Genome-Wide Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Paulownia fortunei (Seem) Hemsl. under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
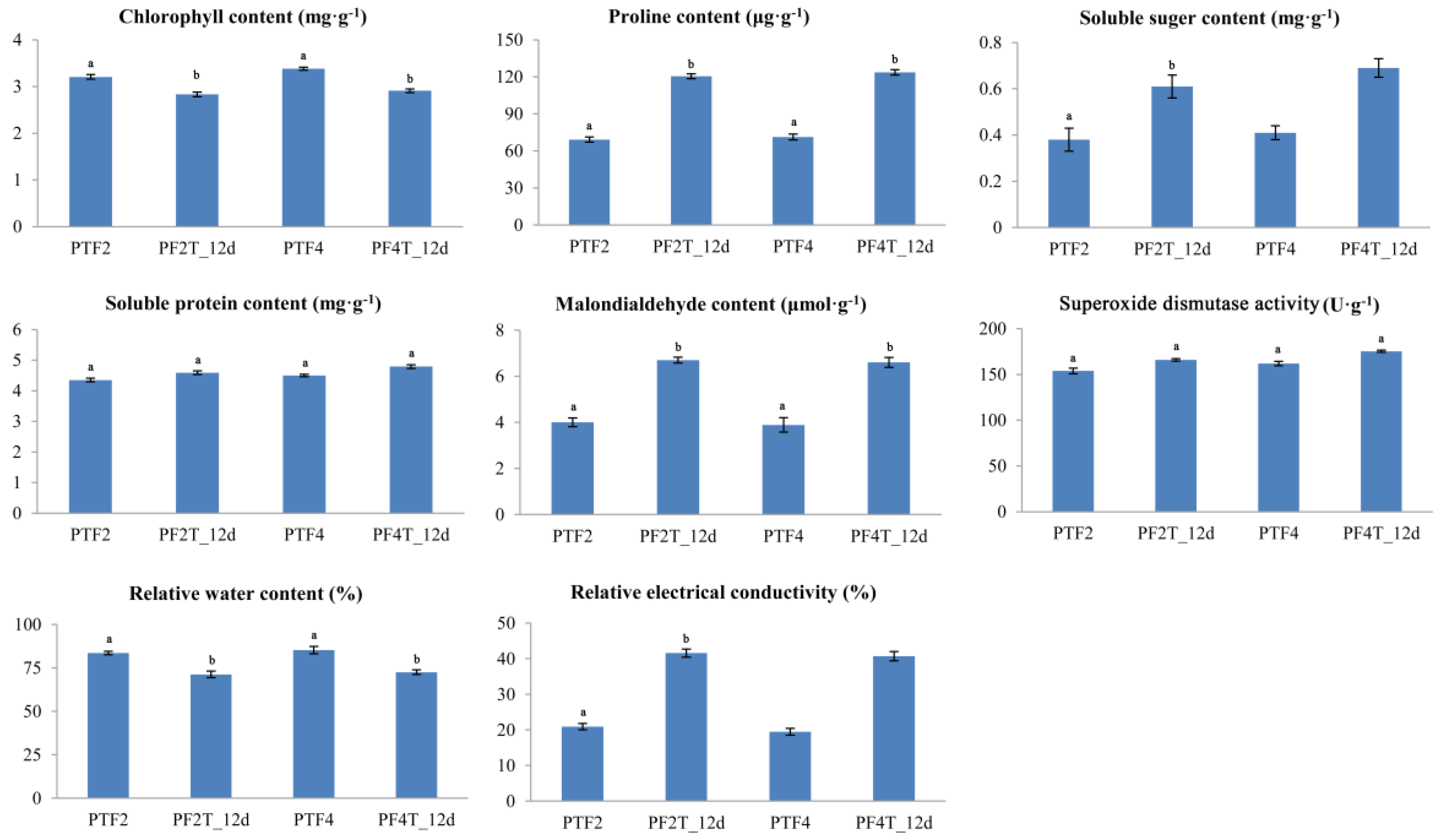
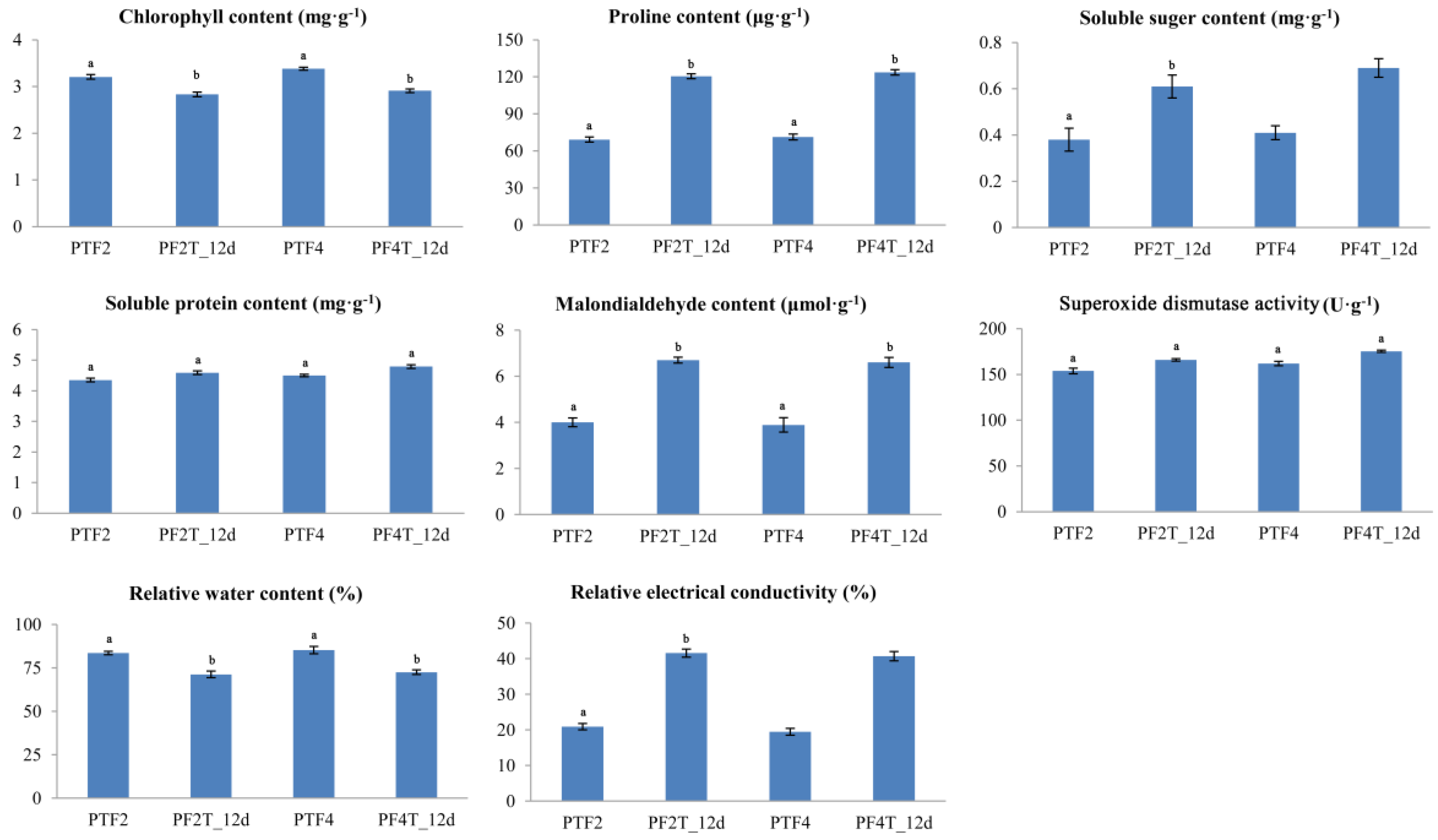
2.2. Physiological Responses of Diploid and Autotetraploid Paulownia fortunei to Drought Stress
2.3. Construction, Processing, and Annotation of the P. fortunei cDNA Libraries
2.4. Construction, Processing, and Annotation of the P. fortunei Small RNA Libraries
2.5. Differential Expression Analysis of Conserved and Novel miRNAs
2.6. Identification of miRNA Target Genes Associated with Drought Stress
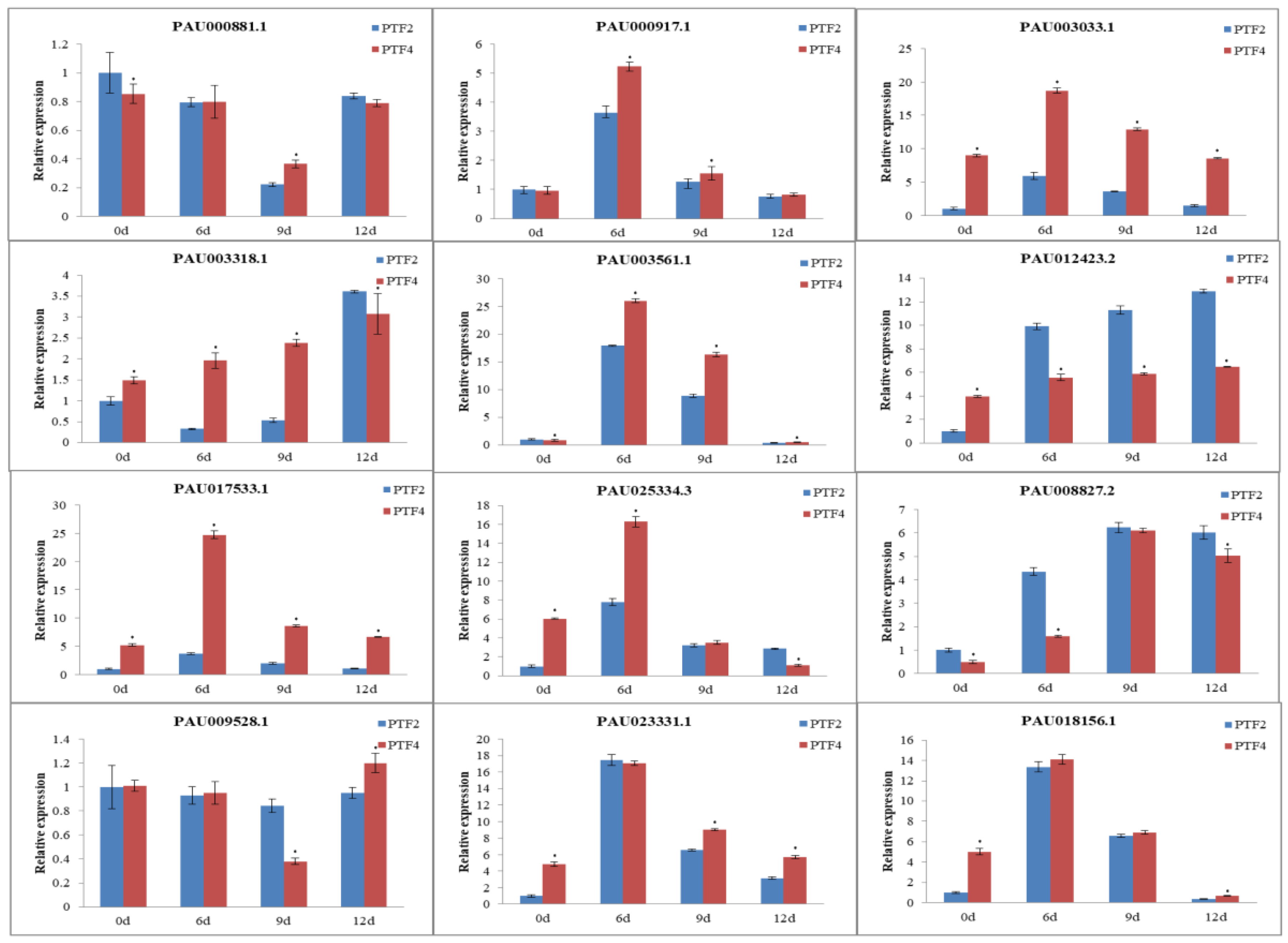
2.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) for Verification of the Sequencing Data
3. Results
3.1. Physiological Responses of Diploid and Autotetraploid Paulownia fortunei to Drought Stress
3.2. Transcriptome Data Mapping to the P. fortunei Genome
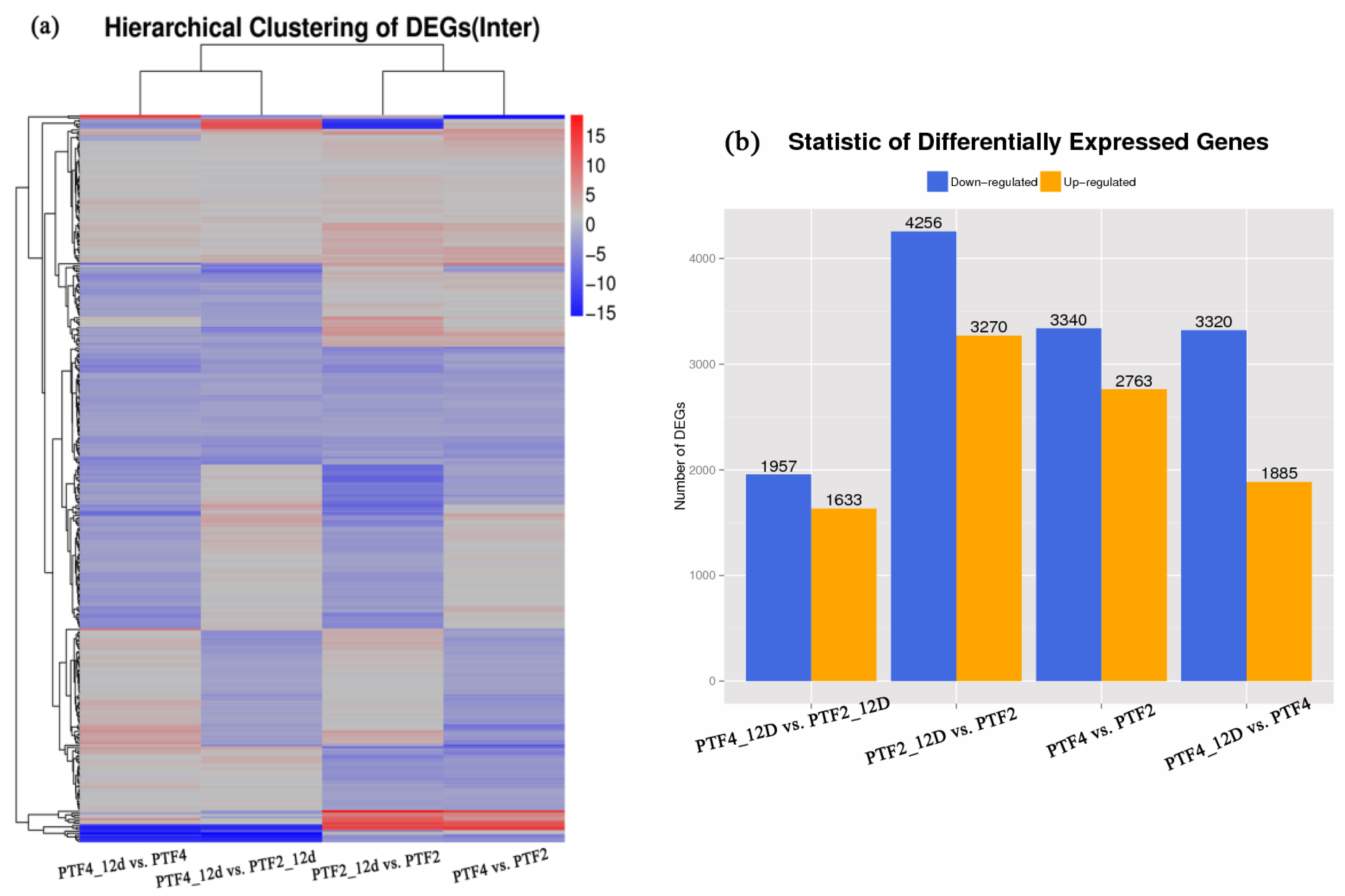
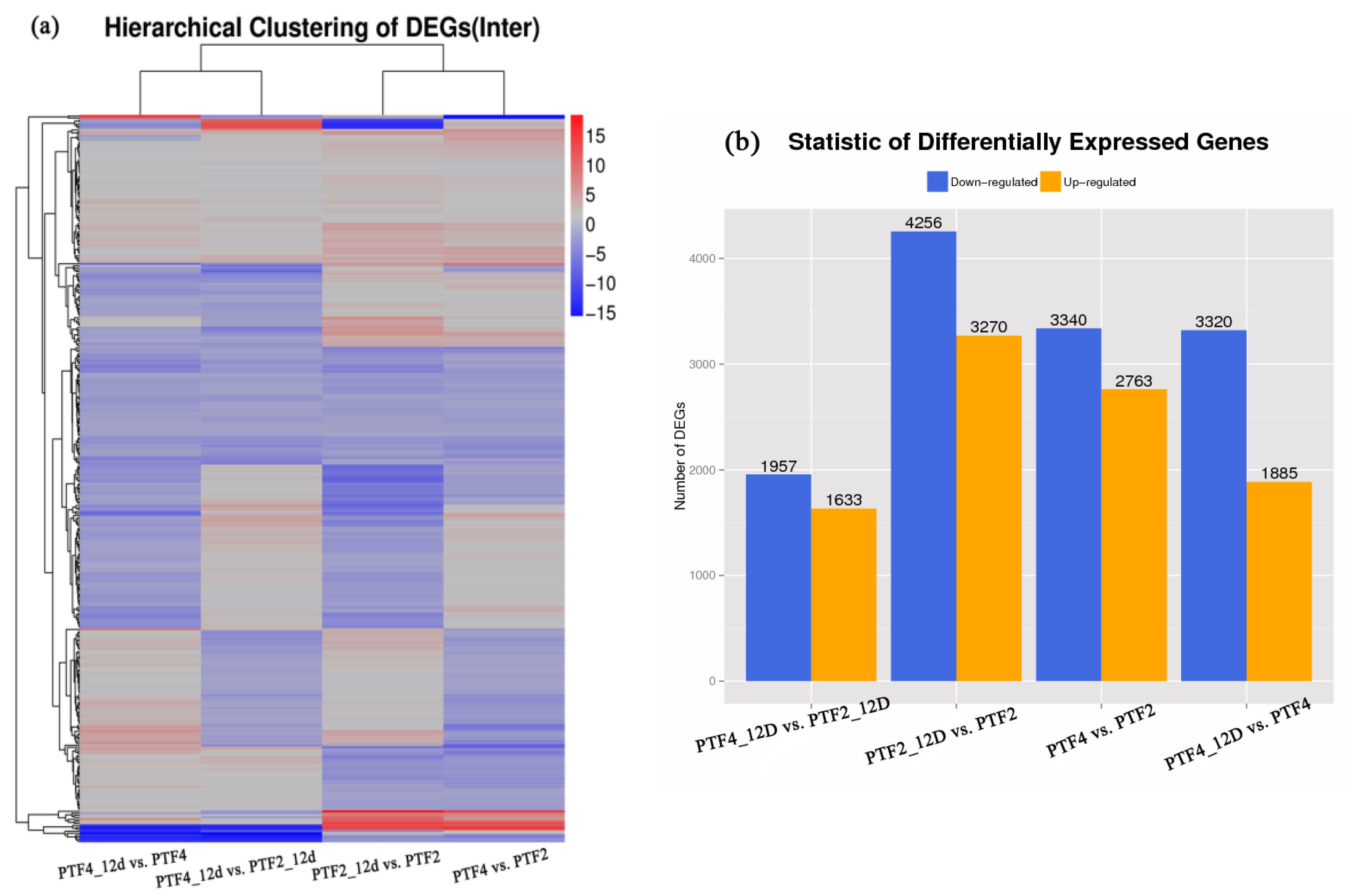
3.3. Comparison of Gene Expression Profiles between the Two P. fortunei Genotypes
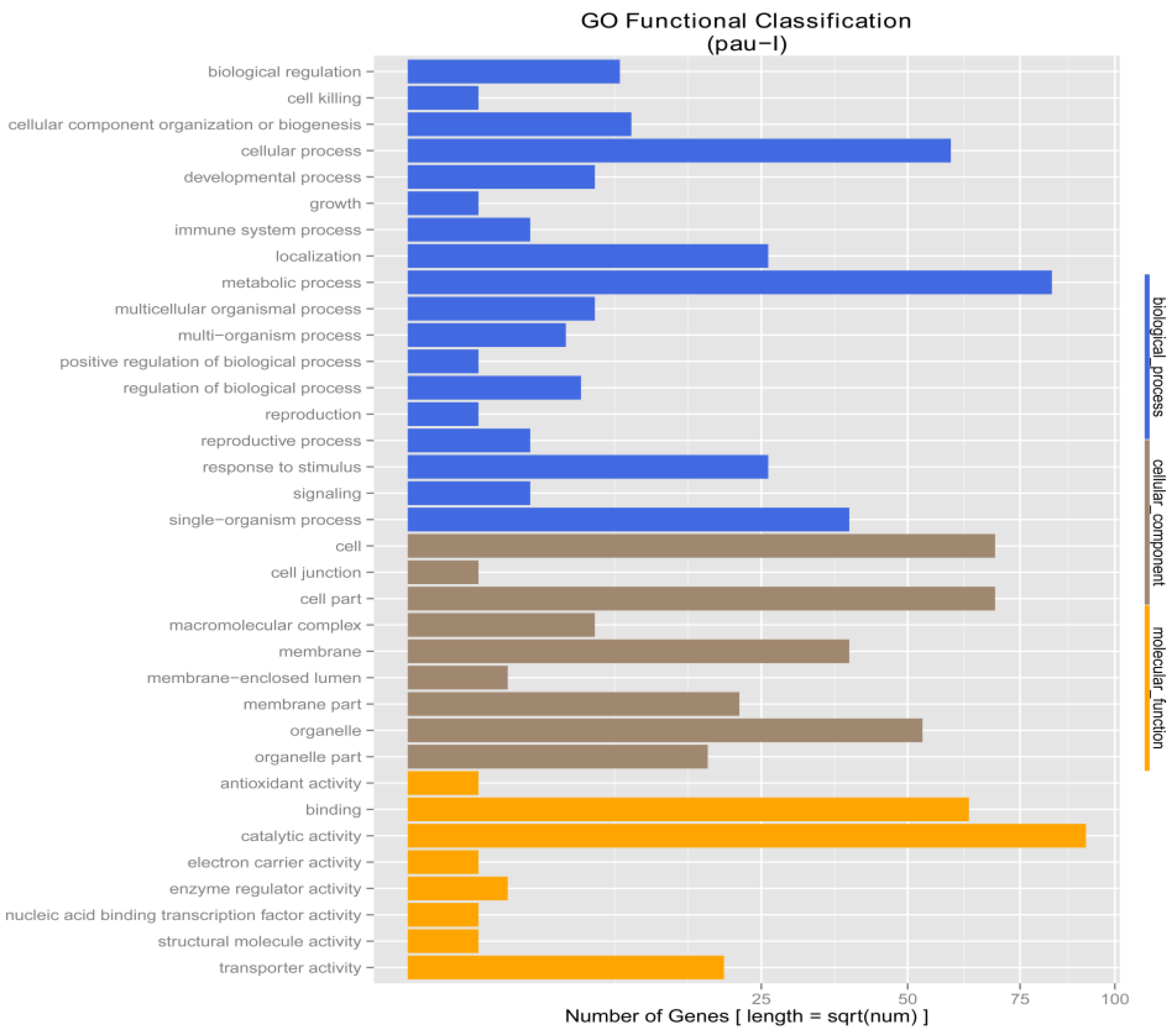
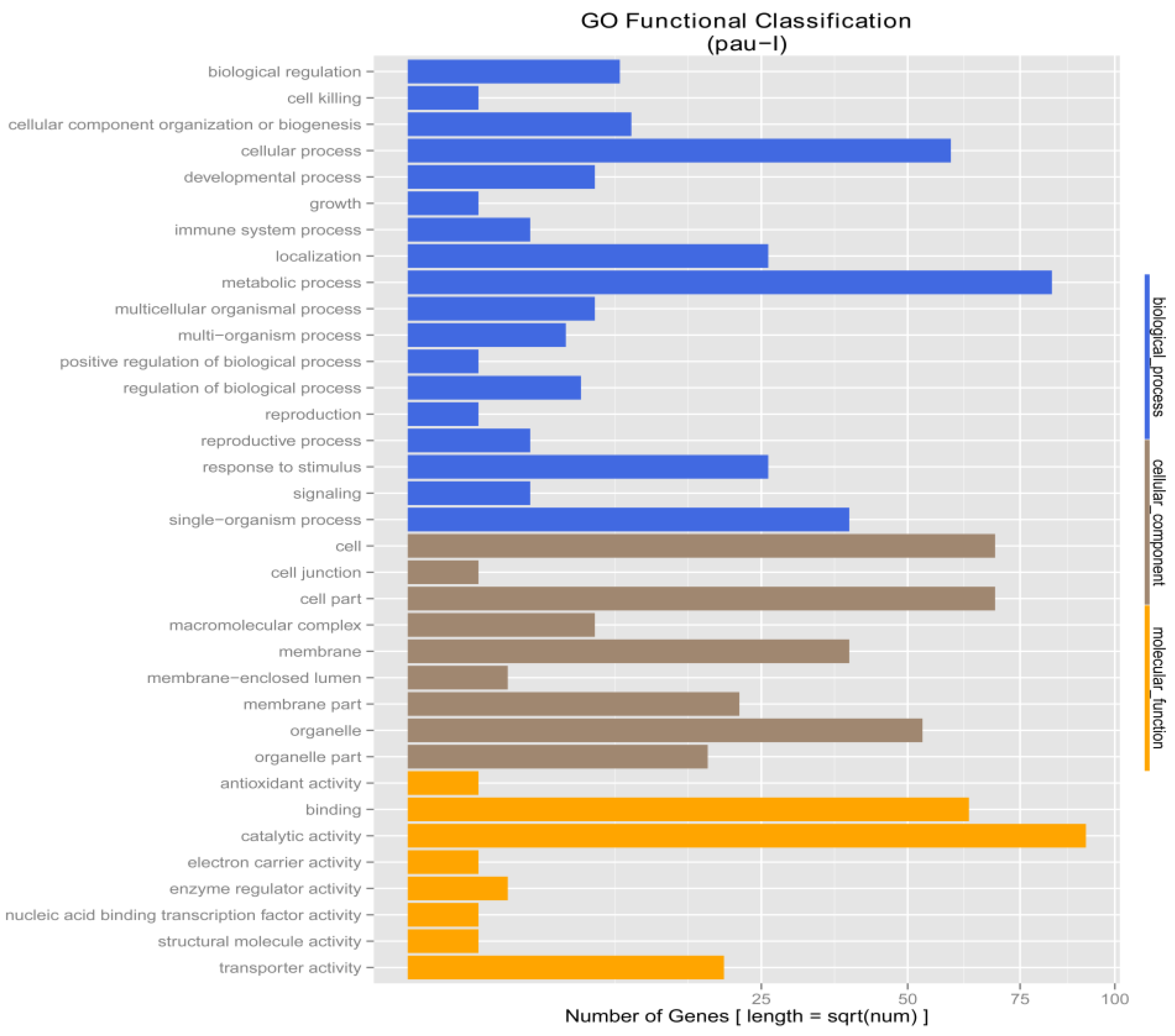
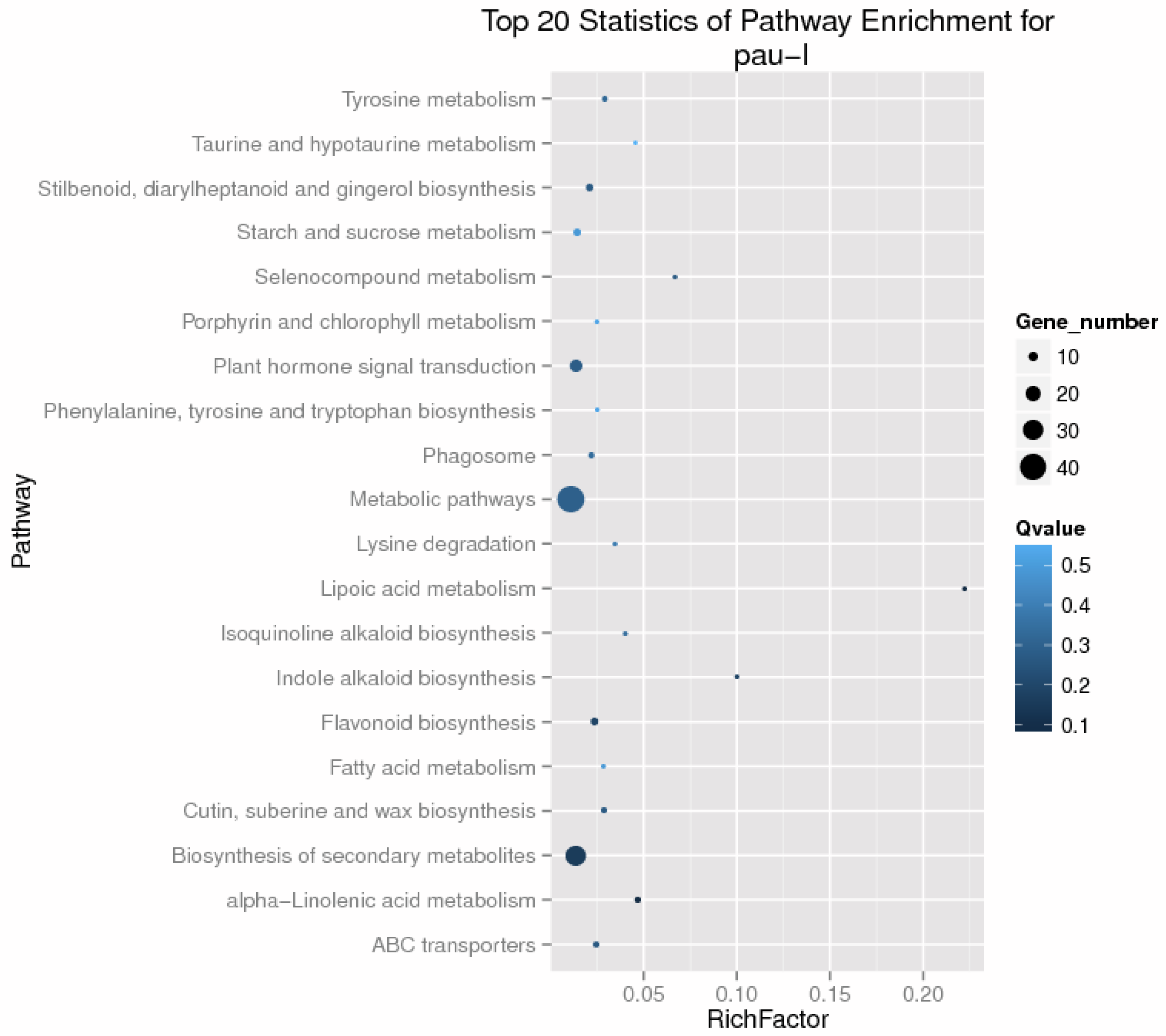
3.4. Drought Responsive Genes in the Two P. fortunei Genotypes
3.5. Analysis of Small RNA Library Data
3.6. Identification of Conserved and Novel miRNAs
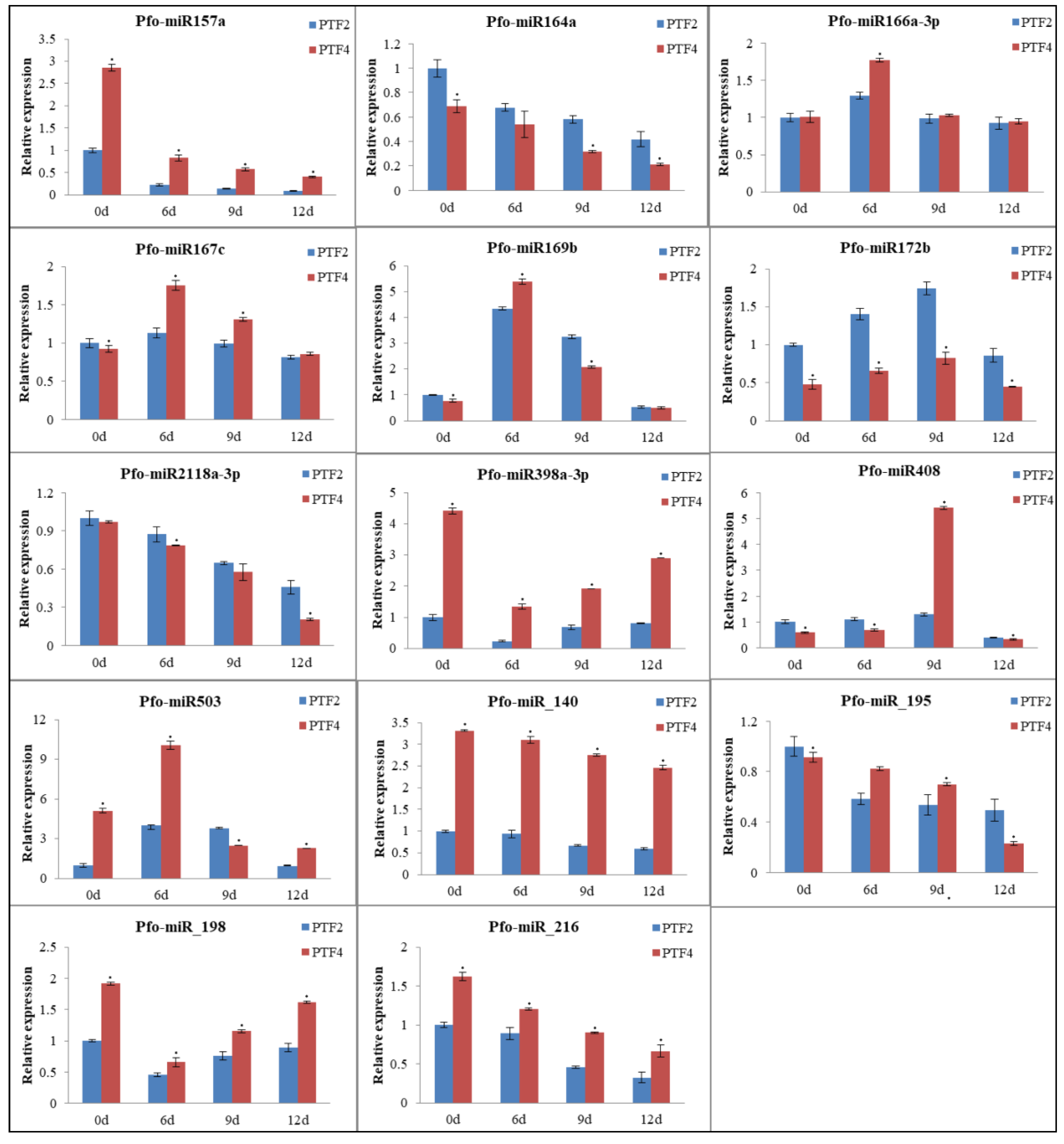
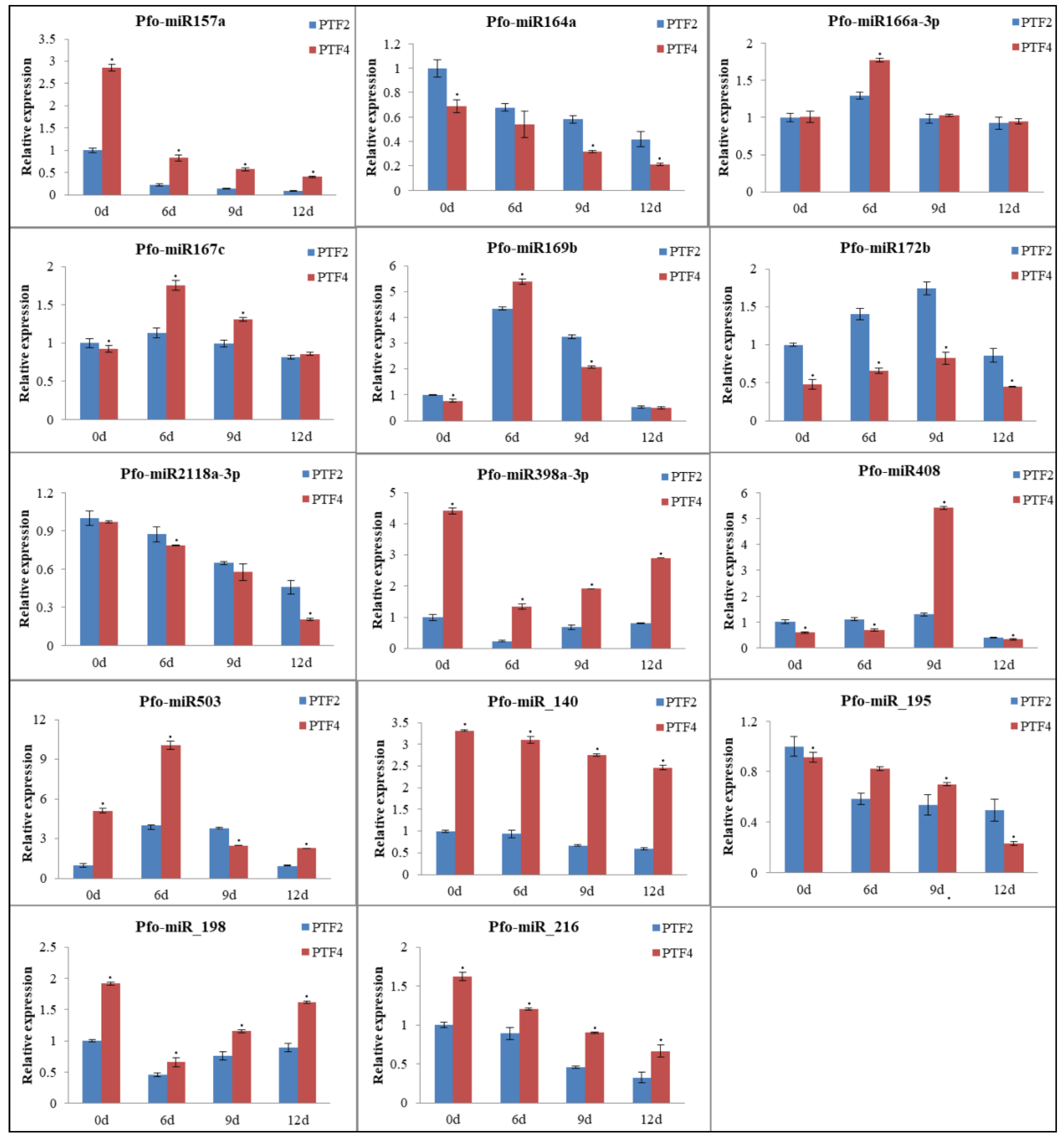
3.7. Analysis of miRNA Expression Profiles in the Two P. fortunei Genotypes
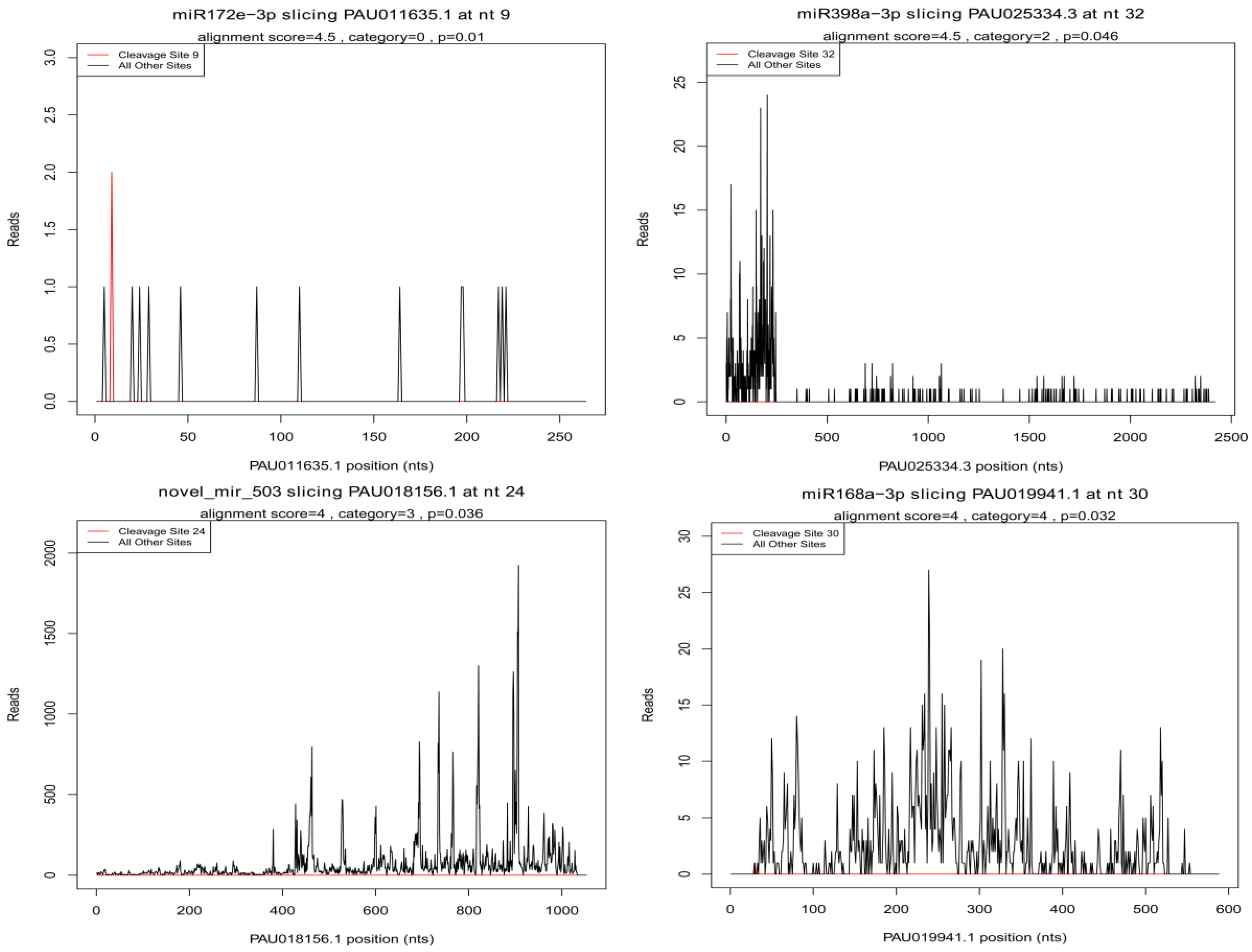
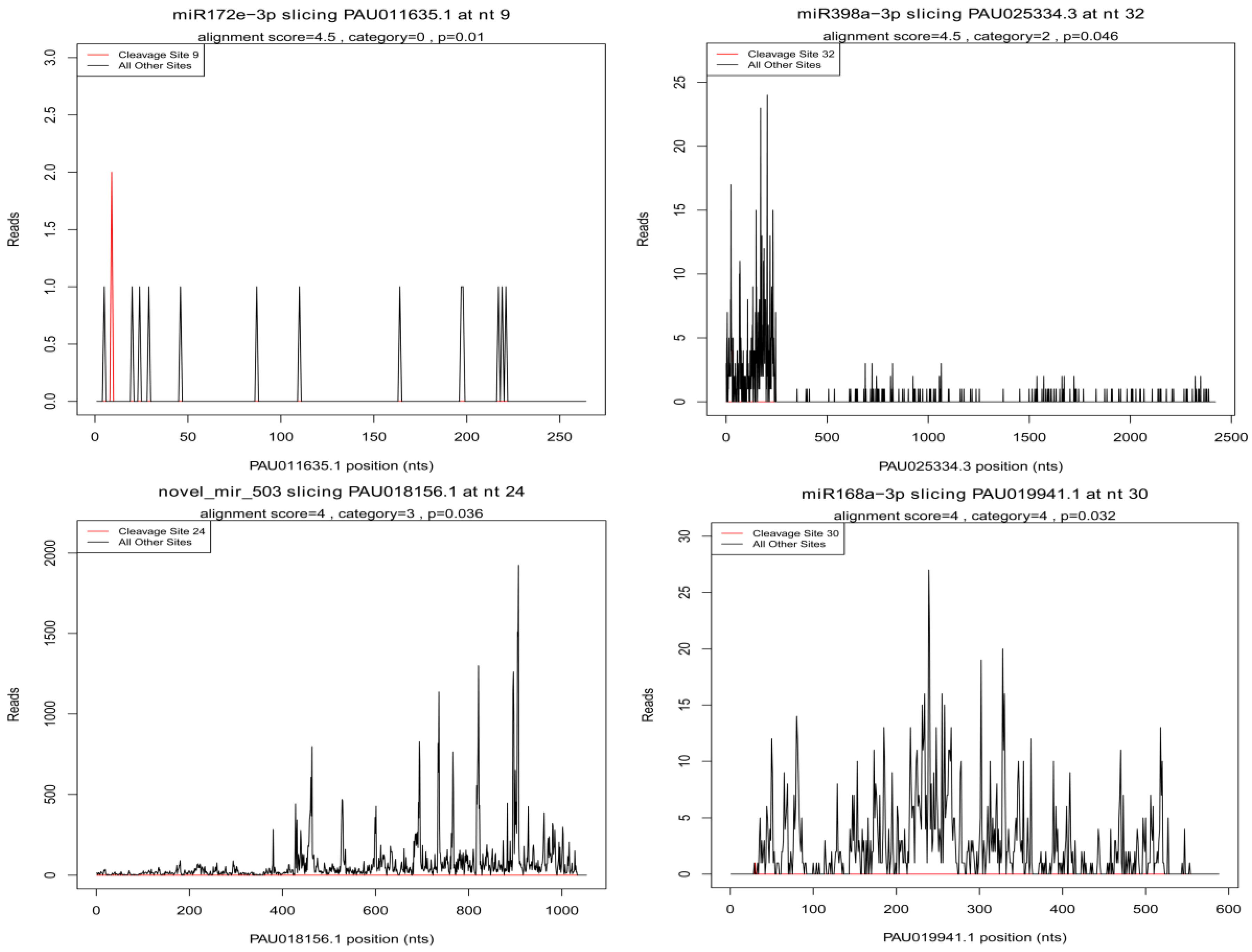
3.8. Identification of miRNA Target Genes Associated with Drought Stress
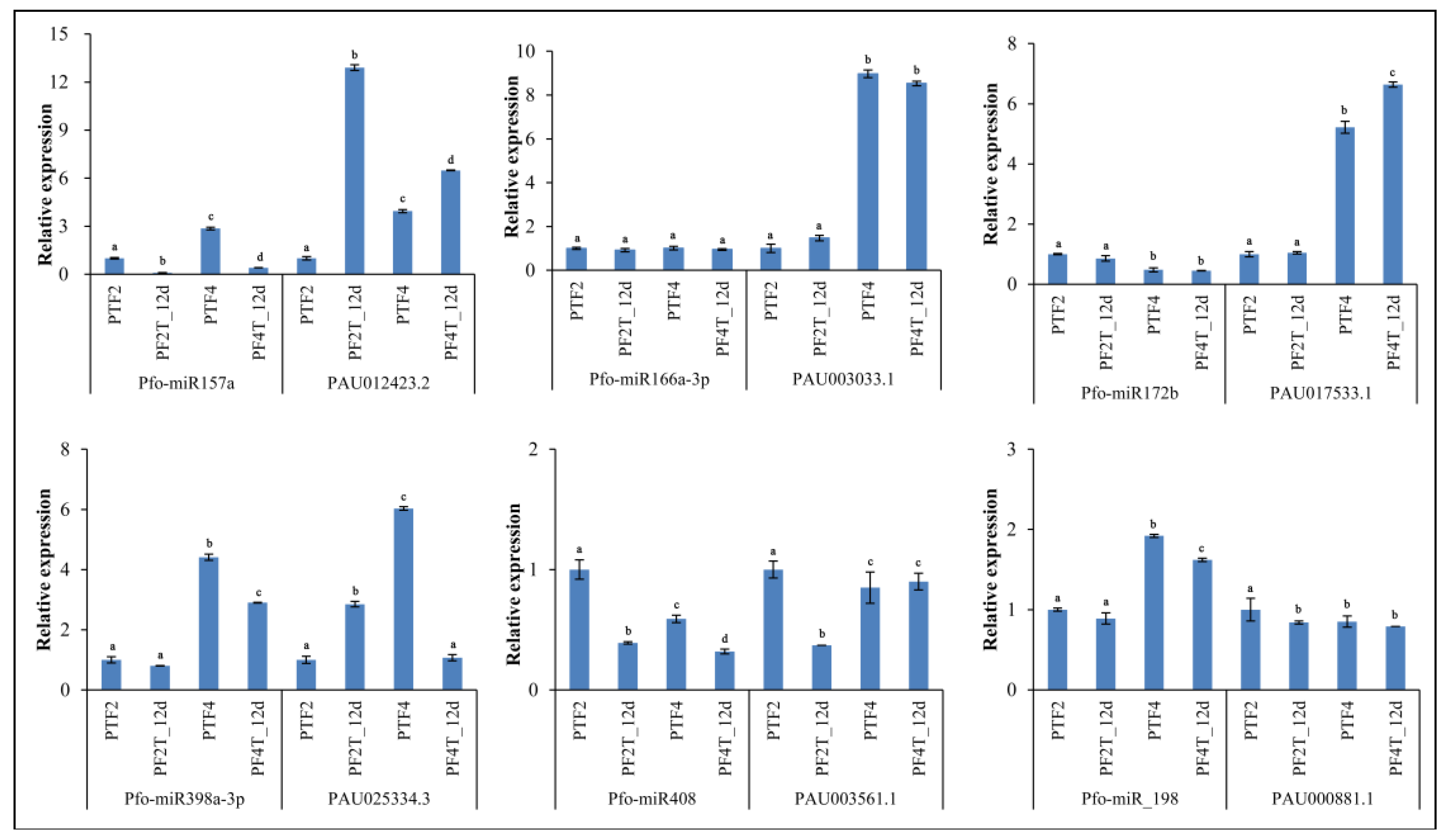
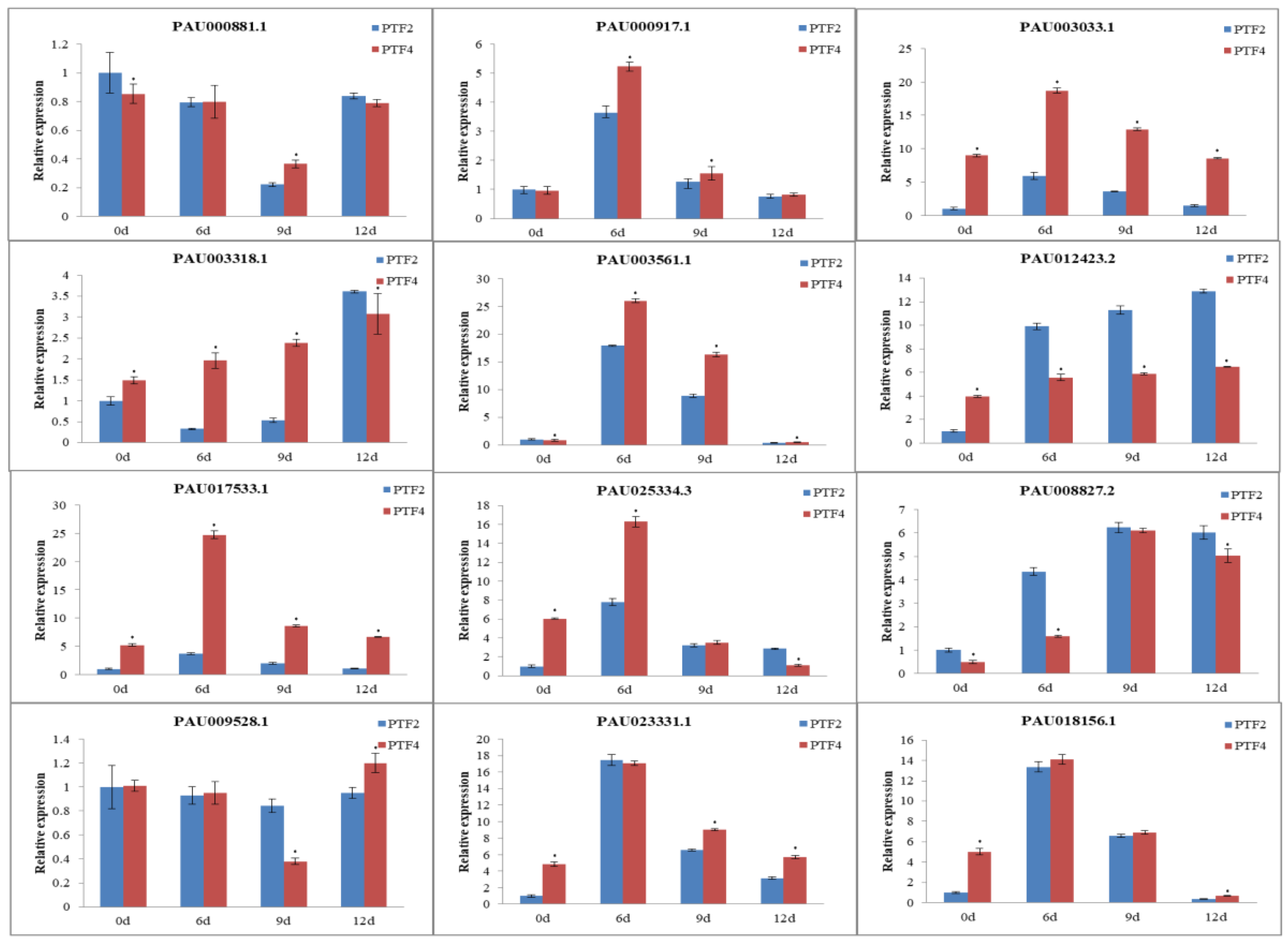
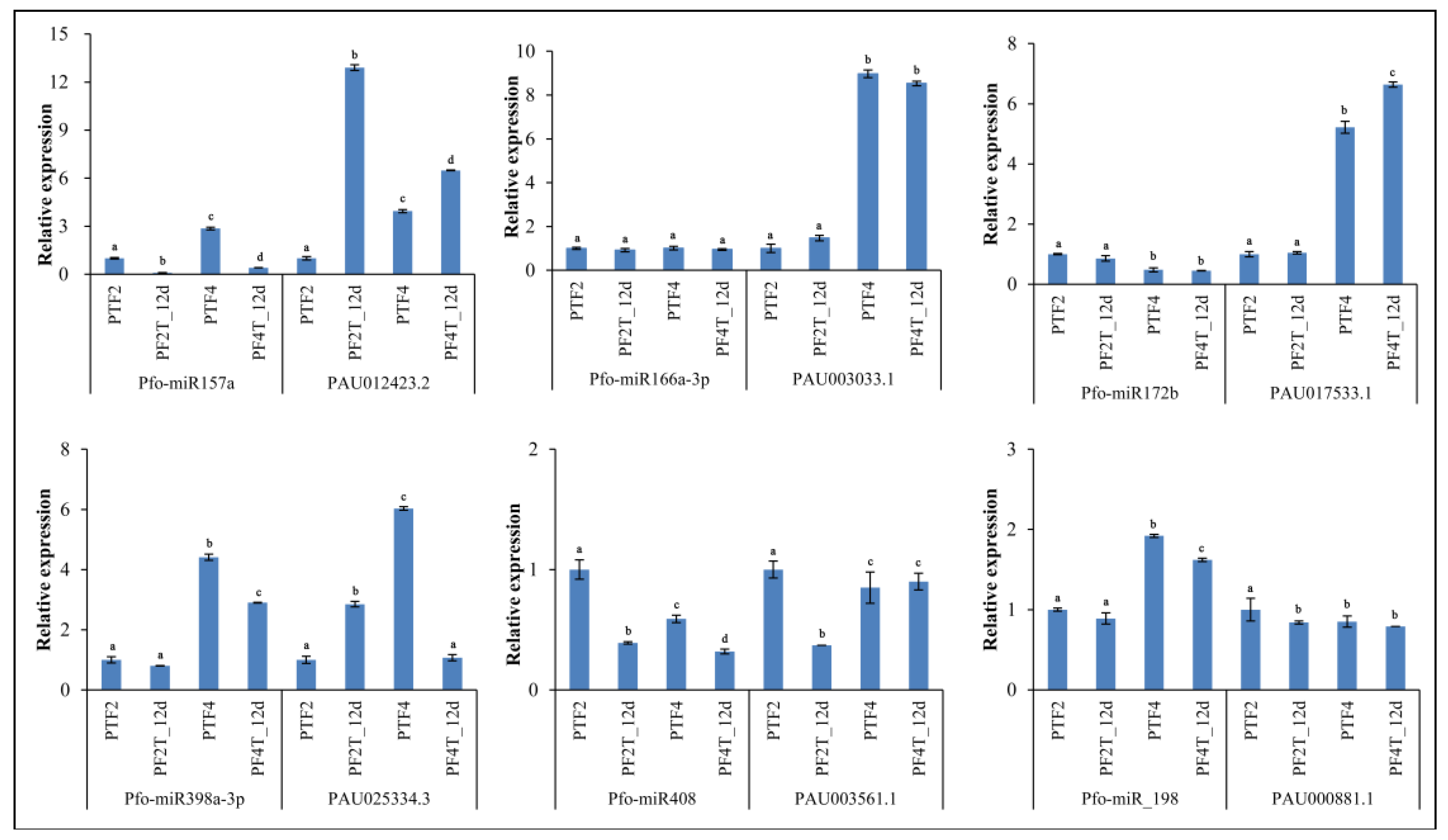
3.9. Confirmation of Genes, miRNAs, and miRNA Targets by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, G.T.; Ma, S.L.; Bai, L.P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, R.; Fan, G.; Zhao, Z.; Deng, M. Study on the physiological response of tetraploid paulownia to drought. J. Henan Agric. Univ. 2013, 47, 543–551. [Google Scholar]
- Raes, J. Duplication and divergence: The evolution of new genes and old ideas. Ann. Rev. Genet. 2004, 38, 615–643. [Google Scholar]
- Jackson, J.A.; Tinsley, R.C. Parasite infectivity to hybridising host species: A link between hybrid resistance and allopolyploid speciation? Int. J. Parasitol. 2003, 33, 137–144. [Google Scholar] [CrossRef]
- Ramsey, J.; Schemske, D.W. Neopolyploidy in flowering plants. Ann. Rev. Ecol. Syst. 2002, 33, 589–639. [Google Scholar] [CrossRef]
- Ni, Z.; Hu, Z.; Jiang, Q.; Zhang, H. Gmnfya3, a target gene of mir169, is a positive regulator of plant tolerance to drought stress. Plant Mol. Biol. 2013, 82, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, N.; Mi, X.; Wu, L.; Ma, R.; Zhu, X.; Yao, L.; Jin, X.; Si, H.; Wang, D. Identification of mir159s and their target genes and expression analysis under drought stress in potato. Comput. Biol. Chem. 2014, 53 Pt B, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, Ž.; Stanisavljević, N.; Mikić, A.; Radović, S.; Maksimović, V. Water deficit down-regulates mir398 and mir408 in pea (Pisum sativum L.). Plant Physiol. Biochem. 2014, 83, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sorin, C.; Declerck, M.; Christ, A.; Blein, T.; Ma, L.; Lelandais-Brière, C.; Njo, M.F.; Beeckman, T.; Crespi, M.; Hartmann, C. A mir169 isoform regulates specific nf-ya targets and root architecture in arabidopsis. New Phytol. 2014, 202, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant mirnas and sirnas. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.A.; Reyes, J.L. Post-transcriptional gene regulation of salinity and drought responses by plant micrornas. Plant Cell Environ. 2010, 33, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-G.; An, M.; Zhou, S.-F.; She, Y.-H.; Li, W.-C.; Fu, F.-L. Expression profile of maize micrornas corresponding to their target genes under drought stress. Biochem. Genet. 2014, 52, 474–493. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.K.; Xi, B.; Yong, L.; Ding, X.D.; Ying, G.; Hua, C.; Wei, J.; Wu, N.; Zhu, Y.M. Profiling of cold-stress-responsive mirnas in rice by microarrays. Gene 2010, 459, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Nigam, D.; Kumar, S.; Mishra, D.C.; Rai, A.; Smita, S.; Saha, A. Synergistic regulatory networks mediated by micrornas and transcription factors under drought, heat and salt stresses in Oryza sativa spp. Gene 2015, 555, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Z.; Deng, M.; Liu, R.; Niu, S.; Fan, G. Identification and functional analysis of micrornas and their targets in platanus acerifolia under lead (pb) stress. Int. J. Mol. Sci. 2015, 16, 7098–7111. [Google Scholar] [CrossRef] [PubMed]
- Jover-Gil, S.; Candela, H.; Ponce, M.R. Plant micrornas and development. Int. J. Dev. Biol. 2005, 49, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, S.; Kobayashi, M.; Taji, T.; Naramoto, M.; Seki, M.; Kato, T.; Tabata, S.; Kakubari, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid, a key in abscisic acid biosynthesis in arabidopsis. Plant J. 2001, 27, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jia, W.; Yang, J.; Ismail, A.M. Role of aba in integrating plant responses to drought and salt stresses. Field Crop Res. 2006, 97, 111–119. [Google Scholar] [CrossRef]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. Aba-dependent and aba-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Taishi, U.; Kazuo, N.; Takuya, M.; Takashi, K.; Masaru, T.; Kazuo, S.; Kazuko, Y.S. Molecular basis of the core regulatory network in aba responses: Sensing, signaling and transport. Plant Cell Physiol. 2010, 51, 1821–1839. [Google Scholar]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K.; Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 2003, 6, 410–417. [Google Scholar] [CrossRef]
- Hussain, S.S.; Kayani, M.A.; Amjad, M. Transcription factors as tools to engineer enhanced drought stress tolerance in plants. Biotechnol. Progr. 2011, 27, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis atmyc2 (bhlh) and atmyb2 (myb) function as transcriptional activators in abscisic acid signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, B.; Lu, G.; Han, B. Overexpression of a NAC transcription factor enhances rice drought and salt tolerance. Biochem. Biophys. Res. Commun. 2009, 379, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Dixit, S.; Jetter, R.; Thoenes, E.; van Arkel, G.; Pereira, A. The shine clade of ap2 domain transcription factors activates wax biosynthesis, alters cuticle properties, and confers drought tolerance when overexpressed in arabidopsis. Plant Cell 2004, 16, 2463–2480. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Chua, N. Aba induction of mir159 controls transcript levels of two myb factors during. Plant J. Cell Mol. Biol. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Bojović, B.; Stojanović, J. Chlorophyll and carotenoid content in wheat cultivars as a function of mineral nutrition. Arch. Biol. Sci. 2005, 57, 283–290. [Google Scholar] [CrossRef]
- Arnon, D.I. Copper enzymes in isolated chloroplasts. Polyphenoloxidase in beta vulgaris. Plant Phys. 1949, 24, 1–15. [Google Scholar] [CrossRef]
- Bates, L.; Waldren, R.; Teare, I. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Li, H. Principle and Technology of Plant Physiology and Biochemistry; Higher Education Press: Beijing, China, 2000. [Google Scholar]
- Liu, J.; Liu, X. Tutorial of Plant Physiology Experiments; Higher Education Press: Beijing, China, 2011. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Method 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Meth. 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genom. Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Integrating microrna annotation and deep-sequencing data. Nucleic Acid Res. 2010, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bonnie, B.; Bartel, D.P.; David, B.; Bowman, J.L.; Xiaofeng, C.; Carrington, J.C.; Xuemei, C.; Green, P.J. Criteria for annotation of plant micrornas. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous sirna and mirna targets identified by sequencing of the arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microrna-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of micrornas by stem-loop rt-pcr. Nucleic Acid Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Niu, S.; Zhao, Z.; Deng, M.; Xu, E.; Wang, Y.; Yang, L. Identification of micrornas and their targets in paulownia fortunei plants free from phytoplasma pathogen after methyl methane sulfonate treatment. Biochimie 2016, 127, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Livaka, K.J.; Schmittgenb, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(t)) method. Methods 2011, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Compatible solute, transporter protein, transcription factor, and hormone-related gene expression provides an indicator of drought stress in paulownia fortunei. Funct. Integr. Genomics 2014, 14, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Xie, H.; Kong, M.L.; Sun, Q.P.; Li, R.Z.; Pan, J.B. Computational identification of mirnas and their targets in phaseolus vulgaris. Genet. Mol. Res. 2014, 13, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Baohong, Z.; Xiaoping, P.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microrna genes. Plant J. Cell Mol. Biol. 2006, 46, 243–259. [Google Scholar]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that mirnas are different from other rnas. Cell. Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. Cleaveland: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- An, F.M.; Chan, M.T. Transcriptome-wide characterization of mirna-directed and non-mirna-directed endonucleolytic cleavage using degradome analysis under low ambient temperature in phalaenopsis aphrodite subsp. Formosana. Plant Cell Physiol. 2012, 53, 1737–1750. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Wei, H.; Wu, M.; Song, M.; Zhang, J.; Yu, J.; Fan, S.; Yu, S. Comparative expression profiling of mirna during anther development in genetic male sterile and wild type cotton. BMC Plant Biol. 2013, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Niu, S.; Li, X.; Wang, Y.; Zhao, Z.; Deng, M.; Dong, Y. Functional analysis of differentially expressed micrornas associated with drought stress in diploid and tetraploid paulownia fortunei. Plant Mol. Biol. Rep. 2017, 35, 389–398. [Google Scholar] [CrossRef]
- Xiaoshen, Z.; Guoqiang, F.; Zhenli, Z.; Xibing, C.; Gaili, Z.; Minjie, D.; Yanpeng, D. Analysis of diploid and its autotetraploid paulownia tomentosa× p. Fortunei with aflp and msap. Sci. Silv. Sin. 2013, 10, 026. [Google Scholar]
- Zhang, Y.; Cao, X.; ZhaiI, X.; Fan, G. Study on DNA extraction of aflp reaction system for paulownia plants. J. Henan Agric. Univ. 2009, 6, 007. [Google Scholar]
- De la Rosa, R.; Angiolillo, A.; Guerrero, C.; Pellegrini, M.; Rallo, L.; Besnard, G.; Bervillé, A.; Martin, A.; Baldoni, L. A first linkage map of olive (Olea europaea L.) cultivars using rapd, aflp, rflp and ssr markers. Theor. Appl. Genet. 2003, 106, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Araujo, J.; Ferreira, J.; Whetten, R.; O’malley, D.; Liu, B.-H.; Sederoff, R. AFLP genetic maps of eucalyptus globulus and e. Tereticornis. Theor. Appl. Genet. 1998, 96, 727–737. [Google Scholar] [CrossRef]
- Phillips, R.; Vasil, I. DNA-Based Markers in Plants, Advances in Cellular and Molecular Biology of Plants; Kluwer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Gupta, P.K.; Varshney, R. The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 2000, 113, 163–185. [Google Scholar] [CrossRef]
- Huang, X.; Yan, H.-D.; Zhang, X.; Zhang, J.; Frazier, T.P.; Huang, D.; Lu, L.; Huang, L.; Liu, W.; Peng, Y.; et al. De novo transcriptome analysis and molecular marker development of two hemarthria species. Front. Plant Sci. 2016, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Gramazio, P.; Blanca, J.; Ziarsolo, P.; Herraiz, F.J.; Plazas, M.; Prohens, J.; Vilanova, S. Transcriptome analysis and molecular marker discovery in solanum incanum and s. Aethiopicum, two close relatives of the common eggplant (Solanum melongena) with interest for breeding. BMC Genom. 2016, 17, 300. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Khu, D.-M.; Torres-Jerez, I.; Udvardi, M.; Monteros, M.J. Plant transcription factors as novel molecular markers for legumes. In Sustainable Use of Genetic Diversity in Forage and Turf Breeding; Springer: Berlin, Germany, 2010; pp. 421–425. [Google Scholar]
- Lu, T.; Lu, G.; Fan, D.; Zhu, C.; Li, W.; Zhao, Q.; Feng, Q.; Zhao, Y.; Guo, Y.; Li, W. Function annotation of the rice transcriptome at single-nucleotide resolution by rna-seq. Genom. Res. 2010, 20, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; Feng, Y.; Zhan, X.; Shen, X.; Wu, W.; Yu, P.; Zhang, Y.; Chen, D.; Wang, H.; Lin, Z. Identification of transcriptome snps for assessing allele-specific gene expression in a super-hybrid rice xieyou9308. PLoS ONE 2013, 8, e60668. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Stegle, O.; Behr, J.; Steffen, J.G.; Drewe, P.; Hildebrand, K.L.; Lyngsoe, R.; Schultheiss, S.J.; Osborne, E.J.; Sreedharan, V.T. Multiple reference genomes and transcriptomes for arabidopsis thaliana. Nature 2011, 477, 419. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A.; Gregory, B.D.; Reverdatto, S.V.; Chen, H.; Kumar, R.; Hooker, T.; Yazaki, J.; Li, P.; Skiba, N.; Peng, Q. Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the arabidopsis transcriptome. Cell 2007, 131, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.F.; Li, B.; Jia, G.Q.; Zhang, T.F.; Dai, J.R.; Li, J.S.; Wang, S.C. Isolation and characterization of genes encoding grf transcription factors and gif transcriptional coactivators in maize (Zea mays L.). Plant Sci. 2008, 175, 809–817. [Google Scholar] [CrossRef]
- Nadgauda, R.S.; Parasharami, V.A.; Mascarenhas, A.F. Precocious flowering and seeding behaviour in tissue-cultured bamboos. Nature 1990, 344, 335–336. [Google Scholar] [CrossRef]
- Thorsen, S.; Rugulies, R.; Løngaard, K.; Borg, V.; Thielen, K.; Bjorner, J.B. Edl3 is an f-box protein involved in the regulation of abscisic acid signalling in arabidopsis thaliana. Int. Arch. Occup. Environ. Health 2011, 62, 5547–5560. [Google Scholar]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Cominelli, E.; Sala, T.; Calvi, D.; Gusmaroli, G.; Tonelli, C. Over-expression of the arabidopsis atmyb41 gene alters cell expansion and leaf surface permeability. Plant J. 2008, 53, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Kwon, Y.; Kim, J.; Noh, H.; Hong, S.-W.; Lee, H. A dual role for myb60 in stomatal regulation and root growth of arabidopsis thaliana under drought stress. Plant Mol. Biol. 2011, 77, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lai, Z.; Shi, J.; Xiao, Y.; Chen, Z.; Xu, X. Roles of arabidopsis wrky18, wrky40 and wrky60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010, 10, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yan, L.; Liu, Z.Q.; Cao, Z.; Mei, C.; Xin, Q.; Wu, F.Q.; Wang, X.F.; Du, S.Y.; Jiang, T. The mg-chelatase h subunit of arabidopsis antagonizes a group of wrky transcription repressors to relieve aba-responsive genes of inhibition. Plant Cell 2010, 22, 1909–1935. [Google Scholar] [CrossRef] [PubMed]
- Miki, F.; Yasunari, F.; Kyonoshin, M.; Motoaki, S.; Keiichiro, H.; Masaru, O.T.; Lam-Son Phan, T.; Kazuko, Y.S.; Kazuo, S. A dehydration-induced NAC protein, rd26, is involved in a novel aba-dependent stress-signaling pathway. Plant J. 2004, 39, 863–876. [Google Scholar]
- Tran, L.S.; Nakashima, K.; Sakuma, Y.; Simpson, S.D.; Fujita, Y.; Maruyama, K.; Fujita, M.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Isolation and functional analysis of arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 2004, 16, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, Z.; Lai, J.; Zhang, Y.; Yang, C.; Yin, B.; Zhao, Q.; Zhang, L.; Li, Y.; Yang, C.; et al. Dual function of arabidopsis ATAF1 in abiotic and biotic stress responses. Cell Res. 2009, 19, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.A.D.; Durnford, D.G. Evolutionary distribution of light-harvesting complex-like proteins in photosynthetic eukaryotes. Genome 2010, 53, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Liu, R.; Yan, L.; Liu, Z.Q.; Jiang, S.C.; Shen, Y.Y.; Wang, X.F.; Zhang, D.P. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in arabidopsis. J. Exp. Bot. 2011, 63, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Minh-Thu, P.T.; Hwang, D.J.; Jeon, J.S.; Nahm, B.H.; Kim, Y.K. Transcriptome analysis of leaf and root of rice seedling to acute dehydration. Rice 2013, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Q. Natural attributes and agricultural implications of somatic genome variation. Curr. Issue Mol. Biol. 2016, 20, 29–46. [Google Scholar]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lei, C.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of drought-responsive micrornas in medicago truncatula by genome-wide high-throughput sequencing. BMC Genom. 2011, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Scheffler, B.E.; Bauer, P.J.; Campbell, B.T. Identification of the family of aquaporin genes and their expression in upland cotton (Gossypium hirsutum L.). BMC Plant Biol. 2010, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Formey, D.; Iñiguez, L.P.; Peláez, P.; Li, Y.F.; Sunkar, R.; Sánchez, F.; Reyes, J.L.; Hernández, G. Genome-wide identification of the phaseolus vulgaris srnaome using small RNA and degradome sequencing. BMC Genom. 2015, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, G.; Zhang, X.; Mercier, R.; Leng, Q.; Lawton, M. Co-expression of calcium-dependent protein kinase with the inward rectified guard cell k+ channel kat1 alters current parameters in xenopus laevis oocytes. Plant Cell Physiol. 2000, 41, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Maathuis, F.J.; Sanders, D. Mechanisms of potassium absorption by higher plant roots. Physiol. Plant. 1996, 96, 158–168. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Map to Genome | Map to Gene | ||||
|---|---|---|---|---|---|
| Category | Reads Number | Percent | Reads Number | Percent | |
| PF2 | Total Reads | 64,267,312 | 100.00% | 64,267,312 | 100.00% |
| Total Base Pairs | 5,784,058,080 | 100.00% | 5,784,058,080 | 100.00% | |
| Total Mapped Reads | 54,049,716 | 84.10% | 31,664,626 | 49.27% | |
| Perfect Match | 39,381,687 | 61.28% | 23,034,911 | 35.84% | |
| Mismatch | 14,668,029 | 22.82% | 8,629,715 | 13.43% | |
| Unique Match | 46,383,583 | 72.17% | 21,725,268 | 33.80% | |
| Multi-position Match | 7,666,133 | 11.93% | 9,939,358 | 15.47% | |
| Total Unmapped Reads | 10,217,596 | 15.90% | 32,602,684 | 50.73% | |
| PF2T_12D | Total Reads | 66,551,876 | 100.00% | 66,551,876 | 100.00% |
| Total Base Pairs | 5,989,668,840 | 100.00% | 5,989,668,840 | 100.00% | |
| Total Mapped Reads | 55,463,664 | 83.34% | 30,623,746 | 46.01% | |
| Perfect Match | 40,236,126 | 60.46% | 22,138,954 | 33.27% | |
| Mismatch | 15,227,538 | 22.88% | 8,484,792 | 12.75% | |
| Unique Match | 47,298,908 | 71.07% | 20,998,174 | 31.55% | |
| Multi-position Match | 8,164,756 | 12.27% | 9,625,572 | 14.46% | |
| Total Unmapped Reads | 11,088,212 | 16.66% | 35,928,128 | 53.99% | |
| PF4 | Total Reads | 67,073,816 | 100.00% | 67,073,816 | 100.00% |
| Total Base Pairs | 6,036,643,440 | 100.00% | 6,036,643,440 | 100.00% | |
| Total Mapped Reads | 55,033,798 | 82.05% | 33,230,102 | 49.54% | |
| Perfect Match | 33,117,090 | 49.37% | 19,896,750 | 29.66% | |
| Mismatch | 21,916,708 | 32.68% | 13,333,352 | 19.88% | |
| Unique Match | 47,201,196 | 70.37% | 21,955,604 | 32.73% | |
| Multi-position Match | 7,832,602 | 11.68% | 11,274,498 | 16.81% | |
| Total Unmapped Reads | 12,040,018 | 17.95% | 33,843,712 | 50.46% | |
| PF4T_12D | Total Reads | 68,805,144 | 100.00% | 68,805,144 | 100.00% |
| Total Base Pairs | 6,192,462,960 | 100.00% | 6,192,462,960 | 100.00% | |
| Total Mapped Reads | 56,335,628 | 81.88% | 31,595,018 | 45.92% | |
| Perfect Match | 34,095,108 | 49.55% | 18,735,185 | 27.23% | |
| Mismatch | 22,240,520 | 32.32% | 12,859,833 | 18.69% | |
| Unique Match | 47,540,030 | 69.09% | 21,592,724 | 31.38% | |
| Multi-position Match | 8,795,598 | 12.78% | 10,002,294 | 14.54% | |
| Total Unmapped Reads | 12,469,516 | 18.12% | 37,210,124 | 54.08% | |
| Gene ID | TF Family | Included Domain | Excluded Domain |
|---|---|---|---|
| PAU000179.1 | C3H | zf-CCCH | PHD, SNF2_N, zf-C2H2 |
| PAU000931.1 | MYB | Myb_DNA-binding | ARID, Response_reg, SNF2_N |
| PAU000931.1 | Trihelix | trihelix | - |
| PAU001653.1 | GRAS | GRAS | - |
| PAU001788.1 | MYB | Myb_DNA-binding | ARID, Response_reg, SNF2_N |
| PAU001788.1 | G2-like | G2-like | Response_reg |
| PAU002069.1 | GRAS | GRAS | - |
| PAU003364.1 | NAC | NAM | - |
| PAU003475.1 | GRAS | GRAS | - |
| PAU006678.1 | HSF | HSF_DNA-bind | - |
| PAU007281.2 | ABI3VP1 | B3 | AP2, Auxin_resp |
| PAU008439.1 | mTERF | mTERF | - |
| PAU010113.2 | AP2-EREBP | AP2 | - |
| PAU011010.1 | LOB | DUF260 | - |
| PAU012184.1 | bHLH | HLH | - |
| PAU012899.1 | MYB-related | Myb_DNA-binding | ARID, G2-like, Response_reg, SNF2_N,trihelix |
| PAU012899.1 | MYB | Myb_DNA-binding | ARID, Response_reg, SNF2_N |
| PAU017462.1 | NAC | NAM | - |
| PAU018191.1 | MYB-related | Myb_DNA-binding | ARID, G2-like, Response_reg, SNF2_N,trihelix |
| PAU018191.1 | MYB | Myb_DNA-binding | ARID, Response_reg, SNF2_N |
| PAU019436.2 | NAC | NAM | - |
| PAU021504.1 | NAC | NAM | - |
| PAU021792.1 | C2C2-GATA | GATA | - |
| PAU022916.1 | ARF | Auxin_resp | - |
| PAU023755.1 | ARF | Auxin_resp | - |
| PAU025363.1 | WRKY | WRKY | - |
| PAU029472.1 | LIM | LIM | - |
| PAU030808.2 | GRAS | GRAS | - |
| Category | Species | Unique sRNAs | Percent% | Reads Number | Percent% |
|---|---|---|---|---|---|
| miRNA | PF2 | 22,521 | 0.47% | 2,882,549 | 18.56% |
| PF2T-12D | 19,768 | 0.42% | 2,788,081 | 18.89% | |
| PF4 | 19,454 | 0.39% | 2,608,853 | 18.60% | |
| PF4T-12D | 21,358 | 0.46% | 2,317,495 | 14.42% | |
| rRNA | PF2 | 51,685 | 1.09% | 718,451 | 4.62% |
| PF2T-12D | 27,242 | 0.59% | 271,635 | 1.84% | |
| PF4 | 38,987 | 0.78% | 280,929 | 2% | |
| PF4T-12D | 38,819 | 0.83% | 604,494 | 3.76% | |
| repeat | PF2 | 257,952 | 5.44% | 658,140 | 4.24% |
| PF2T-12D | 243,489 | 5.23% | 605,304 | 4.10% | |
| PF4 | 196,217 | 3.94% | 469,620 | 3.35% | |
| PF4T-12D | 175,370 | 3.74% | 511,910 | 3.19% | |
| snRNA | PF2 | 1846 | 0.04% | 3375 | 0.02% |
| PF2T-12D | 1316 | 0.03% | 2821 | 0.02% | |
| PF4 | 1729 | 0.03% | 3247 | 0.02% | |
| PF4T-12D | 1630 | 0.03% | 3582 | 0.02% | |
| snoRNA | PF2 | 647 | 0.01% | 1257 | 0.01% |
| PF2T-12D | 487 | 0.01% | 1083 | 0.01% | |
| PF4 | 635 | 0.01% | 1170 | 0.01% | |
| PF4T-12D | 697 | 0.01% | 1124 | 0.01% | |
| tRNA | PF2 | 15,176 | 0.32% | 569,369 | 3.67% |
| PF2T-12D | 7542 | 0.16% | 293,078 | 1.99% | |
| PF4 | 10,605 | 0.21% | 292,476 | 2.09% | |
| PF4T-12D | 10,210 | 0.22% | 669,113 | 4.16% | |
| unann | PF2 | 4,394,575 | 92.63% | 10,701,772 | 68.89% |
| PF2T-12D | 4,353,098 | 93.56% | 10,797,866 | 73.16% | |
| PF4 | 4,711,849 | 94.62% | 10,368,085 | 73.93% | |
| PF4T-12D | 4,436,314 | 94.71% | 11,959,262 | 74.43% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Niu, S.; Fan, G.; Deng, M.; Wang, Y. Genome-Wide Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Paulownia fortunei (Seem) Hemsl. under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing. Forests 2018, 9, 88. https://doi.org/10.3390/f9020088
Zhao Z, Niu S, Fan G, Deng M, Wang Y. Genome-Wide Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Paulownia fortunei (Seem) Hemsl. under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing. Forests. 2018; 9(2):88. https://doi.org/10.3390/f9020088
Chicago/Turabian StyleZhao, Zhenli, Suyan Niu, Guoqiang Fan, Minjie Deng, and Yuanlong Wang. 2018. "Genome-Wide Analysis of Gene and microRNA Expression in Diploid and Autotetraploid Paulownia fortunei (Seem) Hemsl. under Drought Stress by Transcriptome, microRNA, and Degradome Sequencing" Forests 9, no. 2: 88. https://doi.org/10.3390/f9020088