
Degradation of Aflatoxins by Means of Laccases from Trametes versicolor: An In Silico Insight




Abstract
:
1. Introduction
2. Results
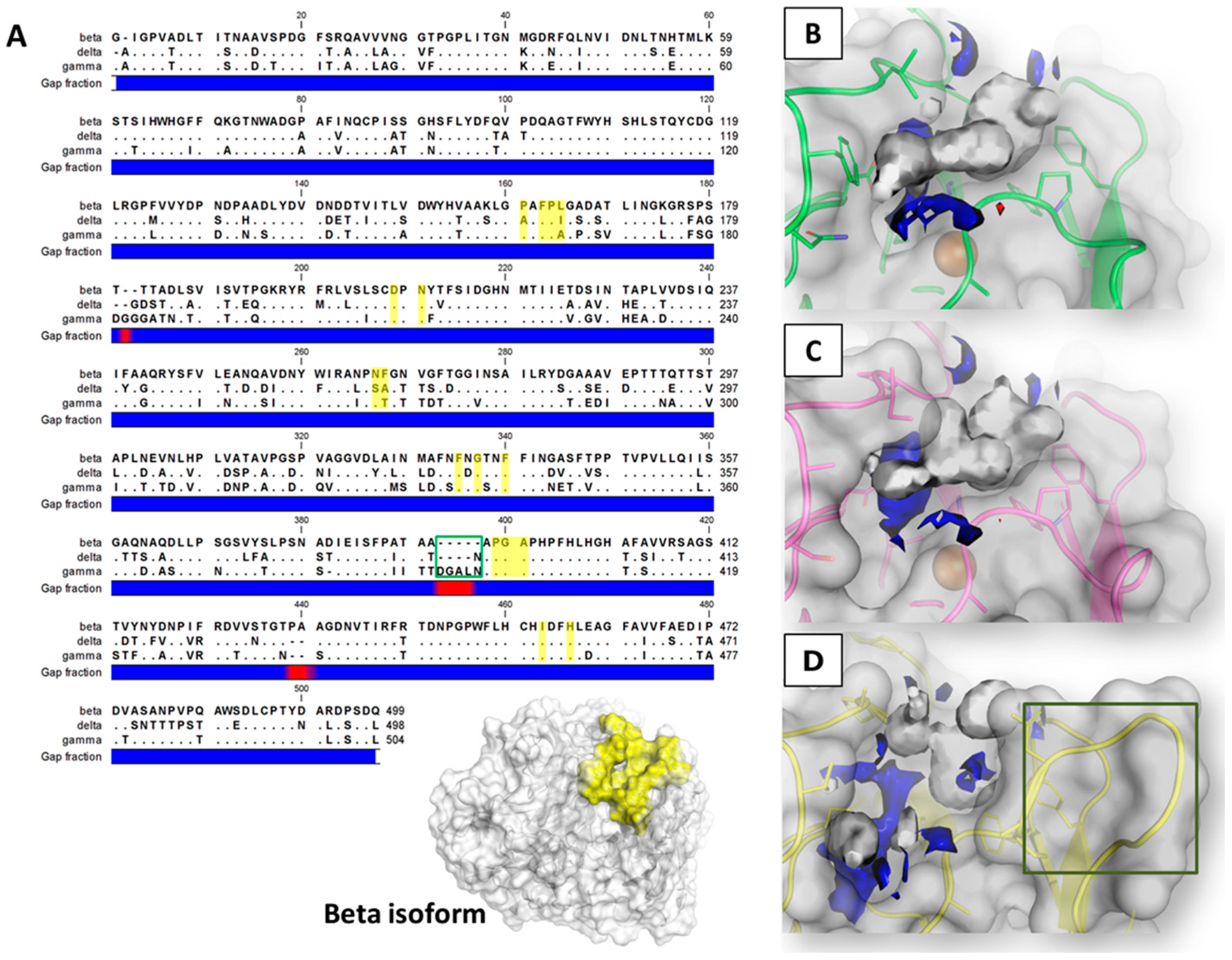
2.1. Sequence Analysis and Pocket Anatomy
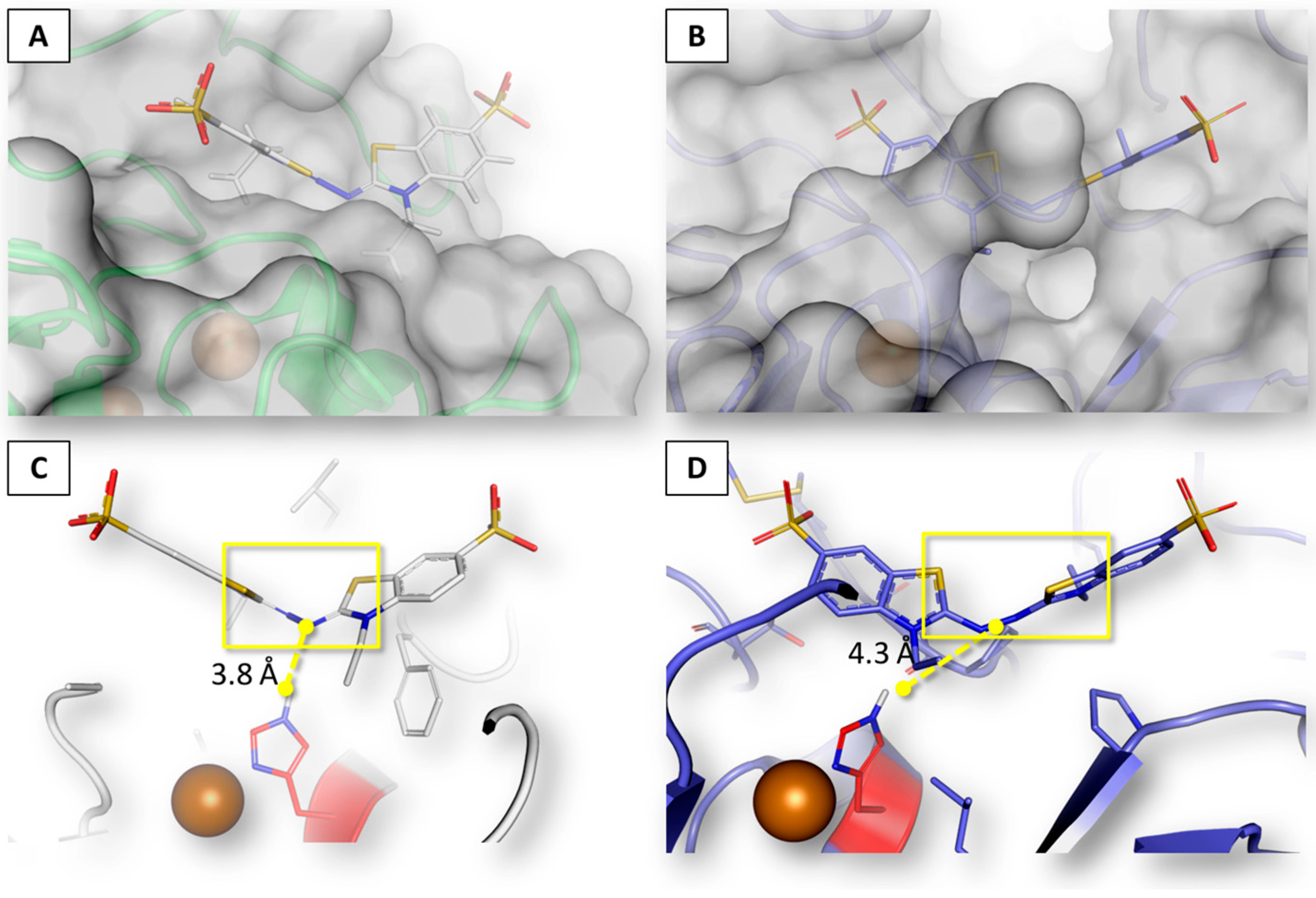
2.2. Assessment of Procedure Reliability
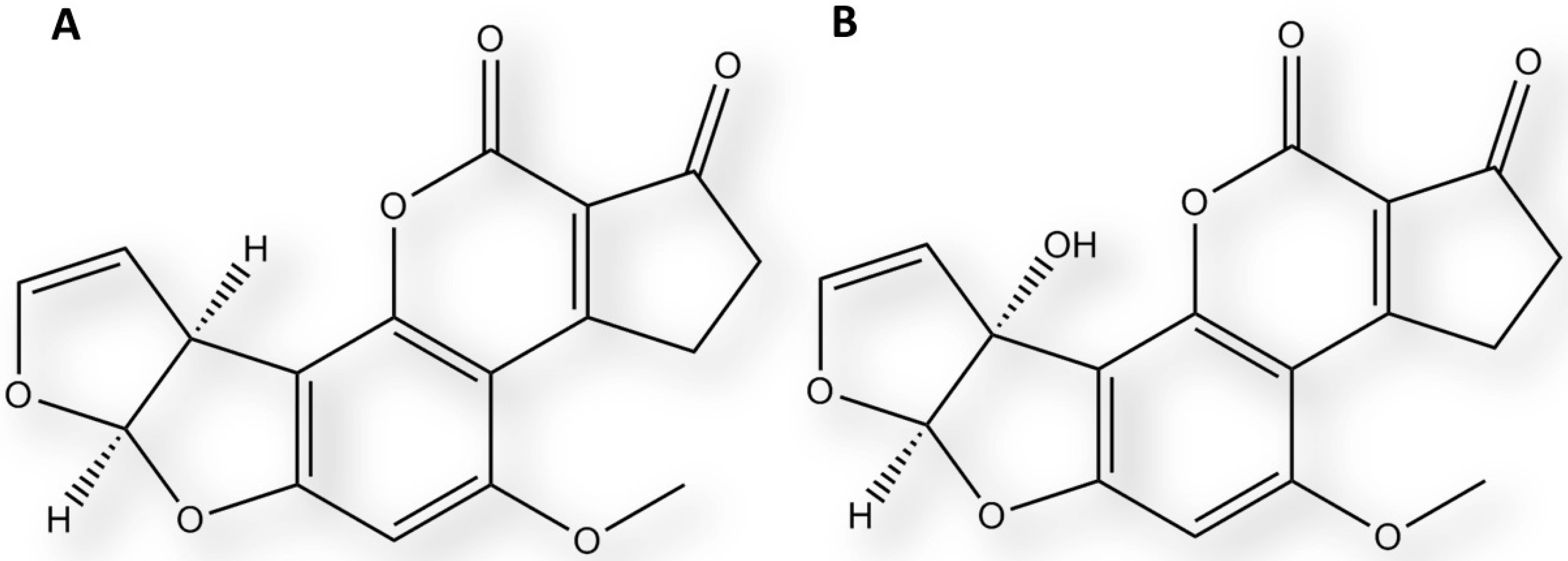
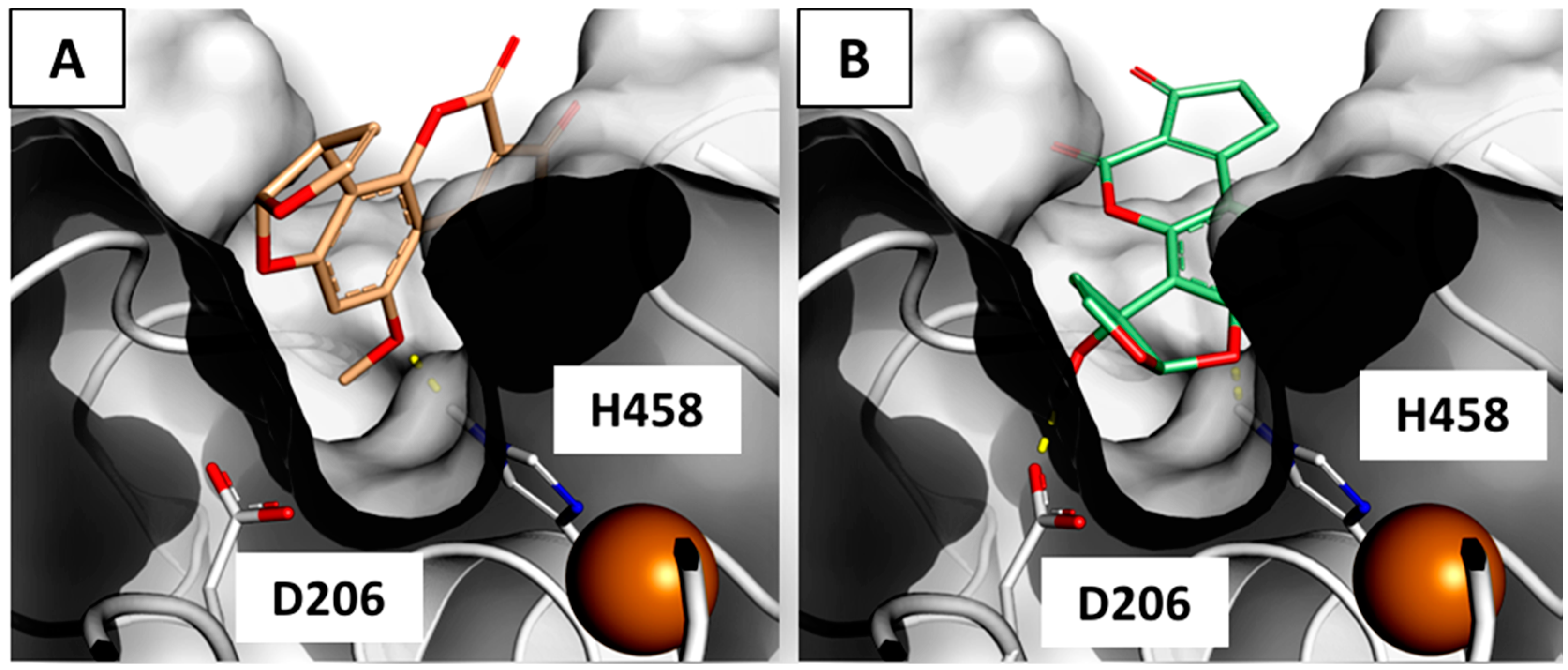
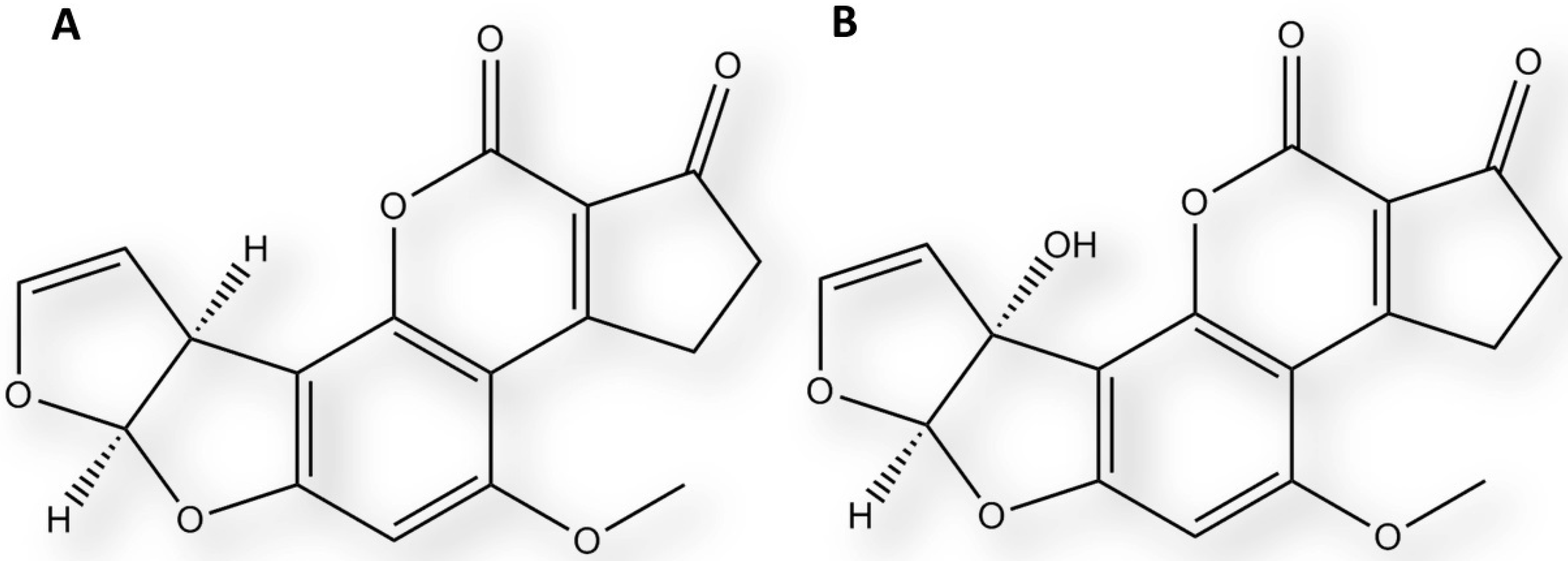
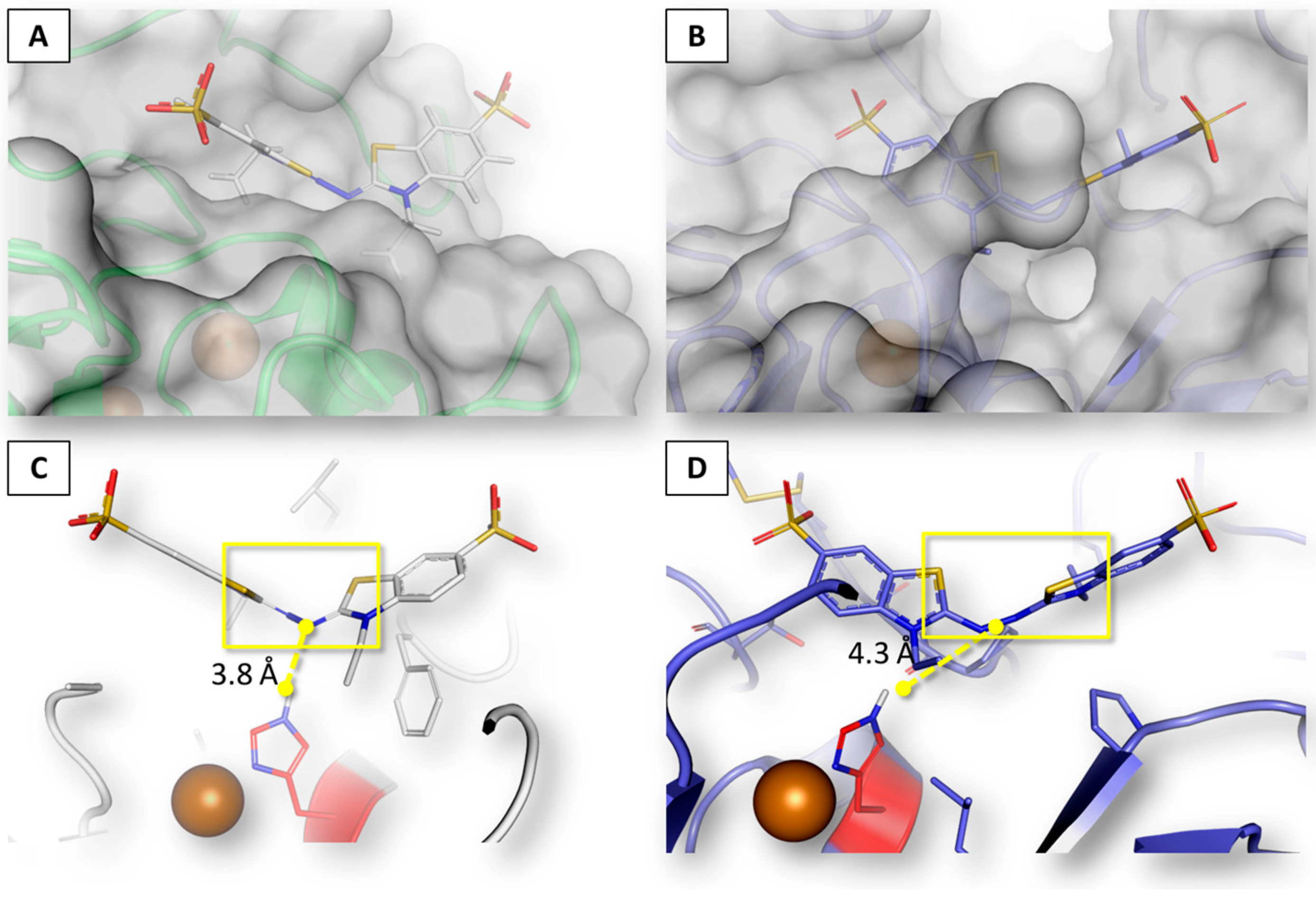
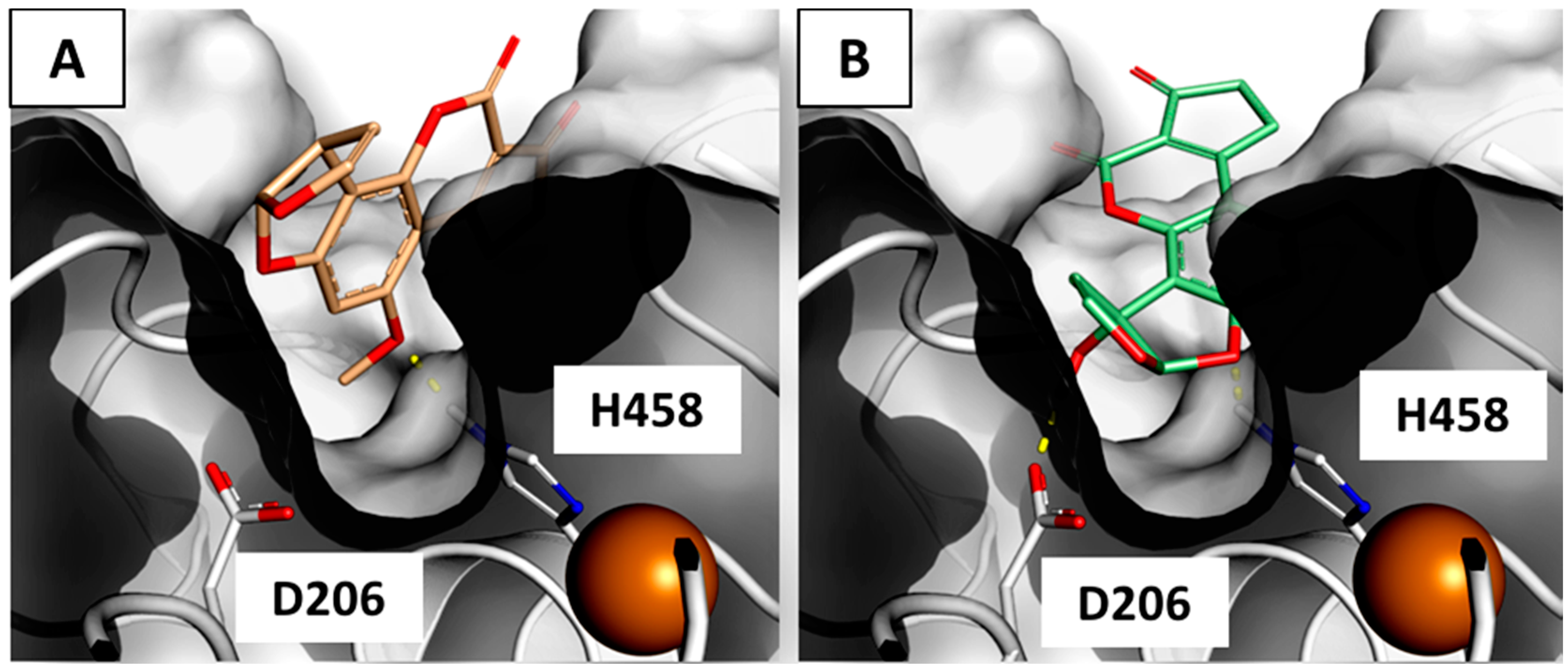
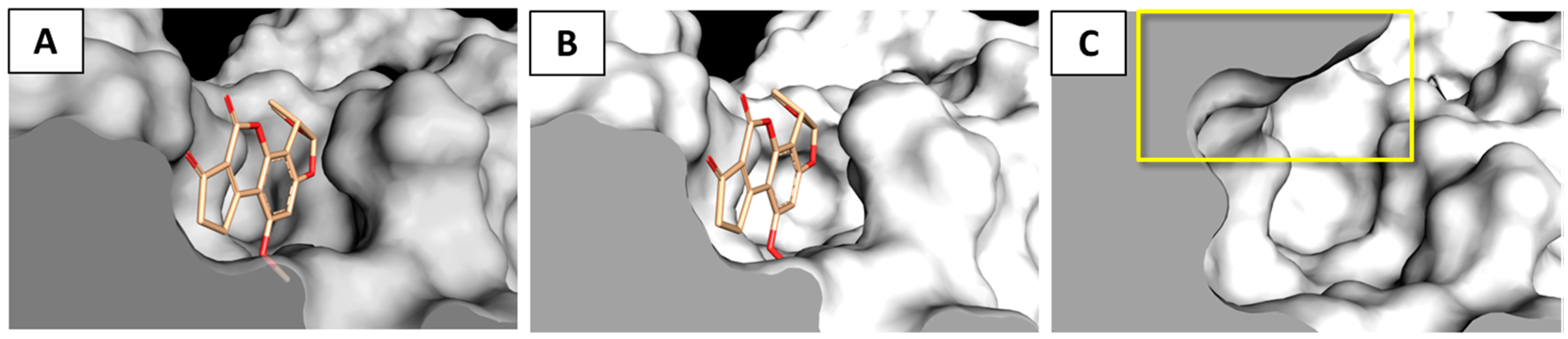
2.3. Interaction of Aflatoxin B1 and M1 within Beta, Delta and Gamma Laccase Isoforms
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Homology Modeling and Sequence Analysis
5.2. Molecular Modeling
5.3. Pharmacophore Models
5.4. Docking Simulations and Re-Scoring Procedures
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellafiora, L.; Perotti, A.; Galaverna, G.; Buschini, A.M.; Dall’Asta, C. On the masked mycotoxin zearalenone-14-glucoside. Does the mask truly hide? Toxicon 2016, 111, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Guclu, H. Aflatoxin regulations in a network of global maize trade. PLoS ONE 2012, 7, e45151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jard, G.; Liboz, T.; Mathieu, F.; Guyonvarc’h, A.; Lebrihi, A. Review of mycotoxin reduction in food and feed: From prevention in the field to detoxification by adsorption or transformation. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1590–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlovsky, P. Detoxification strategies for mycotoxins in plant breeding. In Masked Mycotoxins in Food: Formation, Occurrence and Toxicological Relevance; Dall’Asta, C., Berthiller, F., Eds.; Royal Society of Chemistry: London, UK, 2016. [Google Scholar]
- Dellafiora, L.; Paolella, S.; Dall’Asta, C.; Dossena, A.; Cozzini, P.; Galaverna, G. Hybrid in silico/in vitro approach for the identification of angiotensin I converting enzyme inhibitory peptides from Parma dry-cured ham. J. Agric. Food Chem. 2015, 22, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, C.; Jacobson, M.P. Studying enzyme-substrate specificity in silico: A case study of the Escherichia coli glycolysis pathway. Biochemistry 2010, 49, 4003–4005. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Groopman, J.D.; Pestka, J.J. Public health impacts of foodborne mycotoxins. Annu. Rev. Food Sci. Technol. 2014, 5, 351–372. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). Opinion of the scientific panel on contaminants in the food chain [contam] related to the potential increase of consumer health risk by a possible increase of the existing maximum levels for aflatoxins in almonds, hazelnuts and pistachios and derived products. EFSA J. 2007, 446. [Google Scholar] [CrossRef]
- Raiola, A.; Tenore, G.C.; Manyes, L.; Meca, G.; Ritieni, A. Risk analysis of main mycotoxins occurring in food for children: An overview. Food Chem. Toxicol. 2015, 84, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Prandini, A.; Tansini, G.; Sigolo, S.; Filippi, L.; Laporta, M.; Piva, G. On the occurrence of aflatoxin M1 in milk and dairy products. Food Chem. Toxicol. 2009, 47, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Perrone, G.; Gallo, A.; Logrieco, A.F. Biodiversity of Aspergillus section Flavi in Europe in relation to the management of aflatoxin risk. Front. Microbiol. 2014, 5, 377. [Google Scholar] [CrossRef] [PubMed]
- Mate, D.M.; Alcalde, M. Laccase: A multi-purpose biocatalyst at the forefront of biotechnology. Microb. Biotechnol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Scarpari, M.; Bello, C.; Pietricola, C.; Zaccaria, M.; Bertocchi, L.; Angelucci, A.; Ricciardi, M.R.; Scala, V.; Parroni, A.; Fabbri, A.A.; et al. Aflatoxin control in maize by Trametes versicolor. Toxins (Basel) 2014, 6, 3426–3437. [Google Scholar] [CrossRef] [PubMed]
- Zeinvand-Lorestani, H.; Sabzevari, O.; Setayesh, N.; Amini, M.; Nili-Ahmadabadi, A.; Faramarzi, M.A. Comparative study of in vitro prooxidative properties and genotoxicity induced by aflatoxin B1 and its laccase-mediated detoxification products. Chemosphere 2015, 135, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, T.; Jolivalt, C.; Briozzo, P.; Caminade, E.; Joly, N.; Madzak, C.; Mouqin, C. Crystal structure of a four-copper laccase complexed with an arylamine: Insights into substrate recognition and correlation with kinetics. Biochemistry 2002, 41, 7325–7333. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Mena, P.; Cozzini, P.; Brighenti, F.; Del Rio, D. Modelling the possible bioactivity of ellagitannin-derived metabolites. In silico tools to evaluate their potential xenoestrogenic behavior. Food Funct. 2013, 4, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Mena, P.; Del Rio, D.; Cozzini, P. Modeling the effect of phase II conjugations on topoisomerase I poisoning: Pilot study with luteolin and quercetin. J. Agric. Food Chem. 2014, 62, 5881–5886. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Dall’Asta, C.; Cozzini, P. Ergot alkaloids: From witchcraft till in silico analysis. Multi-receptor analysis of ergotamine metabolites. Toxicol. Rep. 2015, 2, 535–545. [Google Scholar] [CrossRef]
- Madzak, C.; Mimmi, M.C.; Caminade, E.; Brault, A.; Baumberger, S.; Briozzo, P.; Mougin, C.; Jolivalt, C. Shifting the optimal pH of activity for a laccase from the fungus Trametes versicolor by structure-based mutagenesis. Protein Eng. Des. Sel. 2006, 19, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Cozzini, P.; Dellafiora, L. In silico approach to evaluate molecular interaction between mycotoxins and the estrogen receptors ligand binding domain: A case study on zearalenone and its metabolites. Toxicol. Lett. 2012, 214, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Cozzini, P.; Fornabaio, M.; Marabotti, A.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J. Med. Chem. 2002, 45, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Dall’Asta, C.; Cruciani, G.; Galaverna, G.; Cozzini, P. Molecular modelling approach to evaluate poisoning of topoisomerase I by alternariol derivatives. Food Chem. 2015, 189, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Fornabaio, M.; Cozzini, P.; Mozzarelli, A.; Abraham, D.J.; Kellogg, G.E. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 2. Computational titration and pH effects in molecular models of neuraminidase-inhibitor complexes. J. Med. Chem. 2003, 46, 4487–4500. [Google Scholar] [CrossRef] [PubMed]
- Fornabaio, M.; Spirakis, F.; Mozzarelli, A.; Cozzini, P.; Abraham, D.J.; Kellogg, G.E. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 3. The free energy contribution of structural water molecules in HIV-1 protease complexes. J. Med. Chem. 2004, 47, 4507–4516. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.J.; Kepp, K.P. Setting the stage for electron transfer: Molecular basis of ABTS-binding to four laccases from Trametes versicolor at variable pH and protein oxidation state. J. Mol. Catal. B Enzym. 2014, 100, 68–77. [Google Scholar] [CrossRef]
- Galli, C.; Madzak, C.; Vadalà, R.; Jolivalt, C.; Gentili, P. Concerted electron/proton transfer mechanism in the oxidation of phenols by laccase. ChemBioChem 2013, 14, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Redfern, O.; Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 2007, 8, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Enguita, F.J.; Marçal, D.; Martins, L.O.; Grenha, R.; Henriques, A.O.; Lindley, P.F.; Carrondo, M.A. Substrate and dioxygen binding to the endospore coat laccase from Bacillus subtilis. J. Biol. Chem. 2004, 279, 23472–23476. [Google Scholar] [CrossRef] [PubMed]
- Hahn, I.; Thamhesl, M.; Apfelthaler, E.; Klingenbrunner, V.; Hametner, C.; Krska, R.; Schatzmayr, G.; Moll, W.-D.; Berthiller, F.; Schwartz-Zimmermann, H.E. Characterisation and determination of metabolites formed by microbial and enzymatic degradation of ergot alkaloids. World Mycotoxins J. 2015, 8, 393–404. [Google Scholar] [CrossRef]
- Igawa, T.; Takahashi-Ando, N.; Ochiai, N.; Ohsato, S.; Shimizu, T.; Kudo, T.; Yamaguchi, I.; Kimura, M. Reduced contamination by the Fusarium mycotoxin zearalenone in maize kernels through genetic modification with a detoxification gene. Appl. Environ. Microbiol. 2007, 73, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Munkvold, G.P. Cultural and genetic approaches to managing mycotoxins in maize. Annu. Rev. Phytopathol. 2003, 44, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Gerike, U.; Danson, M.J.; Hough, D.W. Cold-active citrate synthase: Mutagenesis of active-site residues. Protein Eng. 2001, 14, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Leferink, N.G.; Antonyuk, S.V.; Houwman, J.A.; Scrutton, N.S.; Eady, R.R.; Hasnain, S.S. Impact of residues remote from the catalytic centre on enzyme catalysis of copper nitrite reductase. Nat. Commun. 2014, 5, 4395. [Google Scholar] [CrossRef] [PubMed]
- Bleve, G.; Lezzi, C.; Spagnolo, S.; Tasco, G.; Tufariello, M.; Casadio, R.; Mita, G.; Rampino, P.; Grieco, F. Role of the C-terminus of Pleurotus eryngii Ery4 laccase in determining enzyme structure, catalytic properties and stability. Protein Eng. Des. Sel. 2013, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Andberg, M.; Hakulinen, N.; Auer, S.; Saloheimo, M.; Koivula, A.; Rouvinen, J.; Kruus, K. Essential role of the C-terminus in Melanocarpus albomyces laccase for enzyme production, catalytic properties and structure. FEBS J. 2009, 276, 6285–6300. [Google Scholar] [CrossRef] [PubMed]
- Reiss, R.; Ihssen, J.; Richter, M.; Eichhorn, E.; Schilling, B.; Thöny-Meyer, L. Laccase versus laccase-like multi-copper oxidase: A comparative study of similar enzymes with diverse substrate spectra. PLoS ONE 2013, 8, e65633. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Fanelli, F.; Zucca, P.; Liuzzi, V.C.; Quintieri, L.; Cimmarusti, M.T.; Monaci, L.; Haidukowski, M.; Logrieco, A.F.; Sanjust, E.; et al. Aflatoxin B1 and M1 degradation by lac2 from Pleurotus pulmonarius and redox mediators. Toxins (Basel) 2016, 8, E245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambria, M.T.; Di Marino, D.; Falconi, M.; Garavaglia, S.; Cambria, A. Docking simulation and competitive experiments validate the interaction between the 2,5-xylidine inhibitor and Rigidoporus lignosus laccase. J. Biomol. Struct. Dyn. 2010, 27, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Suresh, P.S.; Kumar, A.; Kumar, R.; Singh, V.P. An in silico [correction of insilico] approach to bioremediation: Laccase as a case study. J. Mol. Graph. Model. 2008, 26, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favourable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Carosati, E.; Sciabola, S.; Cruciani, G. Hydrogen bonding interactions of covalently bonded fluorine atoms: From crystallographic data to a new angular function in the GRID force field. J. Med. Chem. 2004, 47, 5114–5125. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, E.G.; Abraham, D.J. Hydrophobicity: Is LogPo/w more than the sum of its parts? Eur. J. Med. Chem. 2000, 37, 651–661. [Google Scholar] [CrossRef]
- Dellafiora, L.; Galaverna, G.; Dall’Asta, C.; Cozzini, P. Hazard identification of cis/trans-zearalenone through the looking-glass. Food Chem. Toxicol. 2015, 86, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, V.A.; Dellafiora, L.; Mollergues, J.; Dall’Asta, C.; Serrant, P.; Marin-Kuan, M.; Lo Piparo, E.; Schilter, B.; Cozzini, P. Hazard assessment through hybrid in vitro/in silico approach: The case of zearalenone. ALTEX 2015, 32, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Marabotti, A.; Spyrakis, F.; Facchiano, A.; Cozzini, P.; Alberti, S.; Kellogg, G.E.; Mozzarelli, A. Energy-based prediction of amino acid-nucleotide base recognition. J. Comput. Chem. 2008, 29, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, G.E.; Burnett, J.C.; Abraham, D.J. Very empirical treatment of solvation and entropy: A force field derived from LogPo/w. J. Comput. Aided Mol. Des. 2001, 15, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, G.E.; Fornabaio, M.; Spyrakis, F.; Lodola, A.; Cozzini, P.; Mozzarelli, A.; Abraham, D.J. Getting it right: Modeling of pH, solvent and “Nearly” Everything else in virtual screening of biological targets. J. Mol. Graph. Model. 2004, 22, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Kellogg, G.E. Hydrophobicity-shake flasks, protein folding and drug discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laccase Isoform | Experimental Affinity Rank 1 | HINT Score |
|---|---|---|
| Beta | 1 | 430 |
| Gamma | 2 | 206 |
| Delta | 3 | 104 |
| Laccase Isoform | ABTS | 2,6-dimethoxyphenol | ||
|---|---|---|---|---|
| Experimental Affinity Rank 1 | HINT Score | Experimental Affinity Rank 1 | HINT Score | |
| Beta wild type | 2 | 430 | 1 | 500 |
| Beta D206A | 1 | 495 | 2 | 203 |
| Laccase Isoform | HINT Scores | |
|---|---|---|
| AFB1 | AFM1 | |
| Beta | 248 | 373 |
| Gamma | −199 | 372 |
| Delta | 291 | 339 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dellafiora, L.; Galaverna, G.; Reverberi, M.; Dall’Asta, C. Degradation of Aflatoxins by Means of Laccases from Trametes versicolor: An In Silico Insight. Toxins 2017, 9, 17. https://doi.org/10.3390/toxins9010017
Dellafiora L, Galaverna G, Reverberi M, Dall’Asta C. Degradation of Aflatoxins by Means of Laccases from Trametes versicolor: An In Silico Insight. Toxins. 2017; 9(1):17. https://doi.org/10.3390/toxins9010017
Chicago/Turabian StyleDellafiora, Luca, Gianni Galaverna, Massimo Reverberi, and Chiara Dall’Asta. 2017. "Degradation of Aflatoxins by Means of Laccases from Trametes versicolor: An In Silico Insight" Toxins 9, no. 1: 17. https://doi.org/10.3390/toxins9010017
APA StyleDellafiora, L., Galaverna, G., Reverberi, M., & Dall’Asta, C. (2017). Degradation of Aflatoxins by Means of Laccases from Trametes versicolor: An In Silico Insight. Toxins, 9(1), 17. https://doi.org/10.3390/toxins9010017