13,14], offering hosts to study superconductivity in non-centrosymmetric structures. Finally, clathrates, which form in different, related structure types, are based on frameworks made primarily of Si, Ge, or Sn (with some M included) with the large atoms are found within the framework cages and are generally known to be semiconducting thermoelectrics, although a small number are known to be superconducting [15,16,17].
The chemical stabilities of many 122-type AT2M2 compounds can be interpreted using Zintl-Klemm concepts [18]. In these compounds, the polyanion T2M2 layers and intermediary cation layers alternate along the stacking (c) axis. From the chemical perspective, the differences in the electronegativity of the layers determine whether insulating, semiconducting, semimetallic, or metallic behavior is observed [19]. Here, we report our recent discovery of the 122-phase BaIr2Ge2. The clathrate-like channel framework of BaIr2Ge2 can be regarded as an intermediate structure between clathrate and the normally layered compounds with 122 stoichiometry. The new structural motif for a heavy metal 122 germanide offers a new platform to study structure-property relationships in such compounds. The crystal structure, basic electronic and magnetic properties, and calculated electronic structure are presented in the following.
2. Experimental
2.1. Synthesis of Monoclinic BaIr2Ge2
The synthesis of BaIr2Ge2 was performed by arc melting methods, similar to that used for the synthesis of BaIrGe3 [20]. Starting materials were barium (>99%, rod, Alfa Aesar, Ward Hill, MA, USA), iridium (99.9%, powder, ~325 mesh, Alfa Aesar) and germanium (99.9999%, pieces, Alfa Aesar). The Ir and Ge were weighed in a 1:1 atomic ratio and were arc-melted together under a high purity, Zr-gettered, argon atmosphere. The monoclinic BaIr2Ge2 phase was obtained by arc-melting the shiny IrGe droplet and Ba pieces (at 50% excess). BaIr2Ge2 was stored in the glove box due to its sensitivity to both air and moisture. To investigate the phase stability at different temperatures, we put the as-cast samples into an alumina crucible, which were subsequently sealed in an evacuated (10-5 torr) quartz tube and were annealed at 800 °C or 1000 °C for four days. After annealing, the BaIr2Ge2 compound decomposed to IrGe and unidentified phases.
2.2. Phase Identification
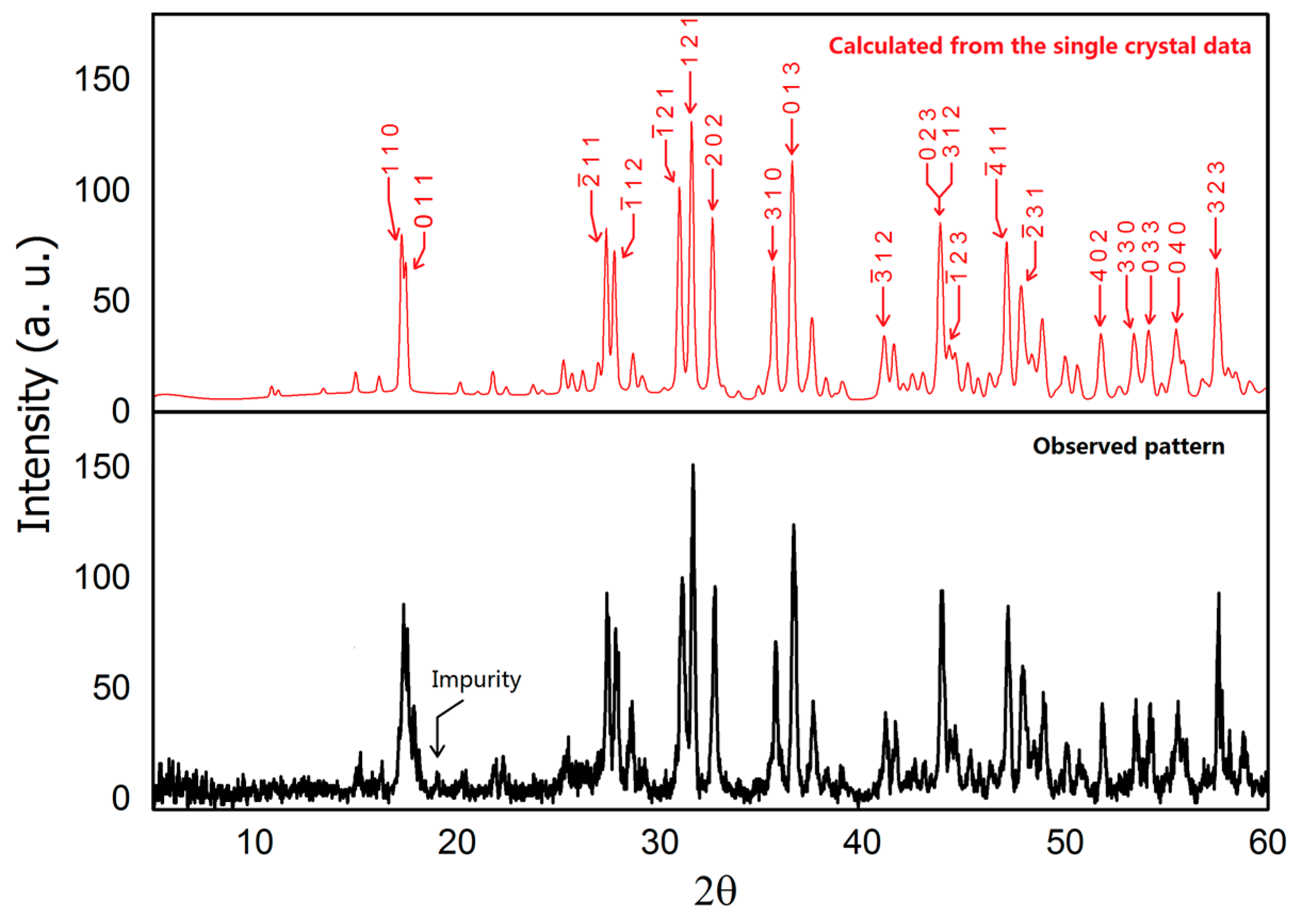
Powder X-ray diffraction data was collected using a Rigaku MiniFlex 600 powder X-ray diffractometer (Rigaku, Tokyo, Japan) equipped with Cu Kα radiation (λ = 1.5406 Å, Ge monochromator). A Bragg angle 2θ ranging from 5° to 60° with a 0.01° step with a fast scanning mode was employed due to the air-sensitivity of BaIr2Ge2. The patterns were analyzed using the LeBail method with Jana2006 [21]. (Lower panel in Figure 1) The calculated pattern in Figure 1 (upper panel) was generated using the crystal structure determined from the single crystal X-ray diffraction results.
2.3. Single Crystal Structure Determination
More than five small single crystals (~0.01 × 0.01 × 0.05 mm3) from the arc-melted samples of BaIr2Ge2 were tested to probe the homogeneity of the new phase. A Bruker Apex II diffractometer (Bruker, Billerica, MA, USA) with Mo radiation (λKα = 0.71073 Å) was utilized to analyze the sample. The single crystals protected with glycerol were mounted on a Kapton loop (MiTeGen, New York, NY, USA) and scanned with a 2θ range of 5–65° at room temperature. The exposure time was set as 10 s per frame, and the width of scans was 0.5°. To solve the crystal structure, we used direct methods and full-matrix least-squares on F2 within the SHELXTL package [22]. Bruker SMART software was applied to make data acquisition, intensity extraction, and corrections for Lorentz and polarization effects [23].
2.4. Magnetic Property Measurements
A quantum design physical property measurement system (PPMS) Dynacool (Quantum Design, San Diego, CA, USA) was used to measure the basic magnetic and electronic properties of BaIr2Ge2. A range from 0 T to 9 T magnetic field at 1.8 K was applied to obtain the field-dependent magnetization. Temperature-dependent magnetization measurements were performed under a magnetic field of 5 T. The 4-probe, zero-field, resistance measurements were carried out in the temperature range from 1.8 K to 300 K.
2.5. Electronic Structure Calculations
Crystal orbital hamilton population (COHP) calculations using the tight-binding linear-muffin-tin-orbital (TB-LMTO) method were performed to analyze the atomic interactions in BaIr2Ge2 and its chemical stability. The k-point mesh in the Brillouin zone was set up as 7 × 8 × 7 to perform the calculations. The electronic structures including the density of states (DOS) and the band structure of BaIr2Ge2 were calculated using the Vienna Ab initio Simulation Package (VASP) based on density functional theory (DFT) [24] with the use of the generalized gradient approximation (GGA) [25]. Spin-orbit coupling (SOC) was included for all the atoms. The cutoff energy was set at 500 eV. A 7 × 8 × 7 Monkhorst-Pack k-point mesh with the linear tetrahedron method was used to perform the calculations. The convergence criterion was set to less than 0.1 meV per atom.
3. Results and Discussion
According to previous research, most 122-type compounds involving alkali-earth metals, group 14 elements, and cobalt group elements (Co/Rh/Ir) adopt the body-centered tetragonal ThCr2Si2-type [18,26,27,28,29,30,31,32,33]. However, only two reported compounds, BaRh2Si2 and BaIr2Si2, crystallize in BaRh2Si2-type structure [34]. Different from CaBe2Ge2 and ThCr2Si2, which are of the tetragonal layered unit cell, BaRh2Si2 structure belongs to the monoclinic system. Our synthetic exploration of BaIr2Ge2 has BaRh2Si2 structure type according to the single crystal X-ray diffraction. Their crystal structures will be discussed in a subsequent section.
3.1. Phase Identification and Structure Determination of BaIr2Ge2
The existence of the new BaIr2Ge2 phase was first seen through the analysis of the powder X-ray diffraction data. Single crystal X-ray diffraction was then used to determine the chemical structure and compositions of BaIr2Ge2. The crystal structure determined is similar to that of BaRh2Si2. The powder XRD pattern was successfully indexed and refined using the crystal structure obtained from single crystal XRD. The refined lattice parameters of BaIr2Ge2 are slightly larger than the ones observed in BaIr2Si2, which is reasonable due to the atomic radius difference between Si and Ge. The results of the diffraction investigation are summarized in Table 1 and Table 2 which include the atomic positions, site occupancies, and isotropic thermal displacements. Results for refinements using anisotropic thermal displacements are summarized in Table S2 of the Supporting Information. The BaIr2Ge2 structure crystallizes in the primitive monoclinic space group P21/c (No. 14) with 20 atoms per unit cell distributed among five crystallographic sites in each unit cell. Mixed site occupancy models have been tested to show whether the atomic distribution in BaIr2Ge2 is ordered and stoichiometric. The crystal structure, shown in Figure 2, is based on an Ir-Ge channel filled with Ba atoms. The view along the a-axis, illustrated in Figure 2a, emphasizes the Ba-filled Ir-Ge framework. Each of the Ir atoms is surrounded by four Ge atoms, forming irregular tetrahedra; similarly, Ge atoms are surrounded by four Ir atoms. The Ir-Ge distances range from 2.40 Å to 2.50 Å. The Ir@Ge4 and Ge@Ir4 clusters share edges and form the channel along the a-axis. The diameter of the columnar channel is approximately 6.45 Å, which is sufficient for hosting some small chemical molecules (with the Ba removed), such as carbon dioxide and methane.
3.2. Structural Comparison of the Different 122 Phases
The results of the analysis of the bonding interactions using the crystal orbital hamilton population (COHP) method are shown in Figure 2e. The atomic interactions between Ir and Ge in the Ir-Ge polyanion framework dominate the atomic interactions in BaIr2Ge2. The Fermi level is located in the non-bonding parts in the COHP, which indicates that the structure of BaIr2Ge2 is electronically stable.
The combination of alkaline-earth elements, a group of 14 metalloids, and Co group metals (Co/Rh/Ir) in a 122 atomic ratio yields three 122-type phases—the BaRh2Si2-type (Pearson Symbol, mP20), the ThCr2Si2-type (Pearson Symbol, tI10), and the CaBe2Ge2-type (Pearson Symbol, tP10). To estimate the structural preferences, the total energies of BaIr2Ge2 in different 122-type structures were calculated using WIEN2k codes. According to calculations for the total energies of these structures, the BaRh2Si2-type gives the lowest energy for BaIr2Ge2, which agrees with our experimental observations.
3.3. Structural Connections between Clathrate and Layered Compounds
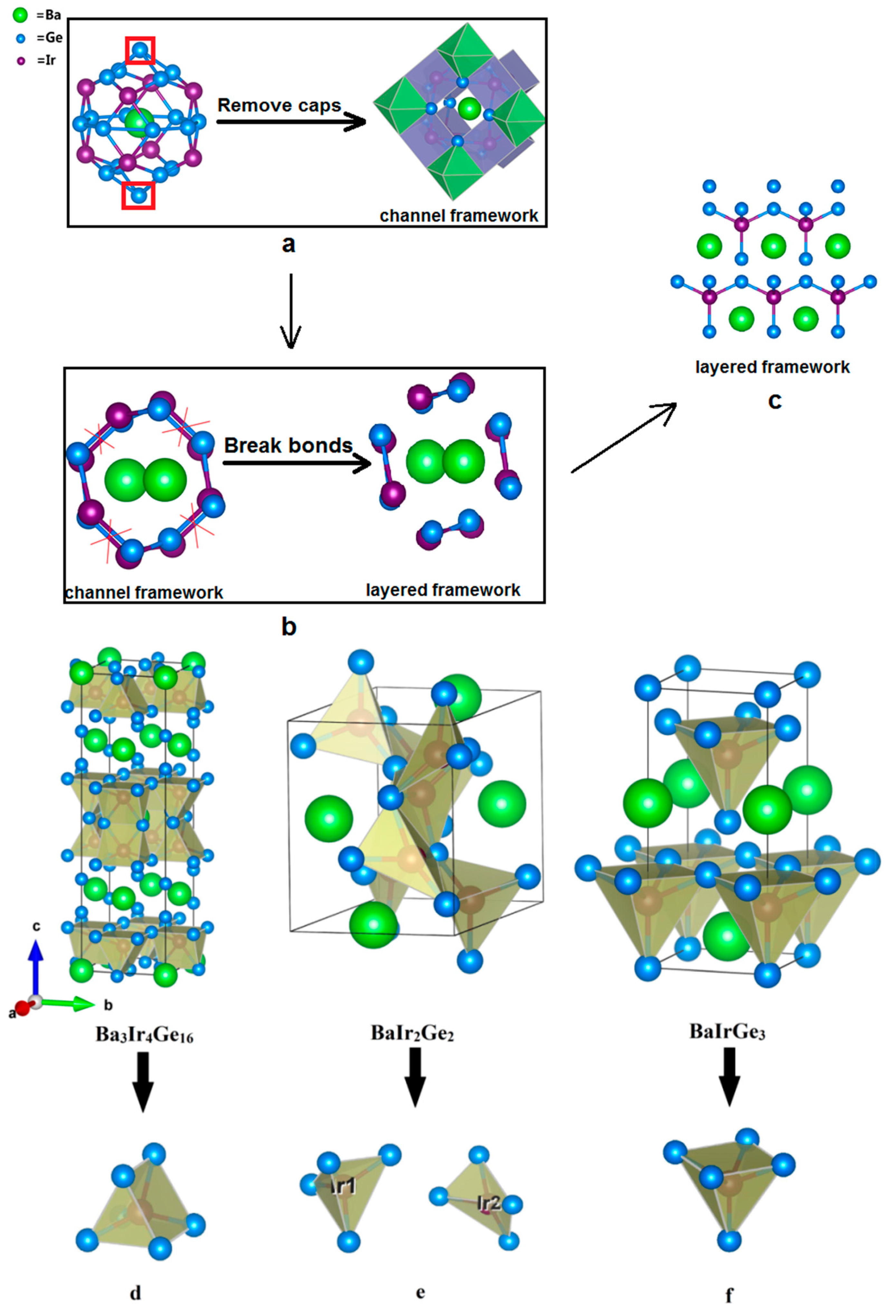
The other interesting ternary compounds in the Ba-Ir-Ge system are the body-centered tetragonal superconductor, Ba3Ir4Ge16 [35] and non-centrosymmetric tetragonal BaIrGe3 [36]. As shown in Figure 3, Ir-centered square pyramids, symmetrical pentahedra, and irregular, non-symmetrical tetrahedra are formed in BaIrGe3, Ba3Ir4Ge16, and BaIr2Ge2, respectively. The crystal structure of the superconductor Ba3Ir4Ge16 reveals that it contains the unique edge-shared crown-shaped Ba@Ge16 polyhedra [35]. The systems with heavy cations “rattling” inside the oversized lattice cavities are called clathrate type structures. Moreover, the open channels around the chains filled with electropositive metals can be considered as clathrate-like structure forms [16]. Accordingly, BaIr2Ge2 can also be regarded as the clathrate-like compound with Ba rattling inside the open chains, which consist of edge-sharing Ir@Ge4 irregular tetrahedra in Figure 3. One can easily see that BaIrGe3 is a layered compound with Ir centered in the vertex-sharing Ge square pyramids along the ab-plane. Therefore, the clathrate-like BaIr2Ge2 structure is likely an intermediate structure between regular clathrate and layered structures.
3.4. Magnetic Properties of Monoclinic BaIr2Ge2
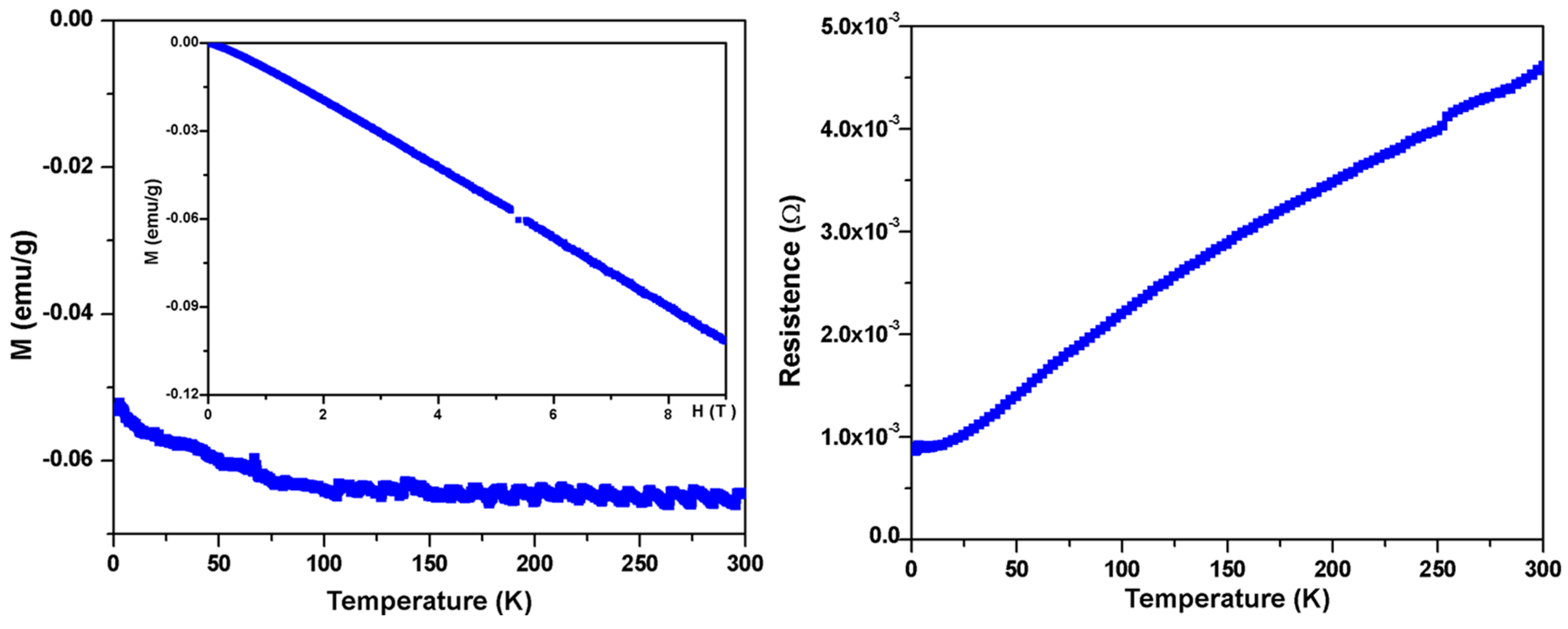
To further study the physical properties of BaIr2Ge2, magnetic measurements were carried out. First, no superconductivity was observed above 1.8 K (in low applied field (20 Oe) measurements). Moreover, the relatively small temperature-independent molar magnetic susceptibility, presented in Figure 4 (Left, Main Panel), is dominated by core diamagnetism; the magnetic susceptibility of the material is around −6 × 10−2 emu/g. The magnetic isotherm measurements in Figure 4 (Left, Inserted) showing diamagnetic behavior at 1.8 K are in agreement with the temperature-dependent magnetization. This indicates that the paramagnetic contribution of conduction electrons to the observed susceptibility is small, which is indirect evidence to support the Zintl-like characteristics of BaIr2Ge2. The resistance measurements in Figure 4 (Right) show a metallic behavior with a residual resistance ratio of ~5.
3.5. Electronic Structure Calculations
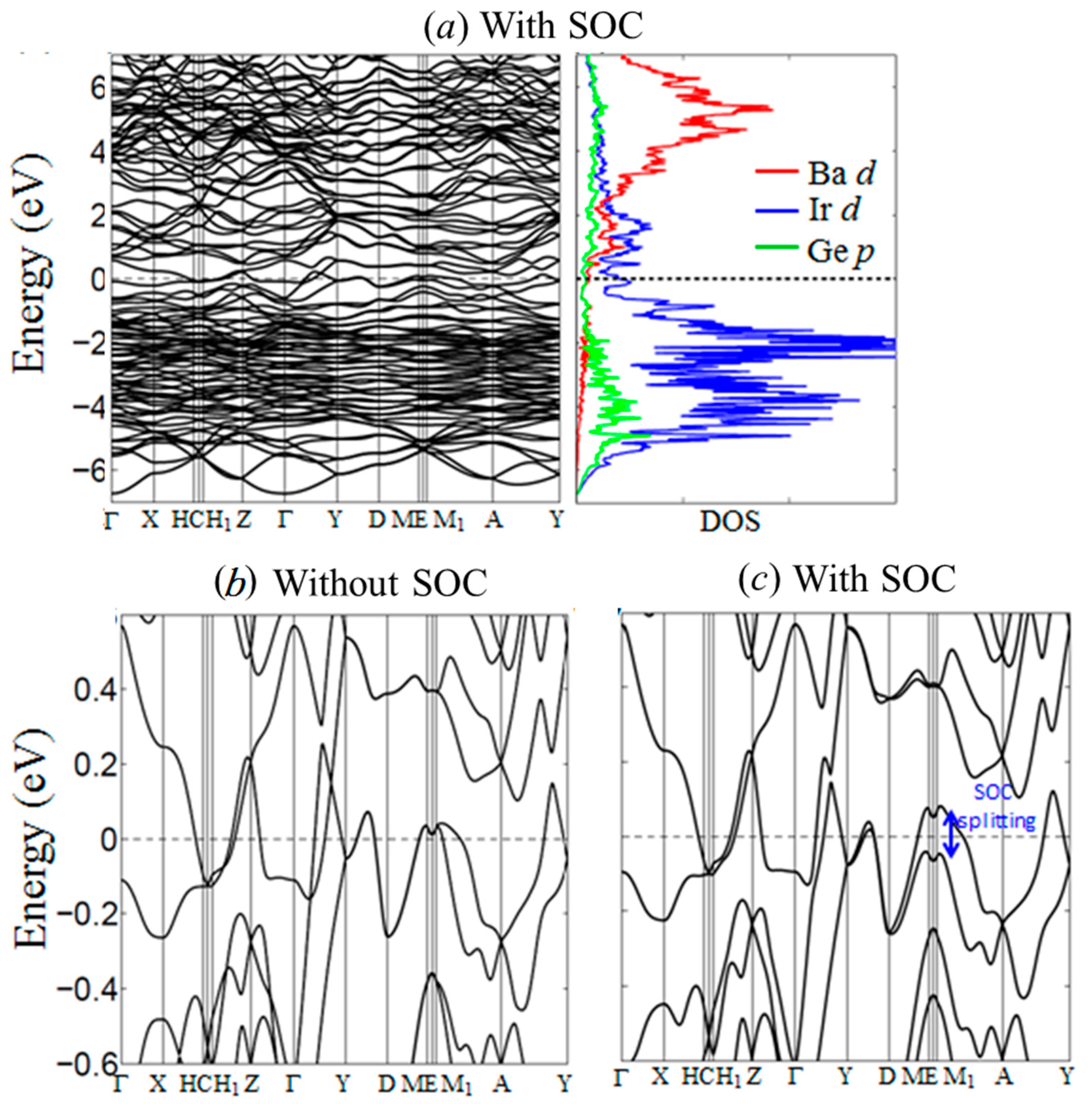
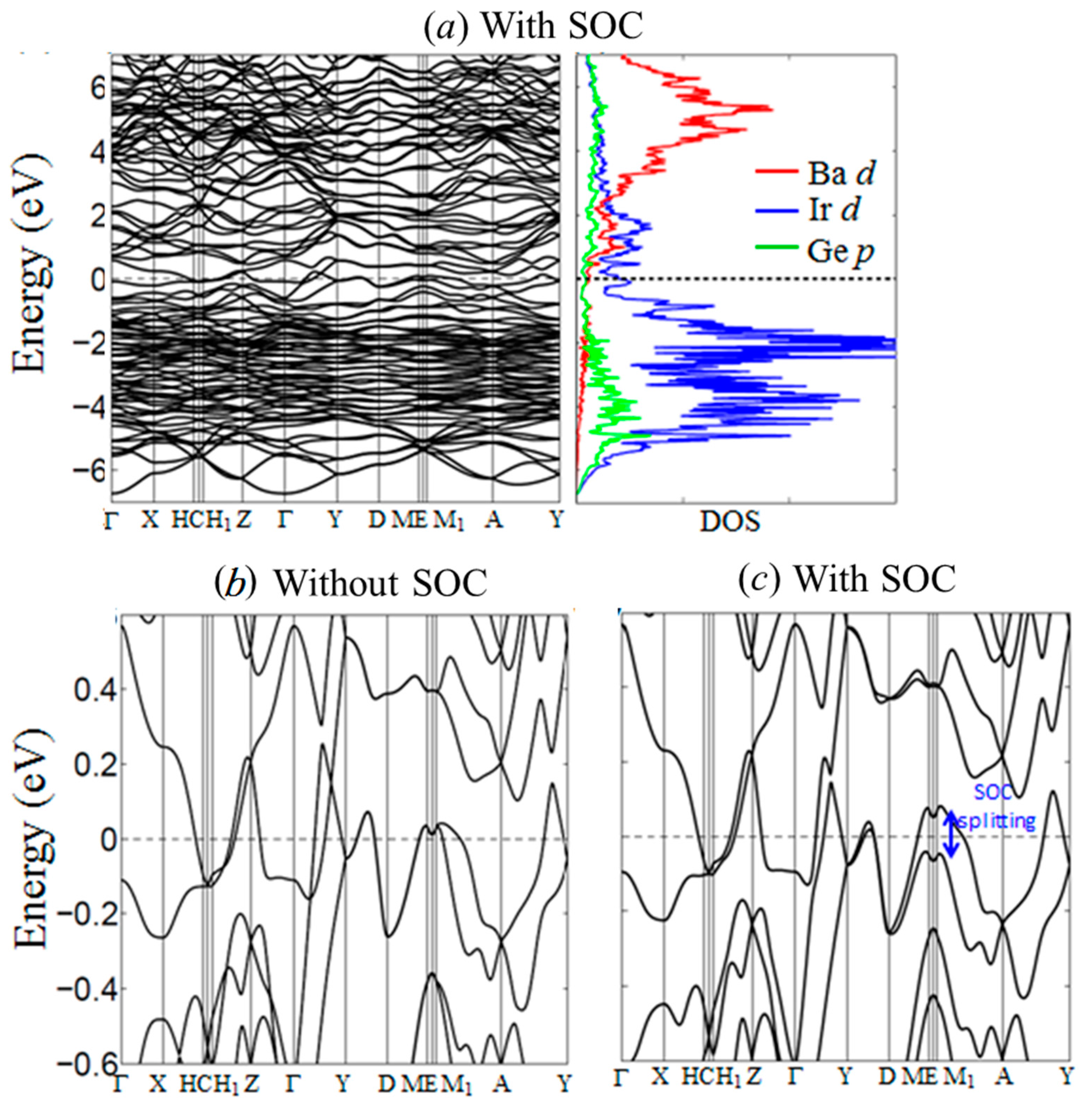
Electronic calculations were carried out to evaluate and analyze the electronic density of states (DOS) and band structure, which can help provide a better understanding of the structural stability and physical properties of BaIr2Ge2. The partial DOS curves (Figure 5a) emphasize the contributions from the valence orbitals of each atom and show that the Fermi level (EF) lies in a broad pseudo gap in the DOS. This is indicative of the chemical stability of the compound, and also, due to the low density of states, is consistent with the diamagnetism and metallic resistivity observed (see below); no saddle point is seen in the band structure in the vicinity of the Fermi level, making the lack of observed superconductivity above 1.8 K consistent (again, see below) with some ideas for what gives rise to superconductivity in many compounds [37]. Based on the comparison of the electronic structure results with and without the inclusion of spin-orbit coupling, the major impact of the SOC near the EF is a band splitting into states derived primarily from the 6s/5d orbitals of the Ir atoms, see Figure 5d. We speculate that doping with electronegative elements like P or A may induce a metal-insulator transition (MIT) in BaIr2Ge2.
4. Conclusions
The new BaIr2Ge2 compound, with monoclinic structure P21/c (S.G.14), was successfully synthesized using arc-melting. The crystal structure of BaIr2Ge2 shows a clathrate-like channel framework of Ir-Ge, which can be regarded as an intermediate structure between clathrate and layered compounds. Electronic structure calculations and chemical bonding interactions of BaIr2Ge2 were investigated to help understand the chemical stability of the compound and its properties. Magnetic measurements indicate diamagnetic susceptibility of BaIr2Ge2 without the observation of superconductivity down to 1.8 K. The resistivity measurements indicate metallic properties for BaIr2Ge2, and electronic structure calculations are consistent with the observed behavior. Future efforts will be focused on inducing a metal-insulator transition in BaIr2Ge2.
Supplementary Materials
The following are available online at www.mdpi.com/1996-1944/10/7/818/s1, Table S1: SEM-EDX, Table S2: Anisotropic Thermal Parameters of BaIr2Ge2 from single crystal diffraction.
Acknowledgments
This research at LSU was supported by LSU-startup funding and the Louisiana Board of Regents Research Competitiveness Subprogram (RCS) under Contract Number LEQSF (2017-20)-RD-A-08. T.-R. Chang was supported by the Ministry of Science and Technology and National Cheng Kung University, Taiwan. T.-R. Chang also thanks National Center for Theoretical Sciences (NCTS), Taiwan for technical support. The physical property characterization at Princeton University was supported by the Department of Energy, Division of Basic Energy Sciences, grant DE-FG02-98ER45706.
Author Contributions
W.X. conceived and designed the experiments; X.G. and M.T.P. performed the experiments; R.J.C. and T.K. contributed physical properties measurements. T.-R.C. contributed electronic structure calculations; all the authors wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Margadonna, S.; Takabayashi, Y.; Mcdonald, M.T.; Kasperkiewicz, K.; Mizuguchi, Y.; Takano, Y.; Fitch, A.N.; Suard, E.; Prassides, K. Crystal structure of the new FeSe1−x superconductor. Chem. Commun. 2008, 43, 5607–5609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-W.; Pardo, V.; Pickett, W.E. Magnetism driven by anion vacancies in superconducting α-FeSe1−x. Phys. Rev. B 2008, 78, 174502. [Google Scholar] [CrossRef]
- Okada, H.; Igawa, K.; Takahashi, H.; Kamihara, Y.; Hirano, M.; Hosono, H.; Matsubayashi, K.; Uwatoko, Y. Superconductivity under high pressure in LaFeAsO. J. Phys. Soc. Jpn. 2008, 77, 113712. [Google Scholar] [CrossRef]
- Rotter, M.; Tegel, M.; Johrendt, D. Superconductivity at 38 K in the iron arsenide (Ba1−xKx)Fe2As2. Phys. Rev. Lett. 2008, 101, 107006. [Google Scholar] [CrossRef] [PubMed]
- Gor’kov, L.P.; Grüner, G. Charge Density Waves in Solids, 1st ed.; North Holland: Amsterdam, The Netherlands, 1989; pp. 1–494. ISBN 9780444600738. [Google Scholar]
- Gruner, G. Density Waves in Solids; reprint; Westview Press: Boulder, CO, USA, 2009; pp. 1–288. ISBN 9780786747795. [Google Scholar]
- Lin, Q.; Miller, G.J.; Corbett, J.D. Ordered BaAl4-type variants in the BaAuxSn4−x system: A unified view on their phase stabilities versus valence electron counts. Inorg. Chem. 2014, 53, 5875–5877. [Google Scholar] [CrossRef] [PubMed]
- Häussermann, U.; Amerioun, S.; Eriksson, L.; Lee, C.-S.; Miller, G.J. The s–p bonded representatives of the prominent BaAl4 structure type: A case study on structural stability of polar intermetallic network structures. J. Am. Chem. Soc. 2002, 124, 4371–4383. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Zheng, C. Making and breaking bonds in the solid state: The thorium chromium silicide (ThCr2Si2) structure. J. Phys. Chem. 1985, 89, 4175–4181. [Google Scholar] [CrossRef]
- Zheng, C.; Hoffmann, R. Donor-acceptor layer formation and lattice site preference in the solid: The CaBe2Ge2 structure. J. Am. Chem. Soc. 1986, 108, 3078–3088. [Google Scholar] [CrossRef]
- Tan, X.; Fabbris, G.; Haskel, D.; Yaroslavtsev, A.A.; Cao, H.; Thompson, C.M.; Kovnir, K.; Menushenkov, A.P.; Chernikov, R.V.; Garlea, V.O.; et al. Transition from localized to strongly correlated electron behavior and mixed valence driven by physical or chemical pressure in ACo2As2 (A = Eu and Ca). J. Am. Chem. Soc. 2016, 138, 2724–2731. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Nishikubo, Y.; Nohara, M. Coexistence of superconductivity and charge density wave in SrPt2As2. J. Phys. Soc. Jpn. 2010, 79, 123710. [Google Scholar] [CrossRef]
- Settaia, R.; Sugitania, I.; Okudaa, Y.; Thamizhavela, A.; Nakashimab, M.; Onukia, Y.; Harimac, H. Pressure-induced superconductivity in CeCoGe3 without inversion symmetry. J. Magn. Magn. Mater. 2007, 310, 844–846. [Google Scholar] [CrossRef]
- Samokhin, K.V.; Zijlstra, E.S.; Bose, S.K. CePt3Si: An unconventional superconductor without inversion center. Phys. Rev. B 2004, 69, 094514. [Google Scholar] [CrossRef]
- Kovnir, K.A.; Zaikina, J.V.; Reshetova, L.N.; Olenev, A.V.; Dikarev, E.V.; Shevelkov, A.V. Unusually high chemical compressibility of normally rigid type-I clathrate framework: Synthesis and structural study of Sn24P19.3BrxI8−x solid solution, the prospective thermoelectric material. Inorg. Chem. 2004, 43, 3230–3236. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Hu, S.; Uher, C.; Hogan, T.; Huang, B.; Corbett, J.D.; Kanatzidis, M.G. Structure and thermoelectric properties of Ba6Ge25−x, Ba6Ge23Sn2, and Ba6Ge22In3: Zintl phases with a chiral clathrate structure. J. Solid State Chem. 2000, 153, 321–329. [Google Scholar] [CrossRef]
- Fukuoka, H.; Kiyoto, J.; Yamanaka, S. Superconductivity and crystal structure of the solid solutions of Ba8−dSi46−xGex (0 ≤ x ≤ 23) with Type I clathrate structure. J. Solid State Chem. 2003, 175, 237–244. [Google Scholar] [CrossRef]
- Klemm, W. Centenary-Lecture-Metalloids and their Compounds with the Alkali Metals. Proc. Chem. Soc. Lond. 1958, 12, 329–341. [Google Scholar]
- Mott, N.F. Metal-Insulator Transitions, 2nd ed.; Taylor & Francis Inc.: London, UK, 1990; pp. 1–34. ISBN 0-85066-783-6. [Google Scholar]
- Kakihana, M.; Akamine, H.; Tomori, K.; Nishimura, K.; Teruya, A.; Nakamura, A.; Honda, F.; Aoki, D.; Nakashima, M.; Amako, Y.; et al. Superconducting, Fermi surface, and magnetic properties in SrTGe3 and EuTGe3 (T: Transition metal) with the Rashba-type tetragonal structure. J. Alloys Compd. 2017, 694, 439–451. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic computing system JANA2006: General features. Z. Kristallogr. Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Bruker. Smart; Bruker AXS Inc.: Madison, WI, USA, 2012; Available online: https://www.bruker.com/products/x-ray-diffraction-and-elemental-analysis/single-crystal-x-ray-diffraction/sc-xrd-software/overview/sc-xrd-software/apex3.html (accessed on 12 July 2017).
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Sato, A. Crystal structure of ternary germanides SrM2Ge2 (M = Ni and Ir). J. Alloys Compd. 2009, 487, 198–201. [Google Scholar] [CrossRef]
- Siggelkow, L.; Hlukhyy, V.; Fässler, T.F. Synthesis, Structure and chemical bonding of CaCo2Si2 and BaCo2Ge2—Two new compounds with ThCr2Si2 Structure type. Z. Anorg. Allg. Chem. 2010, 636, 378–384. [Google Scholar] [CrossRef]
- Venturini, G.; Malaman, B. X-ray single crystal refinements on some RT2Ge2 compounds (R = Ca, Y, La, Nd, U; T = Mn-Cu, Ru-Pd): Evolution of the chemical bonds. J. Alloys Compd. 1996, 235, 201–209. [Google Scholar] [CrossRef]
- Venturini, G.; Malaman, B.; Roques, B. Contribution a la cristallochimie des isotypes de ThCr2Si2 et CaBe2Ge2: II. Variation des distances interatomiques dans des germaniures MM'2Ge2 de type ThCr2Si2 (M = Y, Nd, Ca; M’ = Mn, Cu, Ru, Rh, Pd, Ir). J. Solid State Chem. 1989, 79, 136–145. [Google Scholar] [CrossRef]
- González, J.; Kessens, R.; Schuster, H.U. Darstellung und kristallstruktur neuer AM2X2-verbindungen in den systemen erdalkalimetall-platinmetall-germanium. Z. Anorg. Allg. Chem. 1993, 619, 13–16. [Google Scholar] [CrossRef]
- Hlukhyy, V.; Hoffmann, A.V.; Fässler, T.F. Synthesis, structure and chemical bonding of CaFe2−xRhxSi2 (x = 0, 1.32, and 2) and SrCo2Si2. J. Solid State Chem. 2013, 203, 232–239. [Google Scholar] [CrossRef]
- Doerrscheidt, W.; Niess, N.; Schaefer, H. Neue verbindungen AB2X2 (A = Erdalkalimetall, B = Uebergangselement, X = Element (IV)) im ThCr2Si2-Typ. Z. Naturforsch. B 1976, 31, 890–891. [Google Scholar]
- May, N.; Mueller, W.; Schaefer, H. Ternäre erdalkali-beryllium-silicide und -germanide mit AlB2-struktur. ternary alkaline-earth-beryllium-silicides and -germanides of AlB2-structure. Z. Naturforsch. B 1974, 29, 325–327. [Google Scholar] [CrossRef]
- Langen, D.; Schoolaert, S.; Ploss, H.; Jung, W. Die isotypen verbindungen BaRh2Si2, BaIr2Si2 und BaPt2Ga2-eine monokline verzerrungsvariante der CaRh2B2-strukur. Z. Anorg. Allg. Chem. 1997, 623, 1561–1566. [Google Scholar] [CrossRef]
- Falmbigl, M.; Grytsiv, A.; Rogl, P.; Giester, G. Clathrate formation in the systems Ba-Ir-Ge and Ba-{Rh, Ir}-Si: Crystal chemistry and phase relations. Intermetallics 2013, 36, 61–72. [Google Scholar] [CrossRef]
- Nasir, N.; Melnychenko-Koblyuk, N.; Grytsiv, A.; Rogl, P.; Giester, G.; Wosik, J.; Nauer, G.E. Ternary systems Sr-{Ni, Cu}-Si: Phase equilibria and crystal structure of ternary phases. J. Solid State Chem. 2010, 183, 565–574. [Google Scholar] [CrossRef]
- Baelus, B.J.; Peeters, F.M.; Schweigert, V.A. Saddle-point states and energy barriers for vortex entrance and exit in superconducting disks and rings. Phys. Rev. B 2001, 63, 144517. [Google Scholar] [CrossRef]
Figure 1.
Powder X-ray diffraction pattern of BaIr2Ge2 (Cu Kα radiation, 300 K). Lower—observed pattern; Upper—calculated pattern with marked Miller indices (hkl) based on the single crystal structure.
Figure 1.
Powder X-ray diffraction pattern of BaIr2Ge2 (Cu Kα radiation, 300 K). Lower—observed pattern; Upper—calculated pattern with marked Miller indices (hkl) based on the single crystal structure.
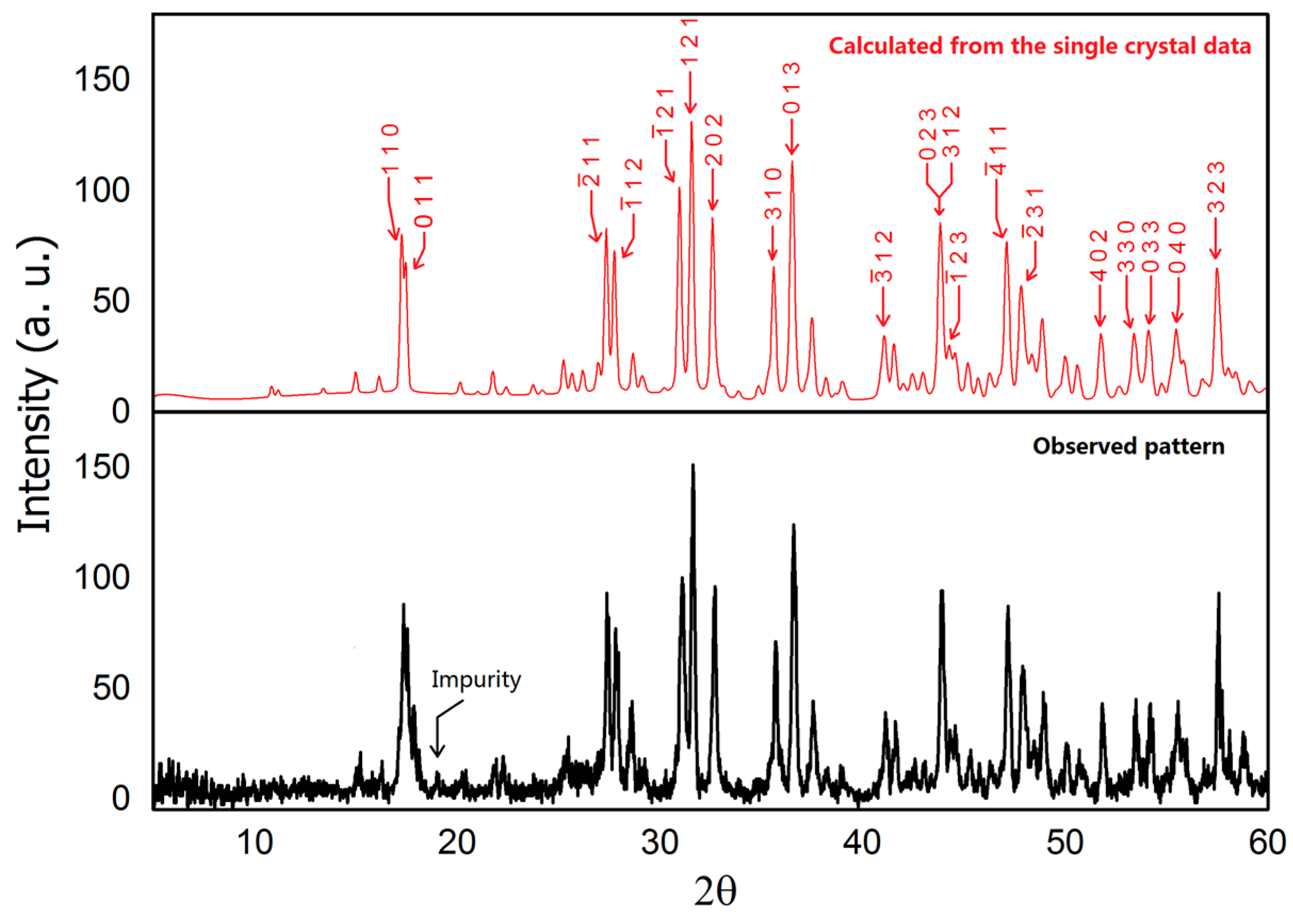
Figure 2.
Crystal structure of monoclinic BaIr2Ge2 refined by single crystal X-ray diffraction. (a) View down the a-axis; (b) View down the b-axis; (c) The IrGe framework. The channels running along the a-axis have a channel of diameter ~6.45 Å; (d) close-up of the framework structure showing the Ir-Ge bond lengths; (e) Crystal orbital hamilton populations (-COHP) calculation emphasis on the Ir-Ge, Ir-Ir, and Ge-Ge interactions.
Figure 2.
Crystal structure of monoclinic BaIr2Ge2 refined by single crystal X-ray diffraction. (a) View down the a-axis; (b) View down the b-axis; (c) The IrGe framework. The channels running along the a-axis have a channel of diameter ~6.45 Å; (d) close-up of the framework structure showing the Ir-Ge bond lengths; (e) Crystal orbital hamilton populations (-COHP) calculation emphasis on the Ir-Ge, Ir-Ir, and Ge-Ge interactions.
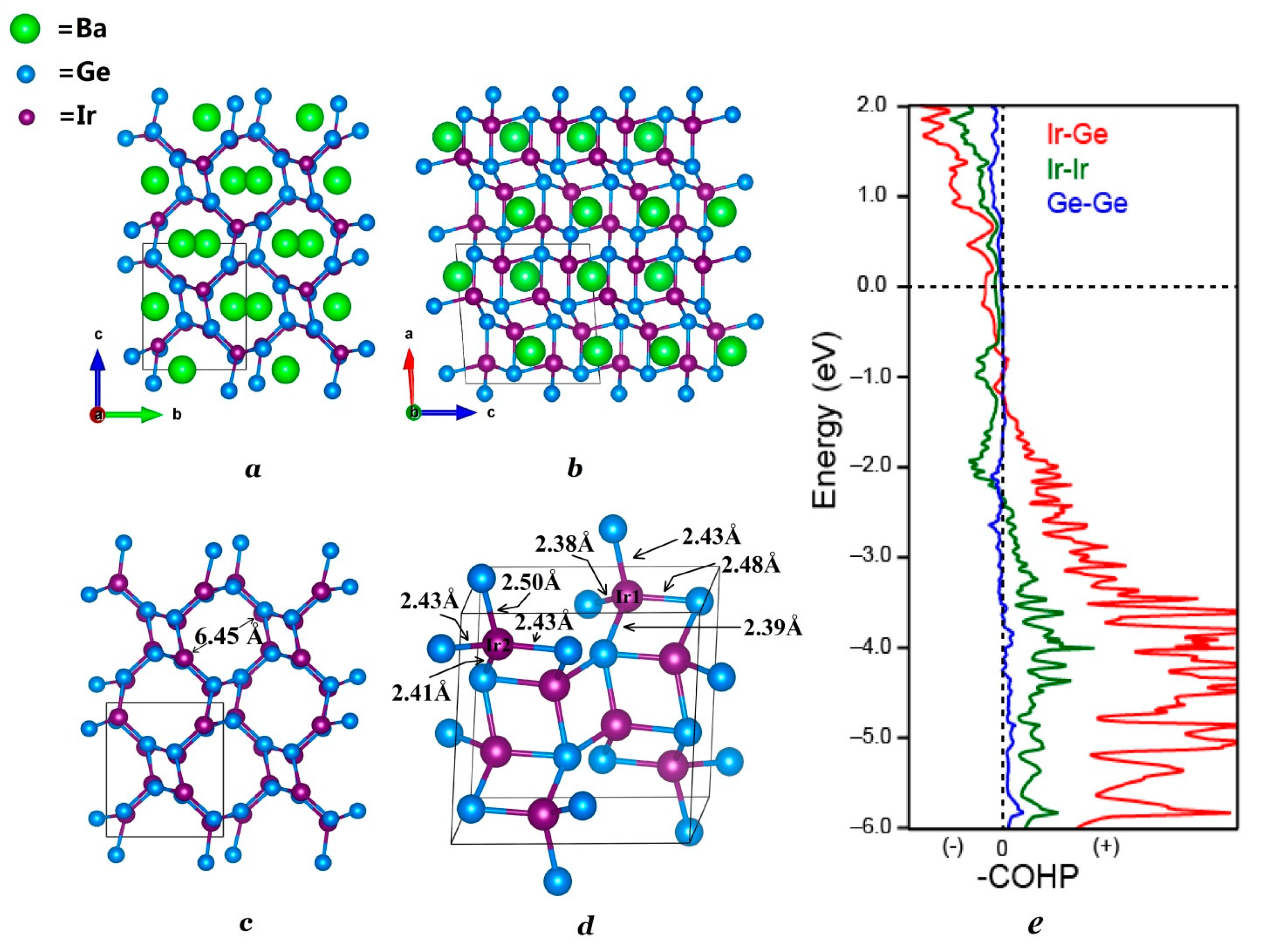
Figure 3.
Structural comparison of 122-type phases. (a) Clathrate structure of Ba3Ir4Ge16; (b) Channel framework of BaIr2Ge2; (c) Layered structure of BaIrGe3; (d) The symmetrical pentahedron in Ba3Ir4Ge16; (e) The irregular, non-symmetrical tetrahedron in BaIr2Ge2; (f) The square pyramid in BaIrGe3.
Figure 3.
Structural comparison of 122-type phases. (a) Clathrate structure of Ba3Ir4Ge16; (b) Channel framework of BaIr2Ge2; (c) Layered structure of BaIrGe3; (d) The symmetrical pentahedron in Ba3Ir4Ge16; (e) The irregular, non-symmetrical tetrahedron in BaIr2Ge2; (f) The square pyramid in BaIrGe3.
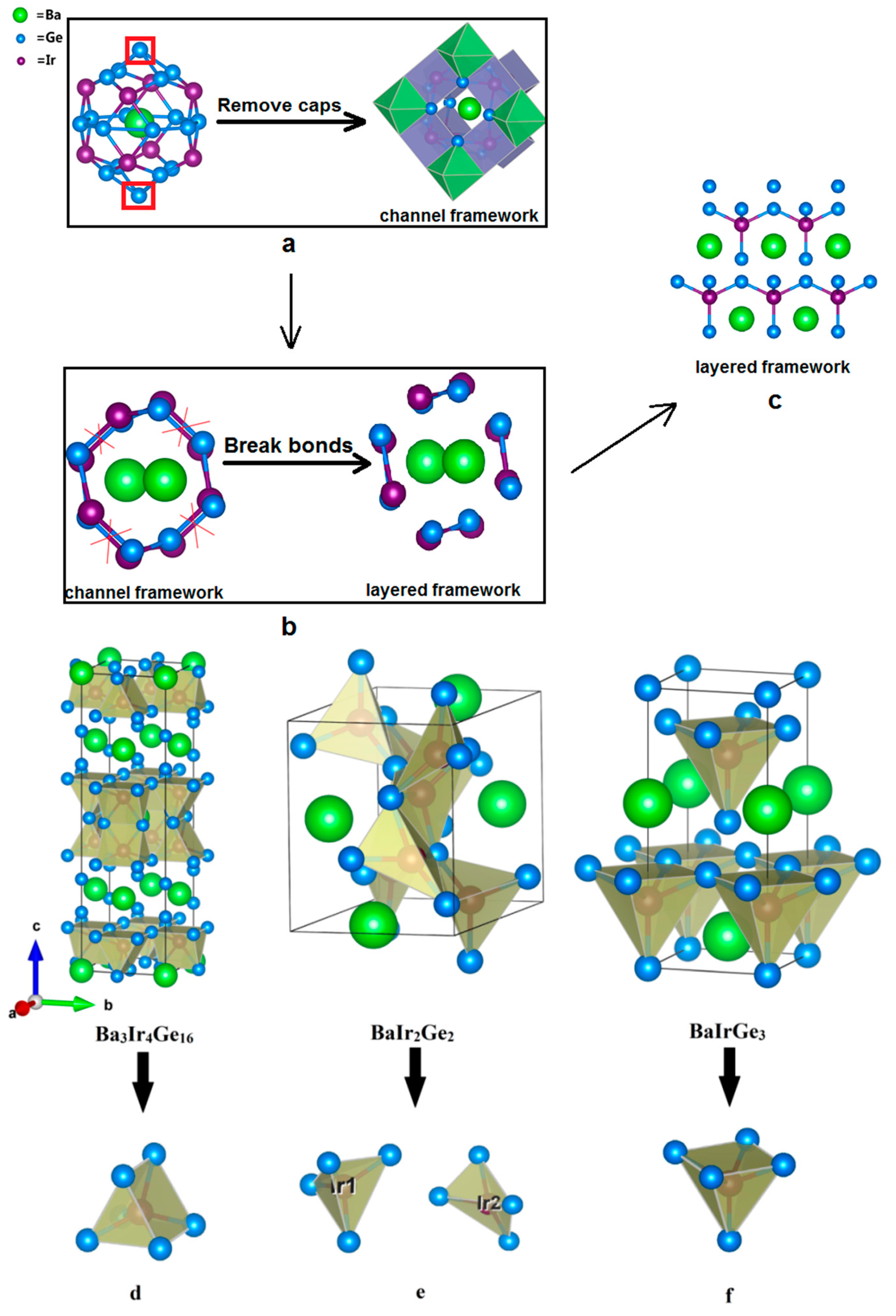
Figure 4.
Basic electronic and magnetic characterization of BaIr2Ge2. (Left: Main Panel) Temperature-dependent magnetic susceptibility (M/μ0H at μ0H = 5 T) and (Left: Inserted) field-dependent magnetization measurements showing the near linearity of M (magnetization) vs. H (magnetic field) for fields beyond 5 T at 1.8 K, justifying the use of the a high applied field in the measurements; (Right) Zero-field resistance data from 1.8 K to 300 K.
Figure 4.
Basic electronic and magnetic characterization of BaIr2Ge2. (Left: Main Panel) Temperature-dependent magnetic susceptibility (M/μ0H at μ0H = 5 T) and (Left: Inserted) field-dependent magnetization measurements showing the near linearity of M (magnetization) vs. H (magnetic field) for fields beyond 5 T at 1.8 K, justifying the use of the a high applied field in the measurements; (Right) Zero-field resistance data from 1.8 K to 300 K.
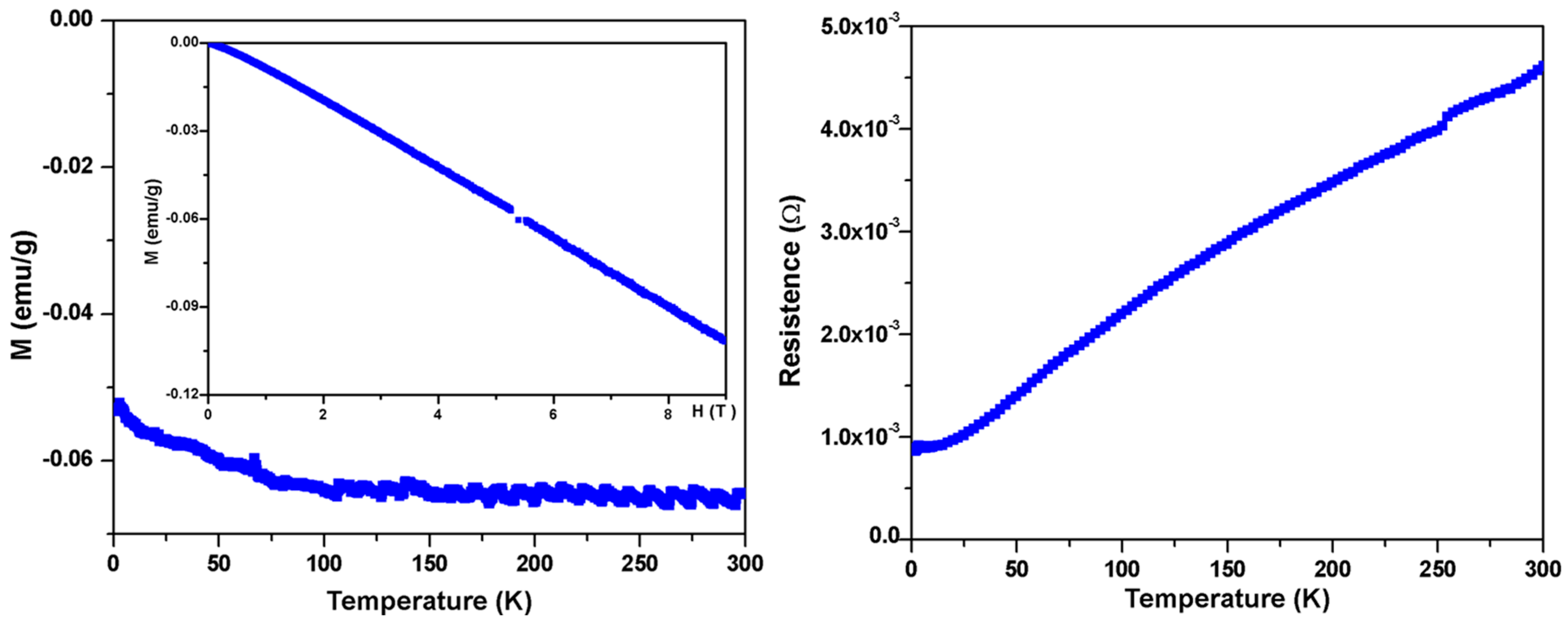
Figure 5.
Calculated electronic band structure and density of states (DOS) of BaIr2Ge2 using generalized gradient approximation (GGA). (a) Band structure and DOS with spin-orbit coupling emphasis on the energy range from −7 to +7 eV; (b) Band structure calculated by GGA without spin-orbit coupling; (c) Band structure calculated by GGA with spin-orbit coupling.
Figure 5.
Calculated electronic band structure and density of states (DOS) of BaIr2Ge2 using generalized gradient approximation (GGA). (a) Band structure and DOS with spin-orbit coupling emphasis on the energy range from −7 to +7 eV; (b) Band structure calculated by GGA without spin-orbit coupling; (c) Band structure calculated by GGA with spin-orbit coupling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Single crystal crystallographic data for BaIr2Ge2 at 299 (2) K.
| Refined Formula | BaIr2Ge2 |
|---|---|
| Formula weight (F.W.) (g/mol) | 666.92 |
| Space group; Z | P21/c (No. 14); 4 |
| a (Å) | 8.204 (5) |
| b (Å) | 6.625 (4) |
| c (Å) | 7.959 (5) |
| β (°) | 94.27 (1) |
| V (Å3) | 431.4 (4) |
| Extinction Coefficient | 0.00061 (9) |
| θ range (deg) | 2.489–32.085 |
| hkl ranges | −12 ≤ h ≤ 12 |
| −9 ≤ k ≤ 9 | |
| −11 ≤ l ≤ 10 | |
| No. reflections; Rint | 9731; 0.0925 |
| No. independent reflections | 1483 |
| No. parameters | 47 |
| R1: ωR2 (all I) | 0.0503; 0.0872 |
| Goodness of fit | 0.954 |
| Diffraction peak and hole (e−/Å3) | 3.812; −3.705 |
Table 2.
Atomic coordinates and equivalent isotropic displacement parameters for BaIr2Ge2 in space group P21/c. Ueq is defined as one-third of the trace of the orthogonalized Uij tensor (Å2).
Table 2.
Atomic coordinates and equivalent isotropic displacement parameters for BaIr2Ge2 in space group P21/c. Ueq is defined as one-third of the trace of the orthogonalized Uij tensor (Å2).
| Atom | Wyckoff. | Occ. | x | y | z | Ueq |
|---|---|---|---|---|---|---|
| Ba1 | 4e | 1 | 0.2318 (1) | 0.8818 (2) | 0.4993 (2) | 0.0116 (3) |
| Ir2 | 4e | 1 | 0.6260 (1) | 0.8959 (1) | 0.1069 (1) | 0.0074 (2) |
| Ir3 | 4e | 1 | 0.8532 (1) | 0.6648 (1) | 0.3334 (1) | 0.0077 (2) |
| Ge4 | 4e | 1 | 0.5560 (2) | 0.8515 (3) | 0.8069 (3) | 0.0088 (4) |
| Ge5 | 4e | 1 | 0.9282 (2) | 0.9171 (3) | 0.1356 (3) | 0.0091 (4) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gui, X.; Chang, T.-R.; Kong, T.; Pan, M.T.; Cava, R.J.; Xie, W. Monoclinic 122-Type BaIr2Ge2 with a Channel Framework: A Structural Connection between Clathrate and Layered Compounds. Materials 2017, 10, 818. https://doi.org/10.3390/ma10070818
AMA Style
Gui X, Chang T-R, Kong T, Pan MT, Cava RJ, Xie W. Monoclinic 122-Type BaIr2Ge2 with a Channel Framework: A Structural Connection between Clathrate and Layered Compounds. Materials. 2017; 10(7):818. https://doi.org/10.3390/ma10070818
Chicago/Turabian StyleGui, Xin, Tay-Rong Chang, Tai Kong, Max T. Pan, Robert J. Cava, and Weiwei Xie. 2017. "Monoclinic 122-Type BaIr2Ge2 with a Channel Framework: A Structural Connection between Clathrate and Layered Compounds" Materials 10, no. 7: 818. https://doi.org/10.3390/ma10070818
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.