Antimycobacterial Activity: A New Pharmacological Target for Conotoxins Found in the First Reported Conotoxin from Conasprella ximenes

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
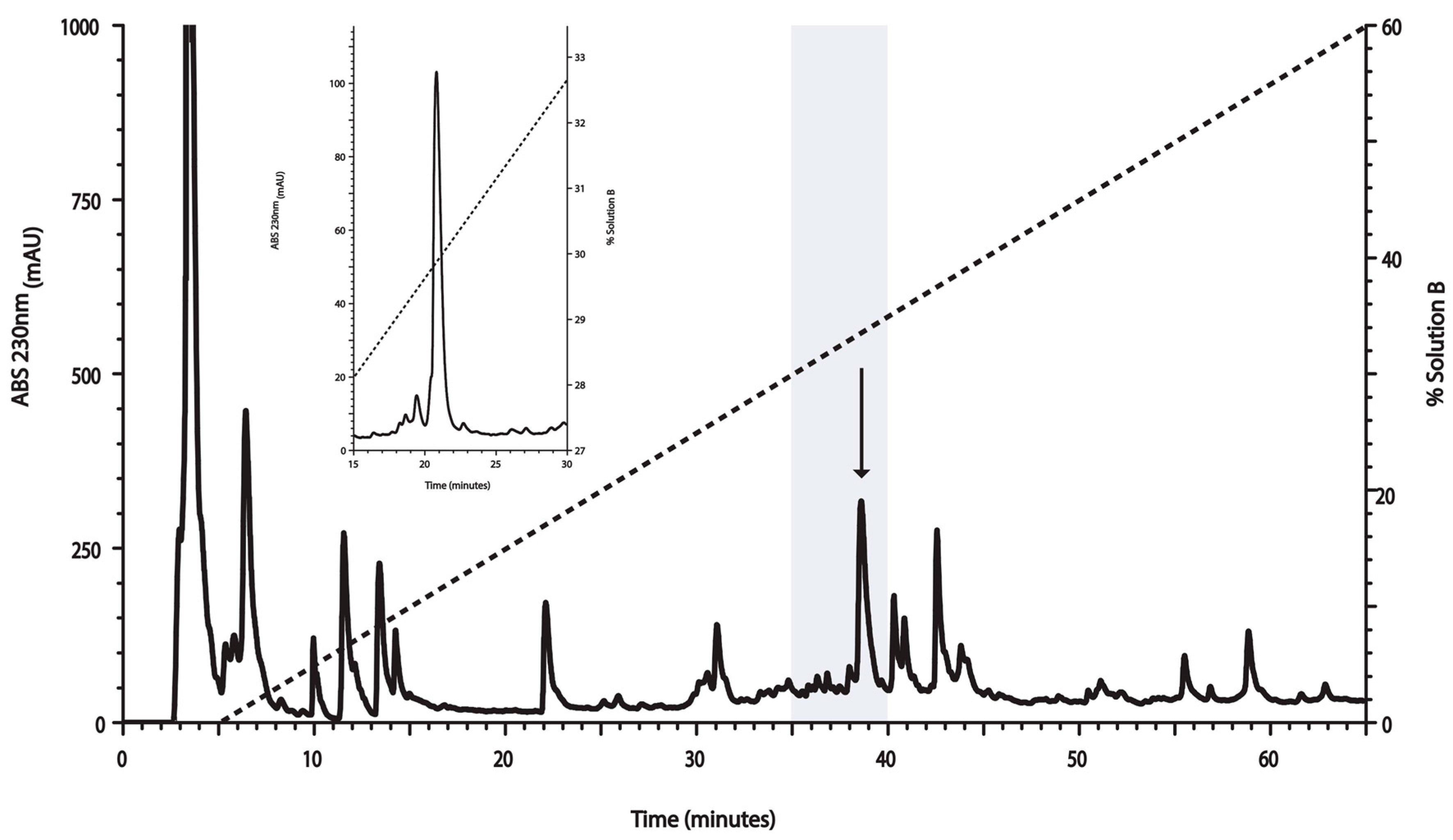
2.1. Venom Fractionation for Anti-Mtb Activity Evaluation
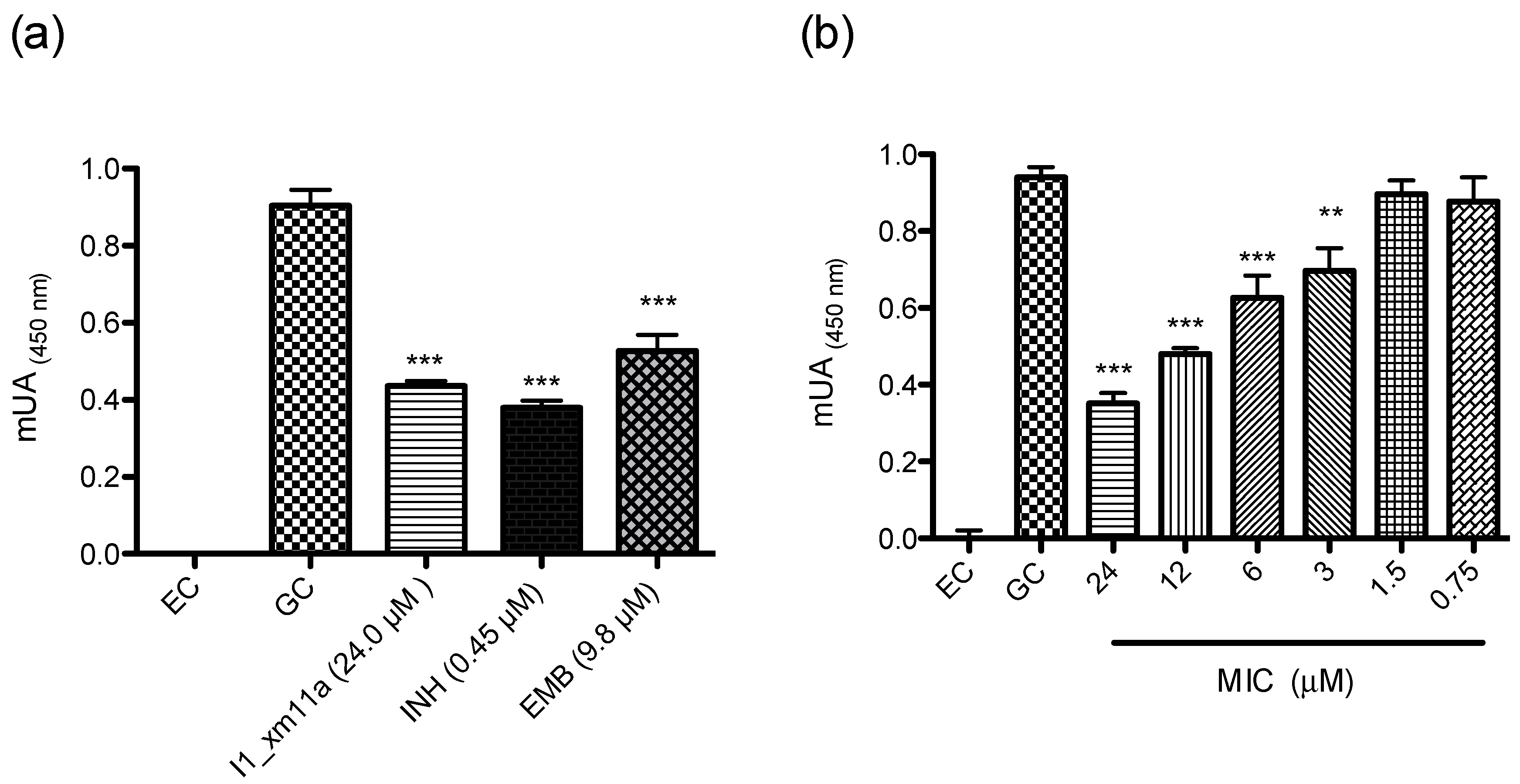
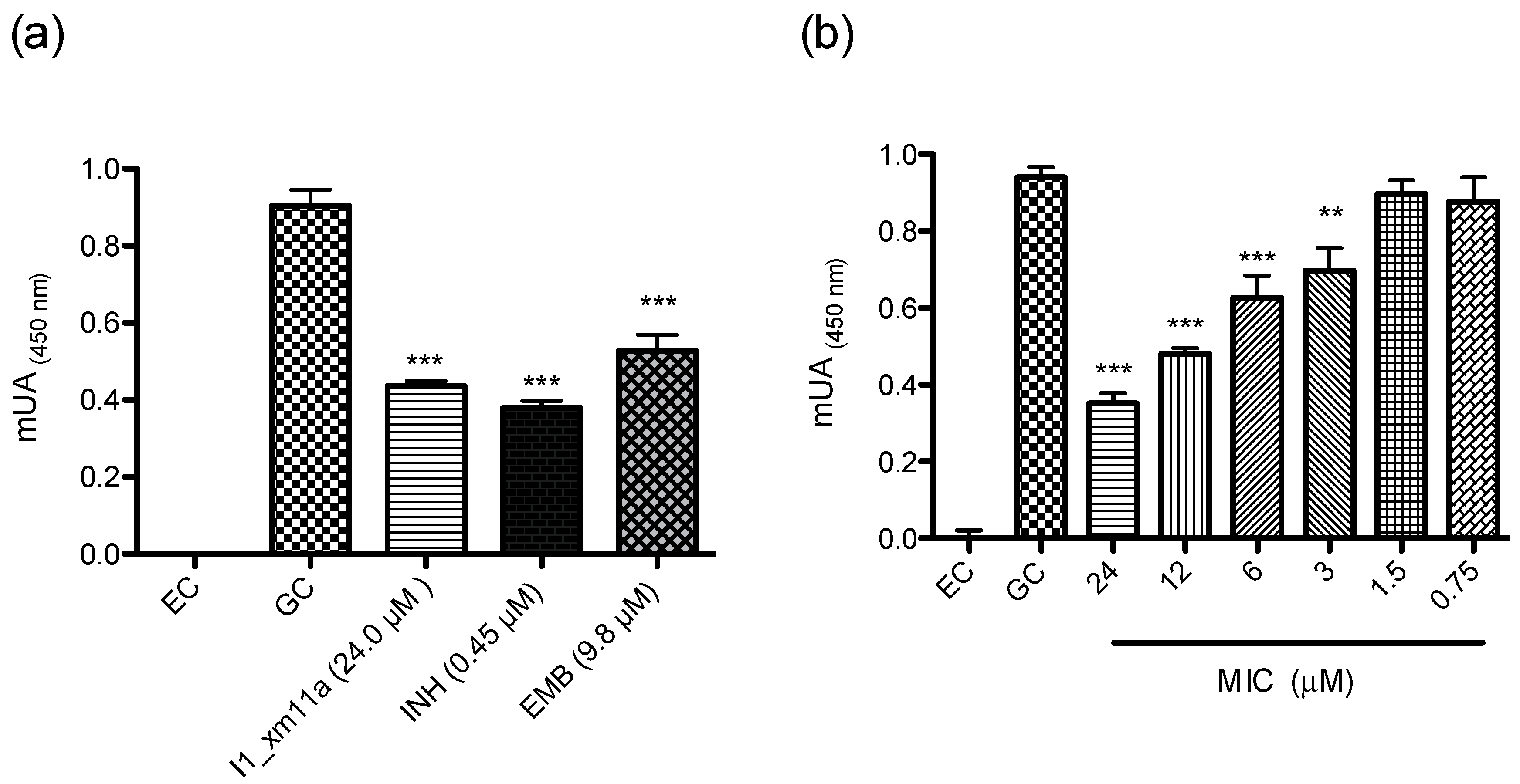
2.2. Antimycobacterial Susceptibility Assay
2.3. Venom Gland Transcriptome Sequence Database
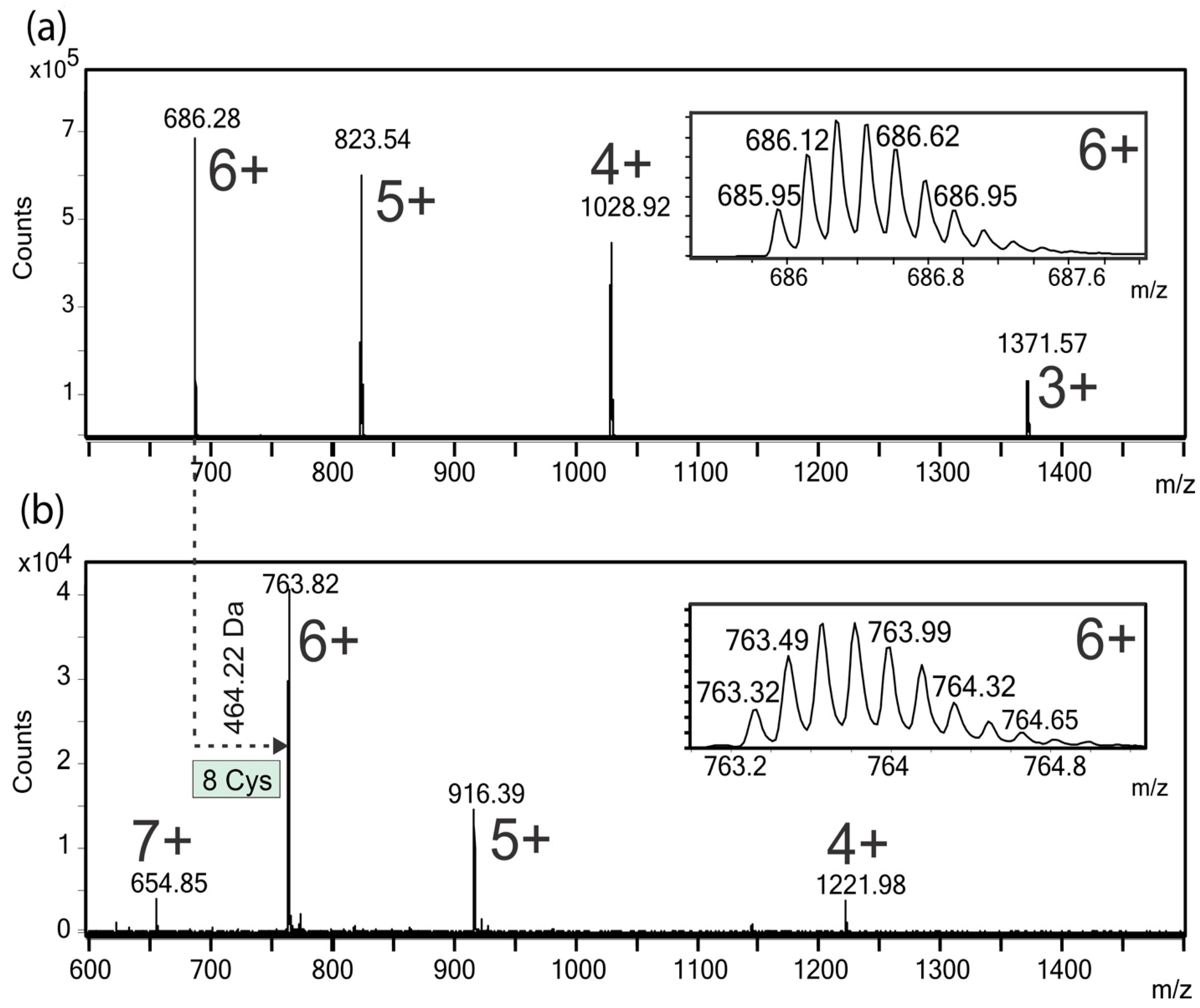
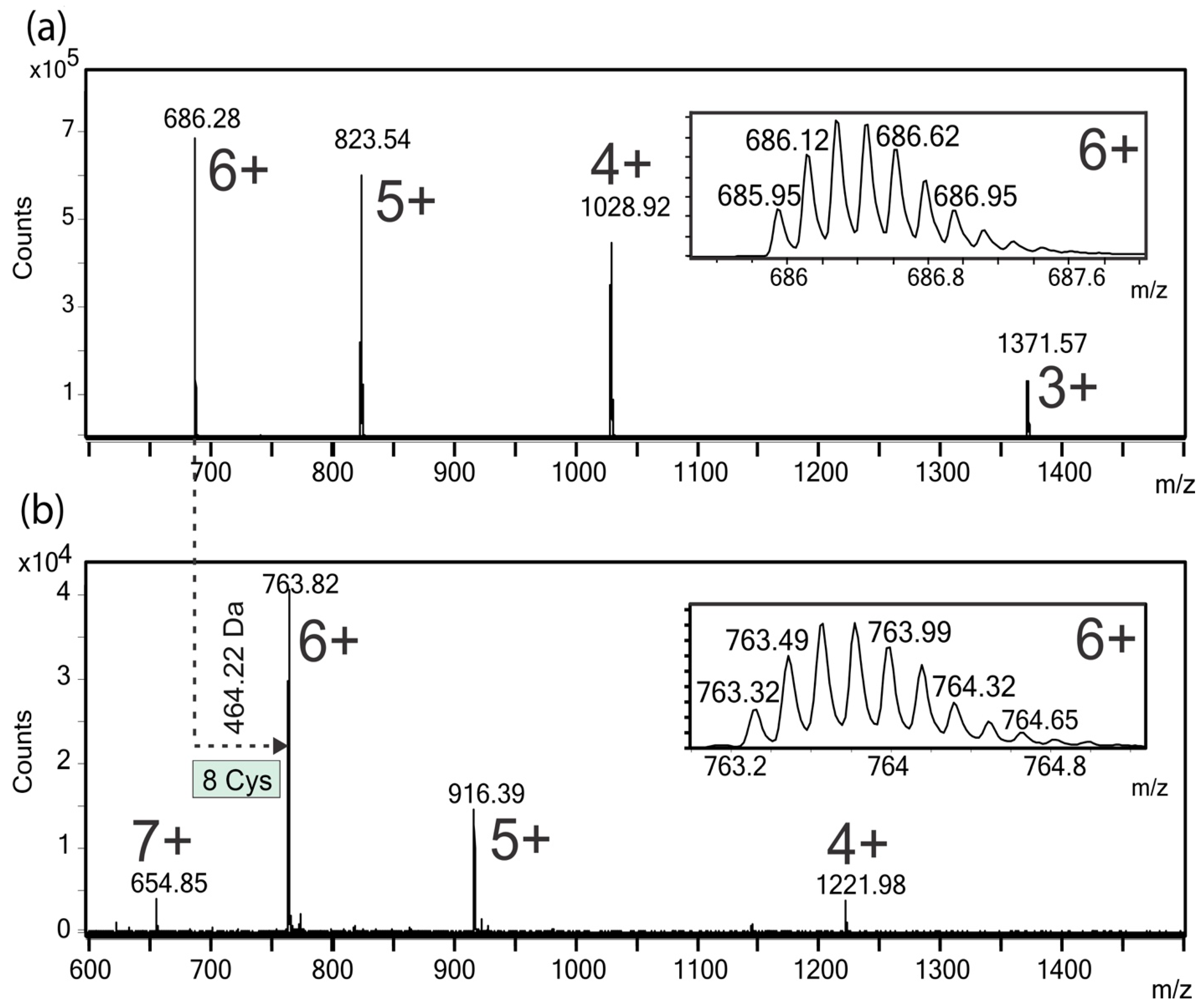
2.4. ESI-MS Analysis of the Native and Carbamidomethylated Peptide
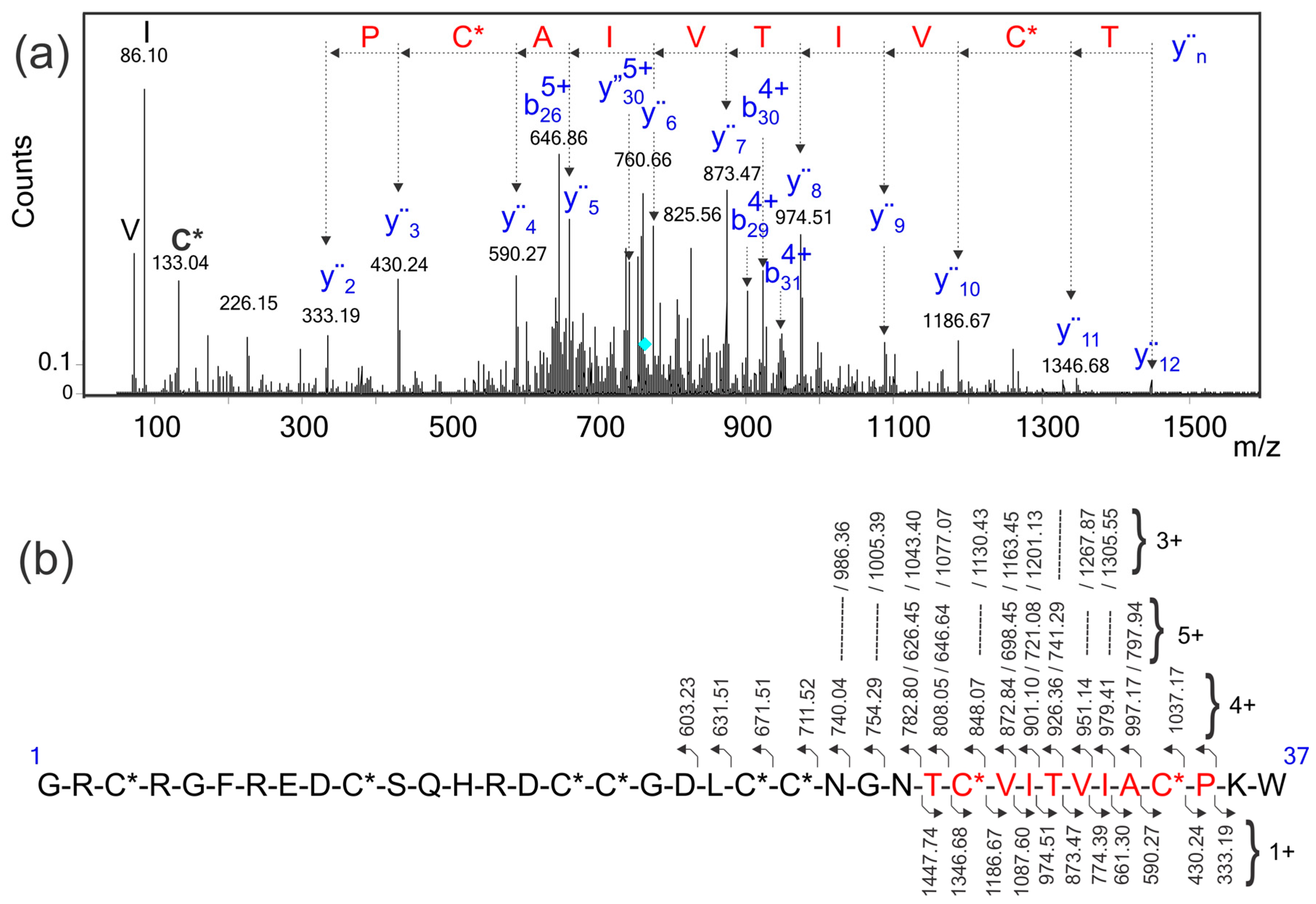
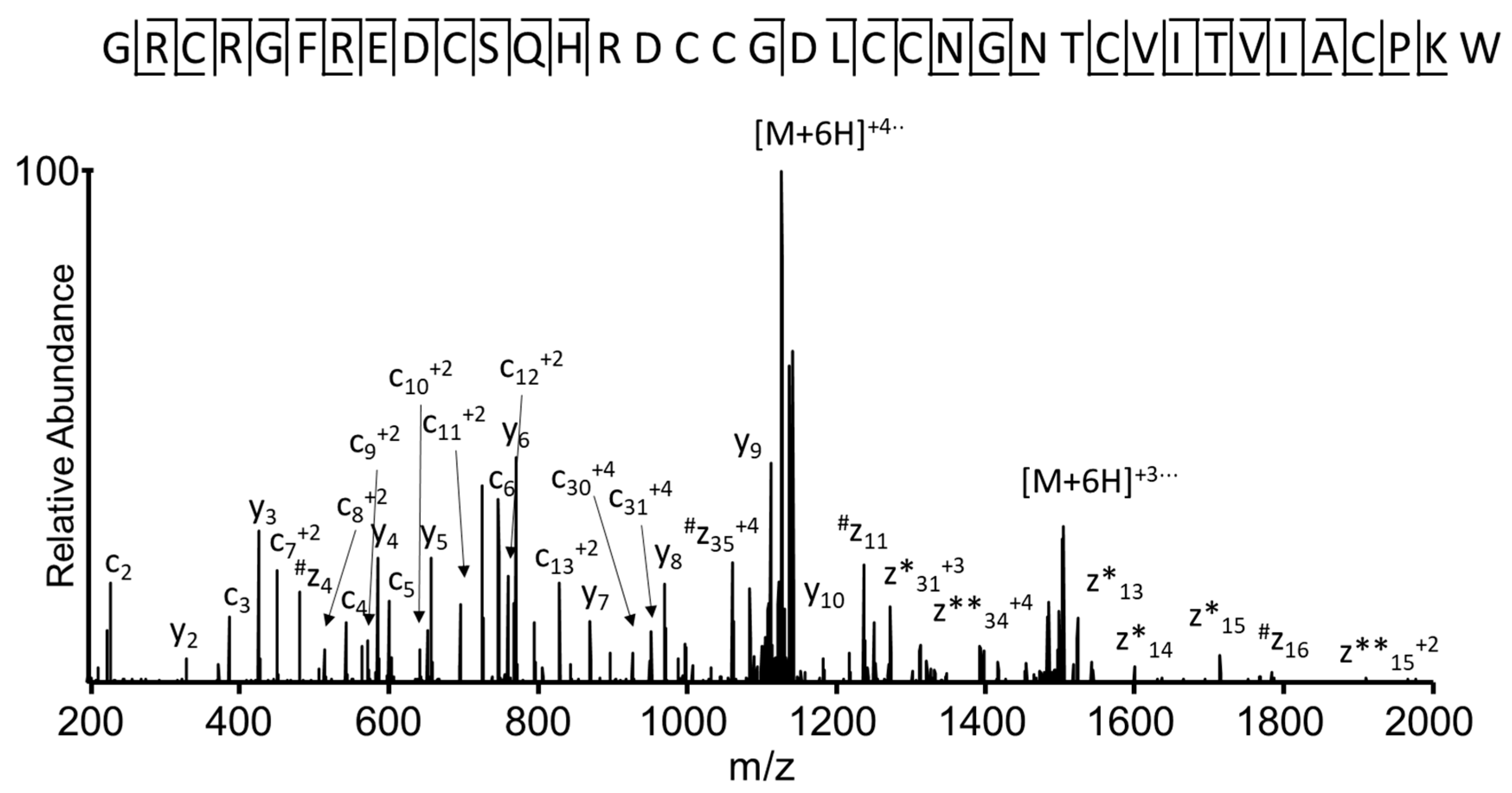
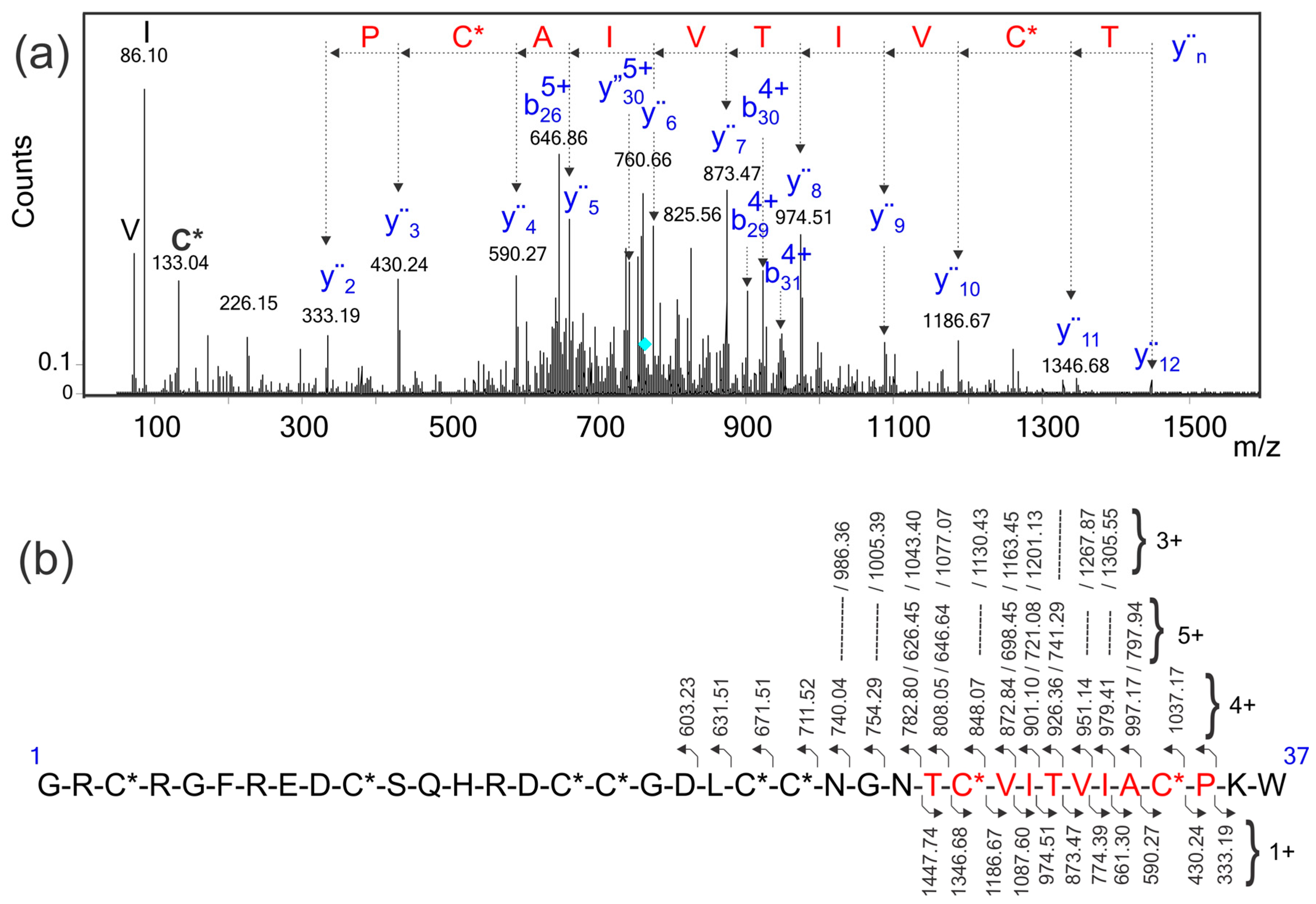
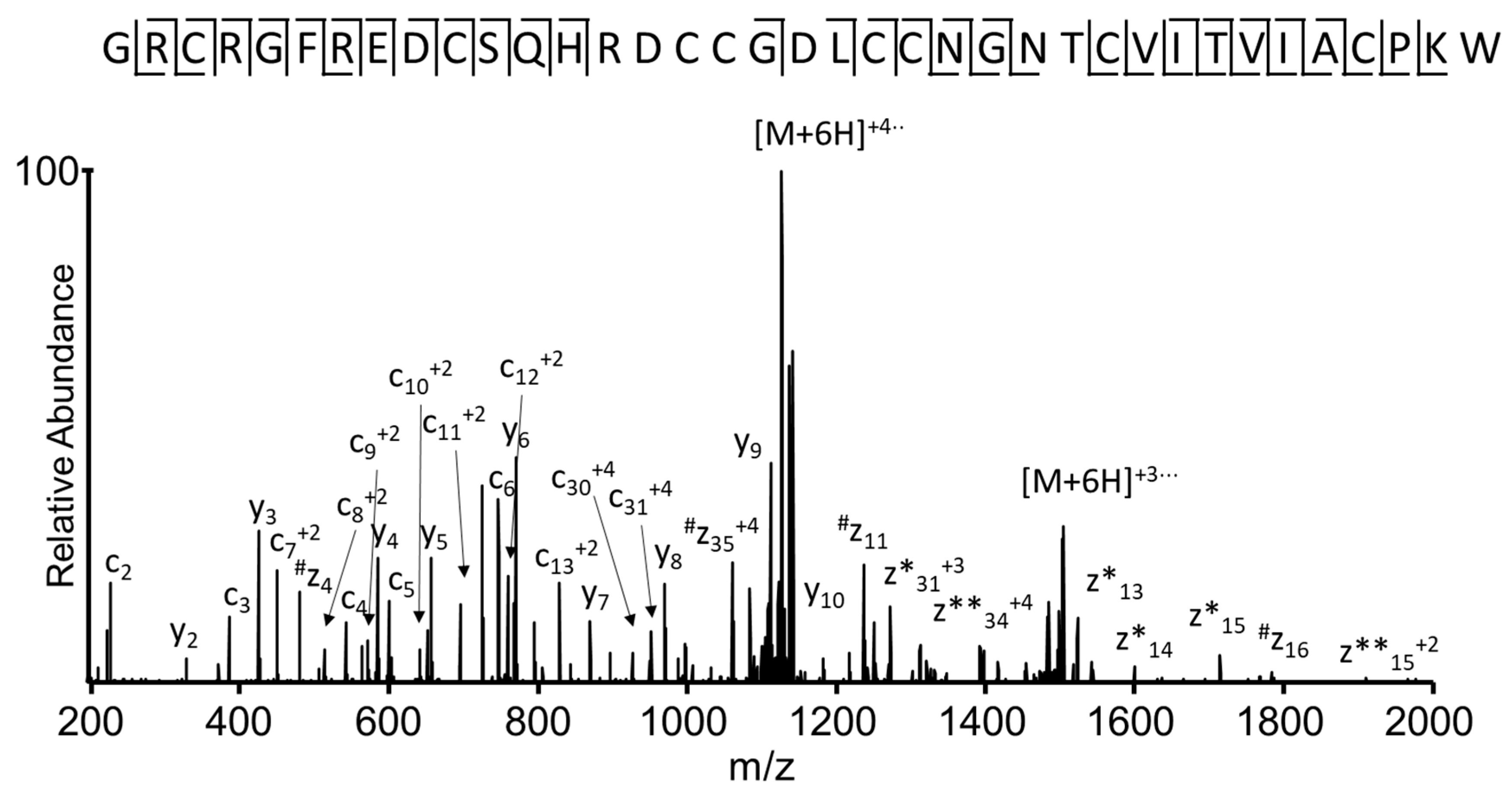
2.5. Identification of the Conotoxin in the Transcriptome Database Using a Sequence Tag Extracted from the MS/MS Spectrum
2.6. Sequence Verification of the I1_xm11a Toxin
2.7. Sequence Analysis of the Identified Conotoxin
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Specimens Collection and Venom Purification
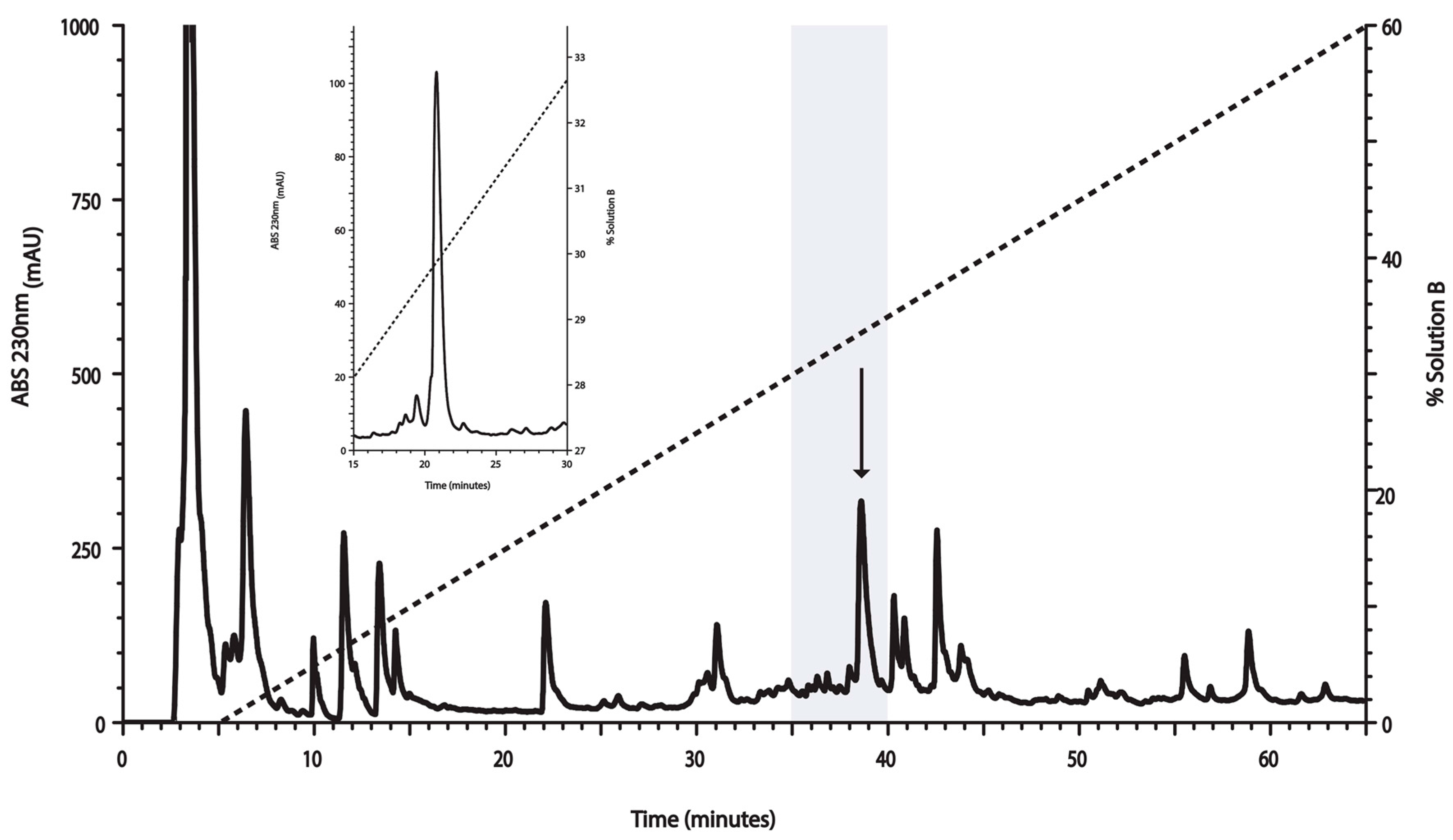
5.2. Peptide Purification
5.3. Antimycobacterial Susceptibility Assay
5.4. Minimal Inhibitory Concentration Assay
5.5. Reduction and S-Alkylation
5.6. LC-MS/MS Analyses
5.7. Transcriptome of the Venom Gland
5.8. Database Search
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2016; Available online: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf (accessed on 21 January 2018).
- Niederweis, M. Nutrient acquisition by mycobacteria. Microbiology 2008, 154, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Seo, C.; Park, Y. Marine Peptides and Their Anti-Infective Activities. Mar. Drugs 2015, 13, 618–654. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus Venom Peptide Pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus Venoms: A Rich Source of Novel Ion Channel-Targeted Peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep Venomics Reveals the Mechanism for Expanded Peptide Diversity in Cone Snail Venom. Mol. Cell. Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Bulaj, G.; Olivera, B.M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 2005, 62, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, J.A.; Kelley, W.P.; Sweedler, J.V. Screening for post-translational modifications in conotoxins using liquid chromatography/mass spectrometry: An important component of conotoxin discovery. Toxicon 2006, 47, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A.; Zubarev, A.R.; Savitski, M.M. Electron Capture/Transfer versus Collisionally Activated/Induced Dissociations: Solo or Duet? J. Am. Soc. Mass Spectrom. 2008, 19, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Mikesh, L.M.; Ueberheide, B.; Chi, A.; Coon, J.J.; Syka, J.E.P.; Shabanowitz, J.; Hunt, D.F. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta Proteins Proteom. 2006, 1764, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, V.H.; Tsaprailis, G.; Smith, L.L.; Breci, L.A. Mobile and localized protons: A framework for understanding peptide dissociation. J. Mass Spectrom. 2000, 35, 1399–1406. [Google Scholar] [CrossRef]
- Kim, M.; Pandey, A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics 2012, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Coon, J.J.; Shabanowitz, J.; Hunt, D.F.; Syka, J.E.P. Electron transfer dissociation of peptide anions. J. Am. Soc. Mass Spectrom. 2005, 16, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Syka, J.E.P.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; van Breukelen, B.; Heck, A.J.R. Facilitating Protein Disulfide Mapping by a Combination of Pepsin Digestion, Electron Transfer Higher Energy Dissociation (EThcD), and a Dedicated Search Algorithm SlinkS. Mol. Cell. Proteom. 2014, 13, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Frese, C.K.; Zhou, H.; Taus, T.; Altelaar, A.F.M.; Mechtler, K.; Heck, A.J.R.; Mohammed, S. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 2013, 12, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; D’Souza, R.C.J.; Cox, J.; Olsen, J.V.; Mann, M. Feasibility of large-scale phosphoproteomics with higher energy collisional dissociation fragmentation. J. Proteome Res. 2010, 9, 6786–6794. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Macek, B.; Lange, O.; Makarov, A.; Horning, S.; Mann, M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Ueberheide, B.M.; Fenyö, D.; Alewood, P.F.; Chait, B.T. Rapid sensitive analysis of cysteine rich peptide venom components. Proc. Natl. Acad. Sci. USA 2009, 106, 6910–6915. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Wilm, M. ErroraTolerant identification of Peptides in Sequence Databases by Peptide Sequence Tags. Anal. Chem. 1994, 66, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Anaya, M.; Camargos, T.S.; Ortiz, E. Scorpion Venom Gland Transcriptomics. In Toxinology; Springer: Dordrecht, The Netherlands, 2013; pp. 1–14. ISBN 978-94-007-6647-1. [Google Scholar]
- King, G. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; Royal Society of Chemistry: London, UK, 2015; ISBN 978-1-84973-663-3. [Google Scholar]
- Perkins, D.N.; Pappin, D.J.C.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Shilov, I.V.; Seymour, S.L.; Patel, A.A.; Loboda, A.; Tang, W.H.; Keating, S.P.; Hunter, C.L.; Nuwaysir, L.M.; Schaeffer, D.A. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteom. 2007, 6, 1638–1655. [Google Scholar] [CrossRef] [PubMed]
- Chrisman, P.A.; Pitteri, S.J.; Hogan, J.M.; McLuckey, S.A. SO2−·electron transfer ion/ion reactions with disulfide linked polypeptide ions. J. Am. Soc. Mass Spectrom. 2005, 16, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Yoshikami, D.; Watkins, M.; Bulaj, G.; Jimenez, E.C.; Olivera, B.M. Characterization of D-amino-acid-containing excitatory conotoxins and redefinition of the I-conotoxin superfamily. FEBS J. 2005, 272, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.C.; Shetty, R.P.; Lirazan, M.; Rivier, J.; Walker, C.; Abogadie, F.C.; Yoshikami, D.; Cruz, L.J.; Olivera, B.M. Novel excitatory Conus peptides define a new conotoxin superfamily. J. Neurochem. 2003, 85, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.S.; Steel, D.; Jacobsen, R.B.; Lirazan, M.B.; Cruz, L.J.; Hooper, D.; Shetty, R.; DelaCruz, R.C.; Nielsen, J.S.; Zhou, L.M.; et al. The T-superfamily of Conotoxins. J. Biol. Chem. 1999, 274, 30664–30671. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of conotoxin gene superfamilies in the venomous snail, conus victoriae. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Jimenez, E.C.; Yoshikami, D.; Imperial, J.S.; Watkins, M.; Morrison, A.; Olivera, B.M. I1-superfamily conotoxins and prediction of single d-amino acid occurrence. Toxicon 2008, 51, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Case, R.J.; Inui, T.; Wang, Y.; Cho, S.; Fischer, N.H.; Franzblau, S.G. New perspectives on natural products in TB drug research. Life Sci. 2005, 78, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Gordien, A.Y.; Gray, A.I.; Ingleby, K.; Franzblau, S.G.; Seidel, V. Activity of Scottish Plant, Lichen and Fungal Endophyte Extracts against Mycobacterium aurum and Mycobacterium tuberculosis. Phyther. Res. 2009, 22. [Google Scholar] [CrossRef]
- Camacho-Corona Mdel, R.; Ramírez-Cabrera, M.A.; Santiago, O.G.; Garza-González, E.; Palacios Ide, P.; Luna-Herrera, J. Activity against drug resistant-tuberculosis strains of plants used in Mexican traditional medicine to treat tuberculosis and other respiratory diseases. Phyther. Res. 2008, 22, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, S.; Zin, N.M.; Wahab, H.A.; Ibrahim, P.; Sulaiman, S.F.; Zahariluddin, A.S.M.; Noor, S.S.M. Antituberculosis potential of some ethnobotanically selected Malaysian plants. J. Ethnopharmacol. 2011, 133, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Barger, A.; Fuhst, C.; Wiedemann, B. Pharmacological indices in antibiotic therapy. J. Antimicrob. Chemother. 2003, 52, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Wei, D.; Babon, J.J.; Yang, X.; Fiedler, B.; Chen, P.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure and Sodium Channel Activity of an Excitatory I 1-Superfamily Conotoxin. Biochemistry 2007, 46, 9929–9940. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, B.; Zhang, M.; Buczek, O.; Azam, L.; Bulaj, G.; Norton, R.S.; Olivera, B.M.; Yoshikami, D. Specificity, affinity and efficacy of iota-conotoxin RXIA, an agonist of voltage-gated sodium channels NaV1.2, 1.6 and 1.7. Biochem. Pharmacol. 2008, 75, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Lugo, P.; Díaz, F.; Re, A.D.; Olivares, F.; González, R.; Dueñas, S.; Licea, A. Thermoregulatory behaviour and thermal tolerance of three species of Conidae in the Eastern Pacific and Gulf of California coasts of Baja California, Mexico. Molluscan Res. 2016, 1–8. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinform. 2012, 1–17. [Google Scholar] [CrossRef]
- Bioinformatics, B. Babraham Institute. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 January 2016).
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bernáldez, J.; Jiménez, S.; González, L.; Ferro, J.; Soto, E.; Salceda, E.; Chávez, D.; Aguilar, M.; Licea-Navarro, A. A New Member of Gamma-Conotoxin Family Isolated from Conus princeps Displays a Novel Molecular Target. Toxins (Basel) 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, G.; Lundberg, U.; Rodríguez-Ulloa, A.; Herrera, M.; Machado, W.; Portela, M.; Palomares, S.; Espinosa, L.A.; Ramos, Y.; Durán, R.; et al. Protein content of the Hylesia metabus egg nest setae (Cramer [1775]) (Lepidoptera: Saturniidae) and its association with the parental investment for the reproductive success and lepidopterism. J. Proteom. 2017, 150, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa-Montiel, A.; Bernáldez, J.; Jiménez, S.; Ueberhide, B.; González, L.J.; Licea-Navarro, A. Antimycobacterial Activity: A New Pharmacological Target for Conotoxins Found in the First Reported Conotoxin from Conasprella ximenes. Toxins 2018, 10, 51. https://doi.org/10.3390/toxins10020051
Figueroa-Montiel A, Bernáldez J, Jiménez S, Ueberhide B, González LJ, Licea-Navarro A. Antimycobacterial Activity: A New Pharmacological Target for Conotoxins Found in the First Reported Conotoxin from Conasprella ximenes. Toxins. 2018; 10(2):51. https://doi.org/10.3390/toxins10020051
Chicago/Turabian StyleFigueroa-Montiel, Andrea, Johanna Bernáldez, Samanta Jiménez, Beatrix Ueberhide, Luis Javier González, and Alexei Licea-Navarro. 2018. "Antimycobacterial Activity: A New Pharmacological Target for Conotoxins Found in the First Reported Conotoxin from Conasprella ximenes" Toxins 10, no. 2: 51. https://doi.org/10.3390/toxins10020051