
Superhard and Superconducting Bilayer Borophene


1
College of Physics and Electronic Engineering, Chongqing Normal University, Chongqing 401331, China
2
Department of Materials Science and Nanoengineering, Rice University, Houston, TX 77005, USA
3
Chemistry Department, Taif University, Taif 21974, Saudi Arabia
4
Department of Chemistry, Rice University, Houston, TX 77005, USA
*
Authors to whom correspondence should be addressed.
Materials 2024, 17(9), 1967; https://doi.org/10.3390/ma17091967
Submission received: 17 March 2024
/
Revised: 14 April 2024
/
Accepted: 19 April 2024
/
Published: 24 April 2024
(This article belongs to the Special Issue Development of Boron-Based Materials)
Abstract
:Two-dimensional superconductors, especially the covalent metals such as borophene, have received significant attention due to their new fundamental physics, as well as potential applications. Furthermore, the bilayer borophene has recently ignited interest due to its high stability and versatile properties. Here, the mechanical and superconducting properties of bilayer- borophene are explored by means of first-principles computations and anisotropic Migdal–Eliashberg analytics. We find that the coexistence of strong covalent bonds and delocalized metallic bonds endows this structure with remarkable mechanical properties (maximum 2D-Young’s modulus of ~570 N/m) and superconductivity with a critical temperature of ~20 K. Moreover, the superconducting critical temperature of this structure can be further boosted to ~46 K by applied strain, which is the highest value known among all borophenes or two-dimensional elemental materials.
1. Introduction
Superconductivity in two-dimensional (2D) materials has attracted perennial and ever-increasing attention over past decades, owing both to fundamental scientific interest and tantalizing applications [1,2,3,4,5]. One of the most important goals in superconductivity research is raising the superconducting transition temperature, Tc. In general, superconducting materials can be classified into two categories: low-temperature superconductors, characterized by critical temperatures (Tc) below 30 K, such as Nb-Ge films [6], and high-temperature superconductors with Tc exceeding 30 K, exemplified by Cu-based oxides [7,8,9]. Within the framework of the conventional Bardeen–Cooper–Schrieffer (BCS) theory [10], it is reasonably anticipated that metals composed of light elements have a better chance to induce a high Tc, because the Debye temperatures within such metals are usually high enough to trigger a strong phonon-mediated superconducting pairing. More specifically, according to the celebrated McMillan–Allen–Dynes (MAD) formula [11,12]:
Tc should be elevated by increasing the log-averaged characteristic phonon frequency () and the electron–phonon coupling (EPC) parameter, . The light elemental materials typically have high frequency phonon modes, enlarging and thus increasing Tc. Furthermore, large phonon frequency and EPC potential induced by strong covalent bonding in light elemental material, together with and the electronic density of states (DOS) at the Fermi level [13], should result in a higher Tc. In fact, the metals with strong covalent bonding can be grouped as “covalent metals” [13] and their potential to harbor a high Tc in 2D form have been confirmed, e.g., hydrogenated monolayer MgB2 (67 K) [14], doped graphene (8.1 K~30 K) [15,16,17], hydrogenated monolayer borophene (32.4 K) [5] and especially 2D metal borides (1.4 K~72 K) [18,19,20,21,22].
Among covalent metals, 2D boron sheets (borophenes) recently came to the fore of superconducting research, motivated by their inherent metallicity, light weight and significant experimental progress on their synthesis [23,24,25]. Based on the first-principles calculations and MAD formula, Penev et al. [26] reported early on the Tc of the (experimentally synthesized , and borophenes) to be in the range of 11.5 K~20.5 K and Gao et al. [27] found them to span 18.7 K–24.7 K. Using a more accurate and sophisticated anisotropic Migdal–Eliashberg (ME) equation, Zhao et al. [28] found the Tc in those borophenes in 26.2 K–33 K. Apart from intrinsic metallicity and light weight, another impetus for searching high-temperature superconductivity in borophenes is their vast polymorphs and superior mechanical properties, which provide a great benefit to potential superconductors, suggesting effective ways to modulate the superconductivity [29].
The monolayer borophenes have been intensively studied; however, the investigations are scarce [30,31,32] for bilayer or multilayer borophenes, where more intriguing properties and tunability could be manifested, compared to their monolayer counterparts. Very recently, the realization of bilayer borophenes were reported [33,34]. Unfortunately, the first reported bilayer -borophene is expected to be unstable if peeled off from the metal substrate [35], and the -like bilayer synthesized on Cu(111) surface appears complicated due to ambiguity (with more than three hundred atoms in its unit cell and the atomic structure still in debate), preventing an exploration of its properties or further application. It can be anticipated that more stable and versatile bilayer borophenes are still to be unveiled for research.
In this work, we investigate the mechanical and superconducting properties of a bilayer composed of two covalently bonded borophene monolayers, “(BL)-” in Figure 1a–c. The compact atomic structure endows BL- with remarkable stability and tantalizing properties compared to its monolayer counterparts. Its computed high Young’s modulus of 570 N/m is even higher than 346 N/m for graphene (or near 280 N/m “per layer”, for a fair comparison). Based on the anisotropic ME equation, we find its Tc = 20 K and can be boosted to 46 K by strain effect, reaching the highest Tc found among all borophenes or elemental 2D materials known to date. The coexistence of superhard (in basal plane direction) and superconducting properties in one material is rare, making BL- a fascinating superconductor for emergent nanoscale devices such as quantum interferometers, superconducting transistors, superconducting qubits, and wear-resistant parts of superconducting devices [3,36,37].
2. Results and Discussion
2.1. Atomic Structure and Mechanical Properties
BL- and share the same top view (Figure 1a), and BL- can be viewed as AB stacking of two monolayers bonded via interlayer covalent bonds (side view in Figure 1b,c). The unit cell of BL- is a rectangle with lattice constants a = 3.243 Å, b = 2.883 Å (Table 1), which is obtained with a Vienna ab initio Simulation Package [38,39] (computational details can be found in Supplementary Note S1). The total energy of BL- is computed to be lower than all experimental synthesized monolayers, due to the interlayer bonding: −6.38 eV/atom, which is 0.171 eV/atom, 0.125 eV/atom and 0.113 eV/atom lower than that of the synthesized , and , respectively (see Table 1, for atomic structures see Figure S2). Moreover, the slight dynamical instability of is also eliminated by the interlayer bonding of BL-. The energetic stability of BL- was also confirmed by an ab initio evolutionary global structure search in our prior work [40] and reported by Zhou et al. [41]. Given that the monolayer has been fabricated on a Ag(111) substrate, the thermal stability of BL- is also tested on Ag(111) substrate by an AIMD simulation at 500 K, and one can observe that its whole structure is well maintained after 5 ps with a timestep of 1 fs under such a high temperature (Figure S3). Therefore, it is well-expected that BL- will be accessed experimentally, supported by its outstanding energetic and thermal stability, and the progress in bilayer borophene synthesis [42,43].
We obtain the four independent elastic constants for BL-: C11 = 570 N/m, C22 = 322 N/m, C12 = 22 N/m, and C44 = 146 N/m, which apparently satisfy the Born–Huang criteria [46]: and , demonstrating the mechanical stability of BL-. The dependence of in-plane Young’s modulus Y and Poisson’s ratio of BL- are plotted with polar coordinates in Figure 1d,e (computational details can be found in Supplementary Note S2 and Figures S5 and S6). We find that the in-plane Young’s moduli and the Poisson’s ratios of BL- are highly anisotropic. For comparison, the values of elastic constants, Young’s moduli, Poisson’s ratios of , , and graphene are summarized in Table 1. The introduced B3-B3 bonds in BL- boost the Ya and Yb to 570 N/m and 321 N/m, respectively. Although graphene is well-known as the strongest 2D material, along the armchair direction Ya = 570 N/m exceeds the Ya = 364 N/m of graphene, partially due to the strong directional B-B -bonds, making it stand out among 2D materials. In contrast, the Yb = 321 N/m (still comparable to graphene) is lower, due to the much weaker multicenter bonds involved in the zigzag direction.
We find the fracture strain is 13% along the a and 8% along the b direction, according to the strained phonon spectra (Figure S4), before reaching the elastic maximum of 16% and 14% (Figure 1f). Thus, the failure mechanism of BL- is phonon instability, for both directions, which is different from that of (elastic instability in the zigzag direction and phonon instability in the armchair direction) [47]. The fracture strengths of BL- are 43 and 23 N/m along the a and b directions, much above that of (20.26 and 12.98 N/m) [47], black phosphorene (9.99 and 4.44 N/m) [48], and even graphene (40.41 and 36.74 N/m) [49] along the zigzag direction. Considering the ultrahigh Young’s moduli and large fracture strength, one can judge the BL- being in-plane superhard.
2.2. Electronic Properties
The electronic structure of BL- is summarized in Figure 2. Three bands cross the Fermi level and form the Fermi surface, of which one forms a small electron pocket centered around the corner of the first BZ and the other two contribute to the rest of Fermi surface (Figure 2b). The atomic- and orbital-resolved band structures suggest that the main contribution near the Fermi level is from the orbital of B1/B3 atoms and the pz orbital of all B atoms (Figure 2f,g, more details in Figure S7), which is also confirmed by the charge distribution in the states within ±0.3 eV from the Fermi level (Figure 2c–e). Using Wannier90 (v 3.1.0) code [50], the Wannier-interpolated band structures are also provided and show excellent agreement with those by first-principles calculations (Figure 2a), which lays a clear ground for subsequent anisotropic superconductivity calculations, as implemented in electron–phonon Wannier (EPW v 5.4) code [51].
2.3. Isotropic and Anisotropic Superconducting Properties
In the phonon spectrum of BL- (Figure 3a), we find no negative frequencies, demonstrating its dynamic stability. This is in stark contrast to , a layer whose phonon dispersion displays small imaginary frequency near the point [24]. Mechanically intuitive, the BL- is stabilized by the covalent B3-B3 bonds. The () mode- and (q) momentum-resolved EPC is also given, calculated by the following equation:
where Nk represents the total number of k points in the k space, is an EPC matrix element, n/m and represent the indices of electronic bands and phonon mode, and are the eigenvalues with respect to the Fermi level, and is the phonon frequency. We found three modes (B2u and two Ag) have comparatively large EPC near the point (left panel of Figure 3a). In the B2u shear mode at 39.3 meV, the B1 and B2 atoms move in one direction and the B3 atoms move in the opposite (Figure 3b). The first Ag mode at 102.6 meV corresponds to a bond stretching contributed by B3 atoms (Figure 3c). The second Ag mode at 168.7 meV is also a stretch mode, but predominantly contributed by B1 atoms (Figure 3d). The vibrational contribution of these three modes can also be inferred from the phonon density of states (right panel of Figure 3a). The Eliashberg spectral function is a key quantity, which determines the total EPC by the following equation:
Accordingly, the and cumulative EPC are calculated (middle panel of Figure 3a). We observe three sharp peaks mainly contributed by the three phonon modes analyzed above. Correspondingly, the EPC strength calculated by Equation (3) is 0.59 and the Tc of BL- within the MAD approximation calculated by Equation (1) is 10.1 K.
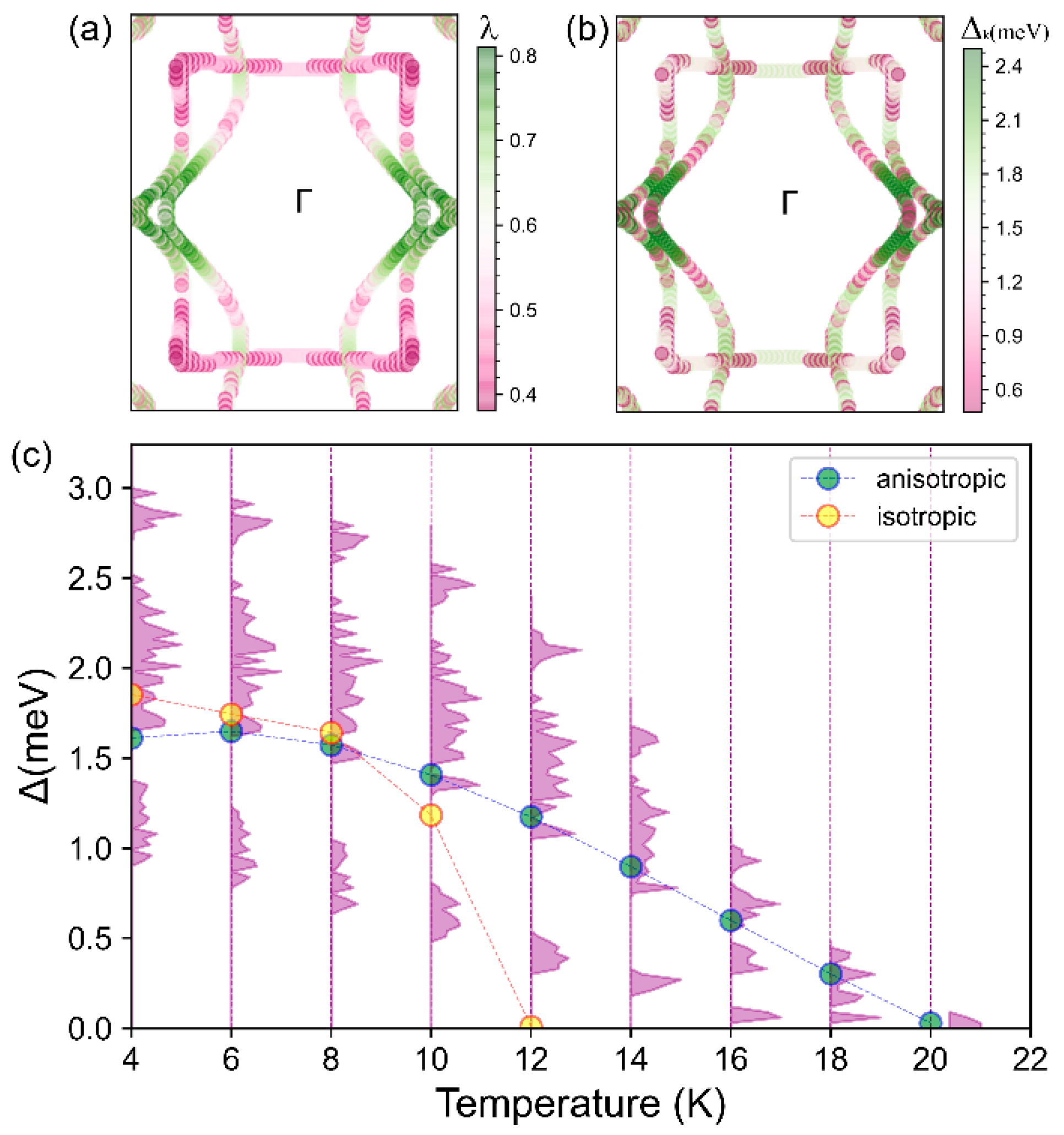
Akin to structure and mechanics anisotropy, the Fermi surface of BL-, formed by multiple bands with different orbital contributions (py and pz orbitals, Figure 2f,g) is also significantly anisotropic. For such a system with a complicated Fermi surface, using the anisotropic ME equation is essential, in order to obtain an anisotropic EPC, and an accurate Tc, compared with the isotropic superconductivity calculated from the MAD formula using an isotropic EPC [28]. As shown in Figure 4a,b, the variation in momentum-dependent EPC parameter and the superconducting gap at 10 K displays similar anisotropy, i.e., the maximum along the direction and the minimum along the direction. We find the superconducting gap ratio = ( = (2.499 − 0.444)/1.470 ≈ 140%, a measure of its strong anisotropy at the Fermi surface, also signifying that the anisotropic ME formula is indispensable for predicting Tc. Figure 4c shows the evolution of the superconducting gap as a function of temperature, based on the ME equations solved in either isotropic or fully anisotropic approximations. The Tc is identified as the lowest temperature at which the vanishing gap is observed. Under the isotropic approximation Tc = 12 K, comparable to the value (10 K) obtained by the MAD formula but is much lower than the value calculated with the fully anisotropic approximation (20 K), further attesting to the anisotropic superconducting nature of BL-, omitted previously [32].
The mechanical robustness of BL- permits a consideration of whether the tensile strain could enhance the Tc, motivated mainly by two reasons. First, according to Equation (2), it can be seen that EPC is inversely proportional to the phonon frequency , which would be helpful to lower, perhaps by tension, weakening the atomic force constants and thus softening the phonon modes. Second, the atomic orbitals overlap is reduced so that the electronic bands become less dispersive, enlarging the , which provides more electrons susceptible to pairing interactions, mediated by dynamic phonons, to certainly contribute to the Tc rise.
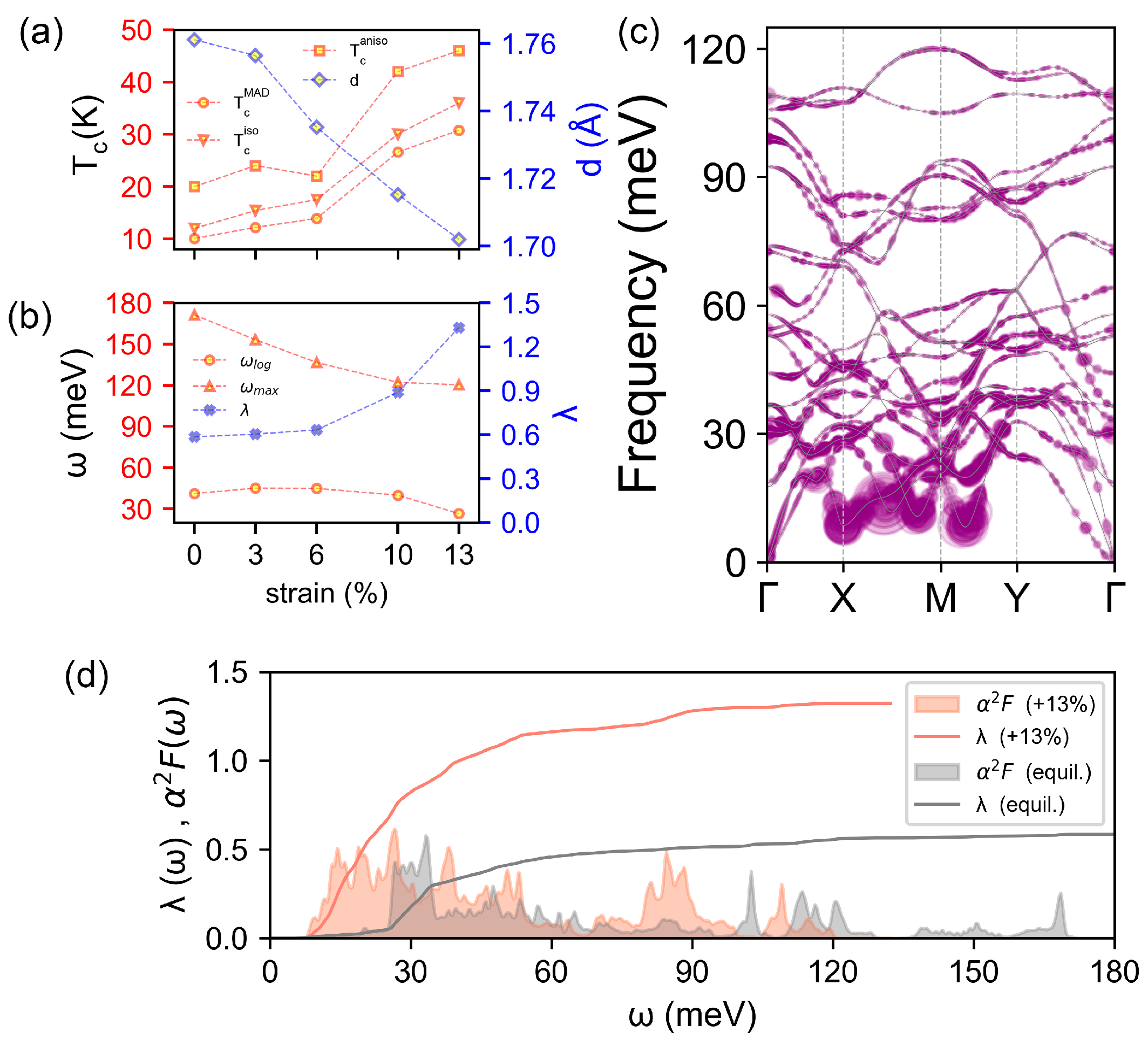
To examine the effect of strain on the BL- superconductivity, the evolution of EPC , the log and maximum frequencies and , as well as Tc, are all calculated within three approximations (i.e., MAD, isotropic ME, anisotropic ME), as a function of tensile strain along the a direction, and presented in Figure 5a,b (for details see Figures S8–S11). As expected, the increment in Tc is accompanied by a decrease in frequency-dependent / and an increase in EPC (Figure 5b). The phonon spectrum under a 13% tension along a (near fracture strain) weighted by , the Eliashberg spectral function and EPC are shown in Figure 5c,d. The red shift of phonon frequency can be clearly observed and the enhanced EPC due to the phonon softening can be seen in all frequency ranges (Figure 5d). In addition, the contribution of low-frequency part becomes more dominant. In particular, four largely softened phonon Kohn anomalies with considerable EPCs around 15 meV appear on the path of X-M-Y. Eventually, these combined effects lead to a significant increase, by strain, in from 0.59 to 1.33 and Tc from 20 K to 46 K. It should be emphasized that 46 K is the highest Tc among all borophenes and elemental 2D materials (Table 2). It should be noted that for bilayer or multilayer materials with positive Poisson ratios, such as BL-, the tensile strain would lead to vertical shrinking (Figure 5a), suggesting that the vertical pressure may also be an effective way to enhance the Tc of BL-, as the tensile strain does.
Before concluding, a few remarks need to be addressed. Superhard materials are usually semiconductors with large bandgaps or insulators, because the majority of electrons are bound into covalent bonds, depleting any excess electrons from electron transport. For instance, diamond is an insulator and the hardest material known. It may seem that metallicity and superhardness cannot coexist in one covalent material. However, this dilemma can be resolved in 2D borophenes, because the electron deficiency nature of boron atom enables strong localized covalent bonds and delocalized multicenter metal bonds in one system, which endows the system with superb mechanical performance and excellent metallicity, as evidenced by the high Young’s moduli and metallicity in BL-.
It is worth mentioning that research on BL borophenes is still in its infancy and has just been ignited by two recent independent experimental investigations. We would also like to point out that in addition to the striking mechanical properties and superconductivity studied in this work, BL- is expected to possess other interesting properties, such as anisotropic plasmonics [52] and ultrahigh thermal conductivity [53], awaiting more research. On the other hand, the excellent mechanical stability is also beneficial for BL- transfer to or even growth on some inert substrates [54], for convenience of characterization. Our results also show that the interlayer bonds, while strengthening the bilayer, do not destroy the σ-bond resonance nor the conducting π-bonds [55], adding a fine tuning-knob to the properties. This also highlights the potential of covalent metals in the quest for high Tc superconductors.
In our investigation, the tensile strain is applied to enhance the superconducting temperature of bilayer borophene. In addition to strain, the phenomenon of disorder, particularly correlated disorder, can also significantly enhance the superconductivity in a material [56,57]. In bilayer borophene, manipulating disorder could pave new paths to enhance superconducting performance. On the other hand, a caveat common for all ≤2D materials, due to the Mermin–Wagner theorem, must be mentioned, since it restricts the stability, both merely structural and of the superconducting phase. Nevertheless, in any BL borophene realization, the sample finite size (and support by a 3D-substrate) should mitigate these concerns, although quantifying the size limitation is far beyond the scope of the present study.
3. Conclusions
In conclusion, we comprehensively investigated mechanical and superconducting properties of a bilayer borophene (BL-), which can be viewed as AB stacking, with interlayer covalent bonding, of earlier realized borophene. The original good stability of and the introduced covalent bond endows BL- with very prominent energy—thermodynamic, thermal, and mechanical stability, suggesting that it has a high chance of occurrence in experiments. The strong directional B-B bonds and very compact atomic configuration give BL- a 2D Young’s modulus along the a direction (569.4 N/m) higher than graphene (346.1 N/m), while ensuring that BL- can sustain an ultimate 13% and 8% tensile strain along the a and b directions, respectively. Furthermore, according to a fully anisotropic solution of the ME equation, we predict BL- is a conventional phonon-mediated 2D superconductor with Tc as high as 20 K. We also highlighted the effects of tensile strain on EPCs and found that the Tc can be boosted to 46 K at a 13% tensile strain along the a direction. To the best of our knowledge, it is the highest value currently known for borophene or elemental 2D materials. Our findings also consolidated the justification that covalent metals, such as borophene, should benefit the search for high Tc superconductors. The concurrent superior mechanical performance and excellent superconductivity is scarce; thus, promising BL- many potential applications, such as quantum interferometers, superconducting transistors, superconducting qubits, and even wear-resistant parts for superconducting devices.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ma17091967/s1. Figure S1: The evolution of Tc with different k and q meshes. Figure S2: The atomic structures of (a) δ6, (b) β12 and (c) χ3. Figure S3: (a) The total potential energy fluctuation of BL- during AIMD simulation at 500 K. (b) The top view and (c) side view of the atomic configuration of BL- on Ag (111) surface after 5 ps of AIMD simulation at 500 K. Figure S4: The phonon spectra of strains under (a) 13% and (b) 14% along a axis, (c) 8% and (d) 9% along b axis. Figure S5: The strain energy per area under different kinds of strains for BL-. Figure S6: The strain energy per area under different kinds of strains for (a) , (b) , (c) and (d) graphene. Figure S7: The atomic and orbital resolved band structures of BL- Figure S8: (a) The electronic band properties of BL- under 3% tensile strain along a direction, red line and blue dashed line denote the results calculated from DFT and Wannier90, respectively. (b) Phonon band structure of BL- weighted by EPC with purple circles, isotropic Eliashberg function and EPC , PHDOS from different kinds of boron atoms’ contribution. (c) Evolution of the superconducting gap as a function of temperature, calculated by solving the ME equations in the isotropic approximation (yellow dots and dashed line interpolation) and with a fully anisotropic solution where the purple shadowed regions indicate the magnitude distribution of the and the light green dots connected with dashed line represents the average value of the entire anisotropic . Figure S9: The results of BL- under 6% tensile strain along a direction. The meaning of (a–c) are the same as Figure S8. Figure S10: The results of BL- under 10% tensile strain along a direction. The meaning of (a–c) are the same as Figure S8. Figure S11: The results of BL- under 13% tensile strain along a direction. The meaning of (a–c) are the same as Figure S8. Refs. [38,39,50,51,58,59,60,61,62,63,64,65] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, C.Z. and B.I.Y.; Methodology, M.S.; Investigation, C.Z. and M.S.; Resources, C.Z., T.A. and B.I.Y.; Writing—original draft, C.Z. and M.S.; Writing—review & editing, T.A. and B.I.Y. All authors have read and agreed to the published version of the manuscript.
Funding
C.Z. acknowledges the financial support from the Scientific and Technology Research Program of Chongqing Municipal Education Commission (KJQN202300515) and the Foundation of Chongqing Normal University (23XLB001). T.A. and B.I.Y. acknowledge the Taif University Research Support Project (TURSPHC2024/1, Saudi Arabia). Work at Rice was supported by the US Office of Naval Research (N00014-22-1-2753).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Saito, Y.; Nojima, T.; Iwasa, Y. Highly crystalline 2D superconductors. Nat. Rev. Mater. 2016, 2, 16094. [Google Scholar] [CrossRef]
- Li, W.; Huang, J.; Li, X.; Zhao, S.; Lu, J.; Han, Z.V.; Wang, H. Recent progresses in two-dimensional Ising superconductivity. Mater. Today Phys. 2021, 21, 100504. [Google Scholar] [CrossRef]
- Qiu, D.; Gong, C.; Wang, S.; Zhang, M.; Yang, C.; Wang, X.; Xiong, J. Recent Advances in 2D Superconductors. Adv. Mater. 2021, 33, e2006124. [Google Scholar] [CrossRef] [PubMed]
- Lilia, B.; Hennig, R.; Hirschfeld, P.; Profeta, G.; Sanna, A.; Zurek, E.; Pickett, W.E.; Amsler, M.; Dias, R.; Eremets, M.I.; et al. The 2021 room-temperature superconductivity roadmap. J. Phys. Condens. Matter 2022, 34, 183002. [Google Scholar]
- Zhong, C.; Li, X.; Yu, P. Strain-tunable Dirac semimetal phase transition and emergent superconductivity in a borophane. Commun. Phys. 2024, 7, 38. [Google Scholar] [CrossRef]
- Gavaler, J.R. Superconductivity in Nb–Ge films above 22 K. Appl. Phys. Lett. 1973, 23, 480–482. [Google Scholar] [CrossRef]
- Wu, M.K.; Ashburn, J.R.; Torng, C.J.; Hor, P.H.; Meng, R.L.; Gao, L.; Huang, Z.J.; Wang, Y.Q.; Chu, C.W. Superconductivity at 93 K in a new mixed-phase Y-Ba-Cu-O compound system at ambient pressure. Phys. Rev. Lett. 1987, 58, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Pęczkowski, P.; Zachariasz, P.; Kowalik, M.; Tokarz, W.; Kumar Naik, S.P.; Żukrowski, J.; Jastrzębski, C.; Dadiel, L.J.; Tabiś, W.; Gondek, Ł. Iron diffusivity into superconducting YBa2Cu3O7−δ at oxygen-assisted sintering: Structural, magnetic, and transport properties. J. Eur. Ceram. Soc. 2021, 41, 7085–7097. [Google Scholar] [CrossRef]
- Peczkowski, P.; Kowalik, M.; Zachariasz, P.; Jastrzebski, C.; Jaegermann, Z.; Szterner, P.; Woch, W.M.; Szczytko, J. Synthesis and Physicochemical Properties of Nd-, Sm-, Eu-Based Cuprate High-Temperature Superconductors. Phys. Status Solidi A 2018, 215, 1700888. [Google Scholar] [CrossRef]
- Bardeen, J.; Cooper, L.N.; Schrieffer, J.R. Theory of Superconductivity. Phys. Rev. 1957, 108, 1175–1204. [Google Scholar] [CrossRef]
- McMillan, W.L. Transition Temperature of Strong-Coupled Superconductors. Phys. Rev. 1968, 167, 331–344. [Google Scholar] [CrossRef]
- Allen, P.B.; Dynes, R.C. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 1975, 12, 905–922. [Google Scholar] [CrossRef]
- Blase, X.; Bustarret, E.; Chapelier, C.; Klein, T.; Marcenat, C. Superconducting group-IV semiconductors. Nat. Mater. 2009, 8, 375–382. [Google Scholar] [CrossRef]
- Bekaert, J.; Petrov, M.; Aperis, A.; Oppeneer, P.M.; Milosevic, M.V. Hydrogen-Induced High-Temperature Superconductivity in Two-Dimensional Materials: The Example of Hydrogenated Monolayer MgB2. Phys. Rev. Lett. 2019, 123, 077001. [Google Scholar] [CrossRef] [PubMed]
- Profeta, G.; Calandra, M.; Mauri, F. Phonon-mediated superconductivity in graphene by lithium deposition. Nat. Phys. 2012, 8, 131–134. [Google Scholar] [CrossRef]
- Lu, H.-Y.; Yang, Y.; Hao, L.; Wang, W.-S.; Geng, L.; Zheng, M.; Li, Y.; Jiao, N.; Zhang, P.; Ting, C.S. Phonon-mediated superconductivity in aluminum-deposited graphene AlC8. Phys. Rev. B 2020, 101, 214514. [Google Scholar] [CrossRef]
- Si, C.; Liu, Z.; Duan, W.; Liu, F. First-principles calculations on the effect of doping and biaxial tensile strain on electron-phonon coupling in graphene. Phys. Rev. Lett. 2013, 111, 196802. [Google Scholar] [CrossRef]
- Zhao, Y.; Lian, C.; Zeng, S.; Dai, Z.; Meng, S.; Ni, J. Two-gap and three-gap superconductivity in AlB2-based films. Phys. Rev. B 2019, 100, 094516. [Google Scholar] [CrossRef]
- Bo, T.; Liu, P.-F.; Yan, L.; Wang, B.-T. Electron-phonon coupling superconductivity in two-dimensional orthorhombic MB6 (M = Mg, Ca, Ti, Y) and hexagonal MB6 (M = Mg, Ca, Sc, Ti). Phys. Rev. Mater. 2020, 4, 114802. [Google Scholar] [CrossRef]
- Zhao, Y.; Lian, C.; Zeng, S.; Dai, Z.; Meng, S.; Ni, J. MgB4 trilayer film: A four-gap superconductor. Phys. Rev. B 2020, 101, 104507. [Google Scholar] [CrossRef]
- Sevik, C.; Bekaert, J.; Petrov, M.; Milošević, M.V. High-temperature multigap superconductivity in two-dimensional metal borides. Phys. Rev. Mater. 2022, 6, 024803. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, S.; Zhao, Y.; Wang, X.; Ni, J. Three-gap superconductivity in two-dimensional InB2/InB4 films. Phys. Rev. B 2021, 104, 174519. [Google Scholar] [CrossRef]
- Kaneti, Y.V.; Benu, D.P.; Xu, X.; Yuliarto, B.; Yamauchi, Y.; Golberg, D. Borophene: Two-dimensional Boron Monolayer: Synthesis, Properties, and Potential Applications. Chem. Rev. 2022, 122, 1000–1051. [Google Scholar] [CrossRef] [PubMed]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef]
- Penev, E.S.; Kutana, A.; Yakobson, B.I. Can Two-Dimensional Boron Superconduct? Nano Lett. 2016, 16, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Li, Q.-Z.; Yan, X.-W.; Wang, J. Prediction of phonon-mediated superconductivity in borophene. Phys. Rev. B 2017, 95, 024505. [Google Scholar] [CrossRef]
- Zhao, Y.; Zeng, S.; Lian, C.; Dai, Z.; Meng, S.; Ni, J. Multigap anisotropic superconductivity in borophenes. Phys. Rev. B 2018, 98, 134514. [Google Scholar] [CrossRef]
- Zhang, Z.; Penev, E.S.; Yakobson, B.I. Two-dimensional materials: Polyphony in B flat. Nat. Chem. 2016, 8, 525–527. [Google Scholar] [CrossRef]
- Gao, N.; Wu, X.; Jiang, X.; Bai, Y.; Zhao, J. Structure and stability of bilayer borophene: The roles of hexagonal holes and interlayer bonding. FlatChem 2017, 7, 48–54. [Google Scholar] [CrossRef]
- Zhong, H.; Huang, K.; Yu, G.; Yuan, S. Electronic and mechanical properties of few-layer borophene. Phys. Rev. B 2018, 98, 054104. [Google Scholar] [CrossRef]
- Yan, L.; Ku, R.; Zou, J.; Zhou, L.; Zhao, J.; Jiang, X.; Wang, B.-T. Prediction of superconductivity in bilayer borophenes. RSC Adv. 2021, 11, 40220–40227. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Q.; Ruan, Q.; Rahn, M.S.; Yakobson, B.I.; Hersam, M.C. Borophene synthesis beyond the single-atomic-layer limit. Nat. Mater. 2022, 21, 35–40. [Google Scholar] [CrossRef]
- Chen, C.; Lv, H.; Zhang, P.; Zhuo, Z.; Wang, Y.; Ma, C.; Li, W.; Wang, X.; Feng, B.; Cheng, P.; et al. Synthesis of bilayer borophene. Nat. Chem. 2022, 14, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Y.; Zhao, X.-Y.; Zan, W.; Mu, Y.; Zhang, Z.; Li, S.-D. Prediction of freestanding semiconducting bilayer borophenes. Nano Res. 2022, 15, 5752–5757. [Google Scholar] [CrossRef]
- Liu, X.; Hersam, M.C. 2D materials for quantum information science. Nat. Rev. Mater. 2019, 4, 669–684. [Google Scholar] [CrossRef]
- Liu, P.; Lei, B.; Chen, X.; Wang, L.; Wang, X. Superior carrier tuning in ultrathin superconducting materials by electric-field gating. Nat. Rev. Phys. 2022, 4, 336–352. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Luo, Y.; Yan, Y.; Schwingenschlögl, U. Ultrahigh Carrier Mobility in the Two-Dimensional Semiconductors B8Si4, B8Ge4, and B8Sn4. Chem. Mater. 2021, 33, 6475–6483. [Google Scholar] [CrossRef]
- Zhou, X.-F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.; Wang, H.-T. Semimetallic Two-Dimensional Boron Allotrope with Massless Dirac Fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef]
- Ebrahimi, M. The birth of bilayer borophene. Nat. Chem. 2022, 14, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Božović, I. Doubling down on borophene electronics. Nat. Mater. 2022, 21, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lü, T.-Y.; Wang, H.-Q.; Feng, Y.P.; Zheng, J.-C. High anisotropy of fully hydrogenated borophene. Phys. Chem. Chem. Phys. 2016, 18, 31424–31430. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals; Clarendon Press: Oxford, UK, 1985. [Google Scholar]
- Wang, H.; Li, Q.; Gao, Y.; Miao, F.; Zhou, X.-F.; Wan, X.G. Strain effects on borophene: Ideal strength, negative Possion’s ratio and phonon instability. New J. Phys. 2016, 18, 073016. [Google Scholar] [CrossRef]
- Wei, Q.; Peng, X. Superior mechanical flexibility of phosphorene and few-layer black phosphorus. Appl. Phys. Lett. 2014, 104, 251915. [Google Scholar] [CrossRef]
- Liu, F.; Ming, P.; Li, J. Ab initio calculation of ideal strength and phonon instability of graphene under tension. Phys. Rev. B 2007, 76, 064120. [Google Scholar] [CrossRef]
- Pizzi, G.; Vitale, V.; Arita, R.; Blügel, S.; Freimuth, F.; Géranton, G.; Gibertini, M.; Gresch, D.; Johnson, C.; Koretsune, T.; et al. Wannier90 as a community code: New features and applications. J. Phys. Condens. Matter. 2020, 32, 165902. [Google Scholar] [CrossRef]
- Noffsinger, J.; Giustino, F.; Malone, B.D.; Park, C.-H.; Louie, S.G.; Cohen, M.L. EPW: A program for calculating the electron–phonon coupling using maximally localized Wannier functions. Comput. Phys. Commun. 2010, 181, 2140–2148. [Google Scholar] [CrossRef]
- Huang, Y.; Shirodkar, S.N.; Yakobson, B.I. Two-Dimensional Boron Polymorphs for Visible Range Plasmonics: A First-Principles Exploration. J. Am. Chem. Soc. 2017, 139, 17181–17185. [Google Scholar] [CrossRef]
- Xiao, H.; Cao, W.; Ouyang, T.; Guo, S.; He, C.; Zhong, J. Lattice thermal conductivity of borophene from first principle calculation. Sci. Rep. 2017, 7, 45986. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Wang, L.; Bets, K.V.; Yakobson, B.I. Step-Edge Epitaxy for Borophene Growth on Insulators. ACS Nano 2021, 15, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhang, X.; Kong, X.; Mitchell, I.; Yan, T.; Kim, S.Y.; Yakobson, B.I.; Ding, F. Theory of sigma bond resonance in flat boron materials. Nat. Comm. 2023, 14, 1804. [Google Scholar] [CrossRef] [PubMed]
- Neverov, V.D.; Lukyanov, A.E.; Krasavin, A.V.; Vagov, A.; Croitoru, M.D. Correlated disorder as a way towards robust superconductivity. Commun. Phys. 2022, 5, 177. [Google Scholar] [CrossRef]
- Ghosal, A.; Randeria, M.; Trivedi, N. Inhomogeneous pairing in highly disordered s-wave superconductors. Phys. Rev. B 2001, 65, 014501. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.J.; Holian, B.L. The Nose–Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Zhong, C.; Wu, W.; He, J.; Ding, G.; Liu, Y.; Li, D.; Yang, S.A.; Zhang, G. Two-dimensional honeycomb borophene oxide: Strong anisotropy and nodal loop transformation. Nanoscale 2019, 11, 2468–2475. [Google Scholar] [CrossRef] [PubMed]
- Cadelano, E.; Palla, P.L.; Giordano, S.; Colombo, L. Elastic properties of hydrogenated graphene. Phys. Rev. B 2010, 82, 235414. [Google Scholar] [CrossRef]
Figure 1.
(a) Planar and (b,c) side views of the atomic structure of BL-δ6. The yellow shaded region is its unit cell, and the green shaded region is the unit cell of δ6. There are three irreducible boron atoms colored as B1 (green), B2 (blue), and B3 (pink). The direction dependence of (d) Young’s modulus and (e) Poisson’s ratio of BL-δ6. (f) Stress–strain curves of BL-δ6, the vertical blue and red dashed lines denote the fractured stress along the a and b directions, respectively.
Figure 1.
(a) Planar and (b,c) side views of the atomic structure of BL-δ6. The yellow shaded region is its unit cell, and the green shaded region is the unit cell of δ6. There are three irreducible boron atoms colored as B1 (green), B2 (blue), and B3 (pink). The direction dependence of (d) Young’s modulus and (e) Poisson’s ratio of BL-δ6. (f) Stress–strain curves of BL-δ6, the vertical blue and red dashed lines denote the fractured stress along the a and b directions, respectively.

Figure 2.
(a) Electronic band structures of BL-δ6 by DFT (red solid lines) and Wannier90 (blue dashed lines). (b) Fermi surface, (c–e) top and side views of the charge density corresponding to the energy range shaded green in (a), and electronic band structures weighted by the py orbital of B1 and B3 atoms (f) and the pz orbital contributions of all boron atoms (g) of BL-δ6.
Figure 2.
(a) Electronic band structures of BL-δ6 by DFT (red solid lines) and Wannier90 (blue dashed lines). (b) Fermi surface, (c–e) top and side views of the charge density corresponding to the energy range shaded green in (a), and electronic band structures weighted by the py orbital of B1 and B3 atoms (f) and the pz orbital contributions of all boron atoms (g) of BL-δ6.

Figure 3.
(a) From left to right: phonon band structure of BL-δ6 weighted by EPC λqν with purple circles, isotropic Eliashberg function α2F and EPC λ(ω), and phonon density of states contributed by different kinds of boron atoms. (b–d) Vibrational modes of the B2u mode and the two Ag modes at Γ, as labeled in (a).
Figure 3.
(a) From left to right: phonon band structure of BL-δ6 weighted by EPC λqν with purple circles, isotropic Eliashberg function α2F and EPC λ(ω), and phonon density of states contributed by different kinds of boron atoms. (b–d) Vibrational modes of the B2u mode and the two Ag modes at Γ, as labeled in (a).

Figure 4.
(a) Momentum-dependent EPC λk across the full BZ and (b) momentum-dependent superconducting band gap ∆k at 10 K and projected onto the Fermi surface. (c) Variation in the superconducting gap ∆k with temperature, calculated by solving the ME equations in the isotropic approximation (yellow dots and dashed line interpolated) and with the fully anisotropic solution, where the purple shadowed regions indicate the magnitude distribution of the ∆k and the light green dots connected with the dashed line represents the average value of the entire anisotropic ∆k.
Figure 4.
(a) Momentum-dependent EPC λk across the full BZ and (b) momentum-dependent superconducting band gap ∆k at 10 K and projected onto the Fermi surface. (c) Variation in the superconducting gap ∆k with temperature, calculated by solving the ME equations in the isotropic approximation (yellow dots and dashed line interpolated) and with the fully anisotropic solution, where the purple shadowed regions indicate the magnitude distribution of the ∆k and the light green dots connected with the dashed line represents the average value of the entire anisotropic ∆k.

Figure 5.
(a) Evolution with tensile strain along the a direction, of the Tc at three approximations (MAD, circles; isotropic ME, triangles; anisotropic ME, squares) and the distance between planes measured as B3-B3 bond length d. (b) Evolution of EPC λ (solid crosses), the log-weighted frequency ωlog, (circles) and maximum phonon frequency ωmax (triangles) versus the strain. (c) Phonon band structure weighted by EPC ωqν under 13% strain. (d) Isotropic Eliashberg α2F and EPC λ with 13% tensile strain along the a direction. Data with no strain are shown in gray, for comparison.
Figure 5.
(a) Evolution with tensile strain along the a direction, of the Tc at three approximations (MAD, circles; isotropic ME, triangles; anisotropic ME, squares) and the distance between planes measured as B3-B3 bond length d. (b) Evolution of EPC λ (solid crosses), the log-weighted frequency ωlog, (circles) and maximum phonon frequency ωmax (triangles) versus the strain. (c) Phonon band structure weighted by EPC ωqν under 13% strain. (d) Isotropic Eliashberg α2F and EPC λ with 13% tensile strain along the a direction. Data with no strain are shown in gray, for comparison.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The space group, lattice constants (Å), elastic constants (N/m), Young’s moduli (N/m), Poisson’s ratios, and cohesive energies (eV/atom) of BL-, , ,
Table 1.
The space group, lattice constants (Å), elastic constants (N/m), Young’s moduli (N/m), Poisson’s ratios, and cohesive energies (eV/atom) of BL-, , ,
| System | Space Group | a | b | C11 | C22 | C12 | C44 | Ya | Yb | Ec | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pmmm | 3.243 | 2.883 | 570.0 | 322.0 | 22.0 | 146.0 | 569.4 | 321.4 | 0.038 | 0.068 | −6.380 | |
| Pmmn | 1.614 | 2.874 | 398.6 | 171.8 | −4.0 | 93.9 | 398.6 | 171.8 | −0.01 | −0.02 | −6.209 | |
| [24] | 1.617 | 2.865 | 398.0 | 170.0 | −7.0 | 94.0 | 398.0 | 170.0 | −0.04 | −0.02 | ||
| Pmmm | 2.931 | 5.065 | 187.0 | 218.6 | 36.8 | 62.7 | 180.8 | 211.4 | 0.168 | 0.197 | −6.255 | |
| [44] | 185.5 | 210.5 | 37.0 | 68.5 | 179.0 | 203.1 | 0.176 | 0.199 | ||||
| Cmmm | 8.407 | 2.912 | 207.3 | 198.5 | 28.2 | 60.0 | 203.3 | 194.7 | 0.135 | 0.142 | −6.267 | |
| [44] | 201.0 | 185.0 | 21.5 | 60.5 | 198.5 | 182.7 | 0.116 | 0.107 | ||||
| Graphene | P6/mmm | 2.468 | 356.9 | 62.2 | 147.3 | 346.1 | 0.174 | |||||
| Graphene [45] | 2.470 | 352.7 | 60.9 | 145.9 | 342.2 | 0.173 |
Table 2.
The EPC λ, Tc calculated with MAD formula and anisotropic ME equation of δ6, β12, χ3, for Li-doped graphene [15] and BL-δ6.
Table 2.
The EPC λ, Tc calculated with MAD formula and anisotropic ME equation of δ6, β12, χ3, for Li-doped graphene [15] and BL-δ6.
| System | (K, ) | (K, ) | |
|---|---|---|---|
| [26] | 1.10 | 20.5 | |
| [28] | 0.82 | 27.0 | |
| [26] | 0.80 | 16.1 | |
| [27] | 0.89 | 18.7 | |
| [28] | 1.01 | 33.0 | |
| [26] | 0.60 | 11.5 | |
| [27] | 0.95 | 24.7 | |
| [28] | 0.79 | 26.2 | |
| Graphene (Li deposition) [15] | 0.61 | 8.1 | |
| 0.59 | 10.1 | 20 | |
| [32] | 0.61 | 11.9 | |
| (13%) | 1.33 | 30.78 | 46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhong, C.; Sun, M.; Altalhi, T.; Yakobson, B.I. Superhard and Superconducting Bilayer Borophene. Materials 2024, 17, 1967. https://doi.org/10.3390/ma17091967
AMA Style
Zhong C, Sun M, Altalhi T, Yakobson BI. Superhard and Superconducting Bilayer Borophene. Materials. 2024; 17(9):1967. https://doi.org/10.3390/ma17091967
Chicago/Turabian StyleZhong, Chengyong, Minglei Sun, Tariq Altalhi, and Boris I. Yakobson. 2024. "Superhard and Superconducting Bilayer Borophene" Materials 17, no. 9: 1967. https://doi.org/10.3390/ma17091967
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.