
Toxin Fused with SUMO Tag: A New Expression Vector Strategy to Obtain Recombinant Venom Toxins with Easy Tag Removal inside the Bacteria

 ,
,
Abstract
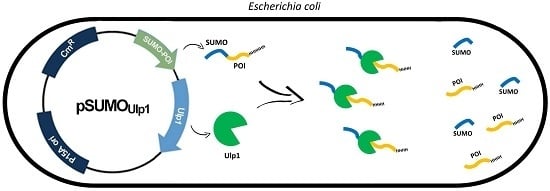
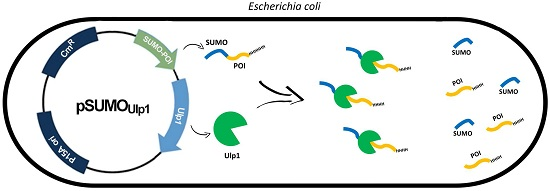
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
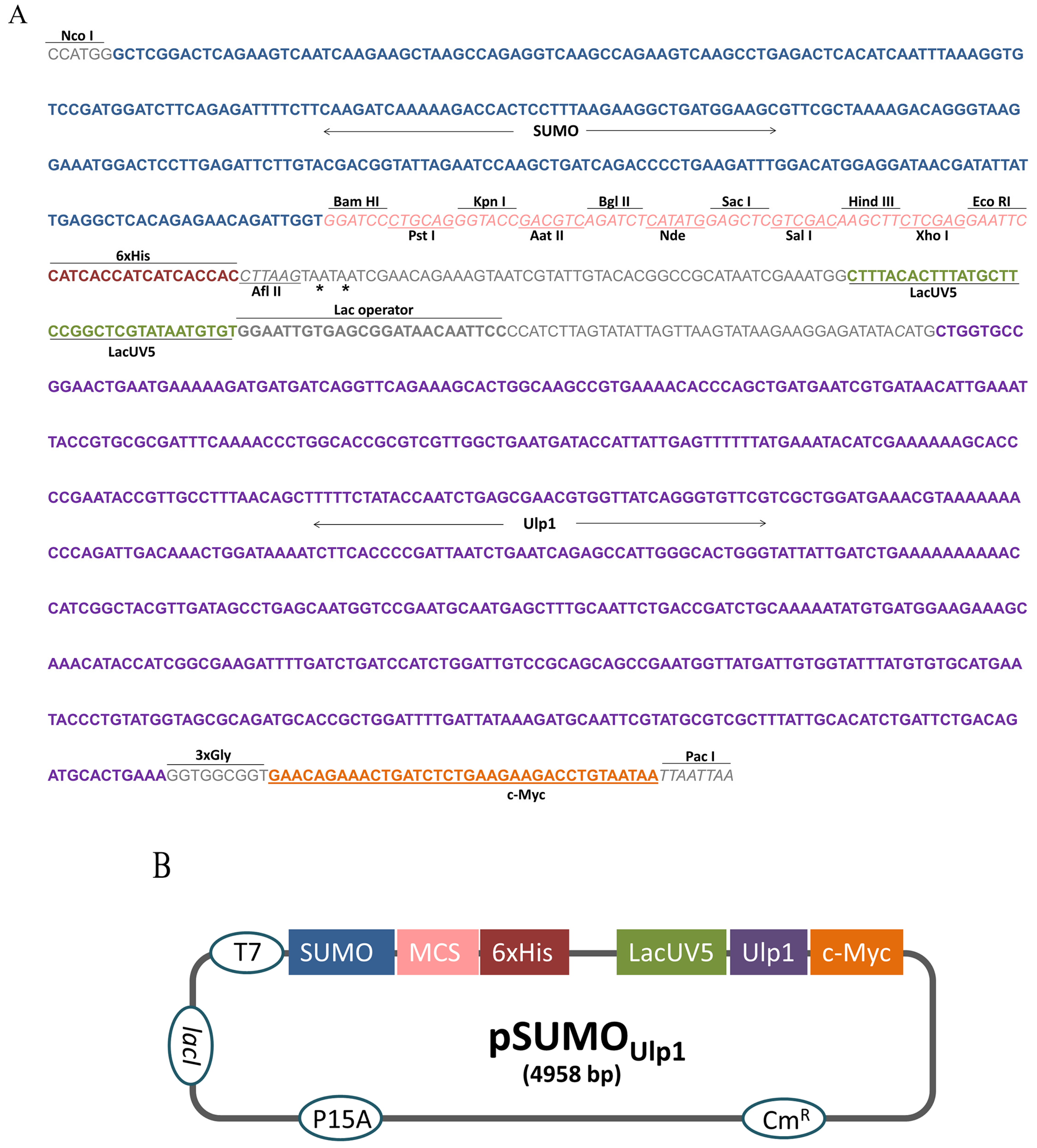
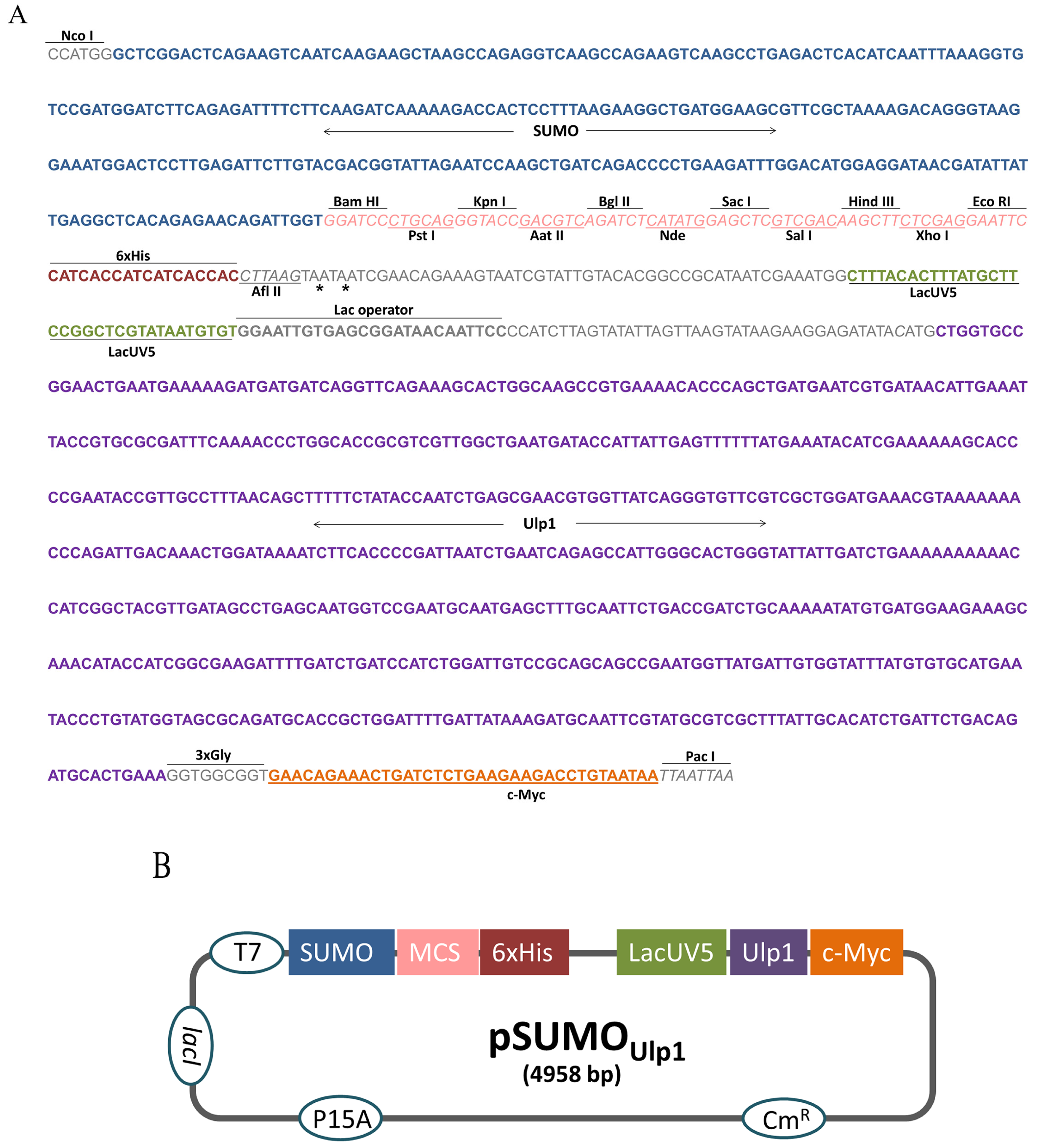
2.1. Construction of the pSUMOUlp1 Vector
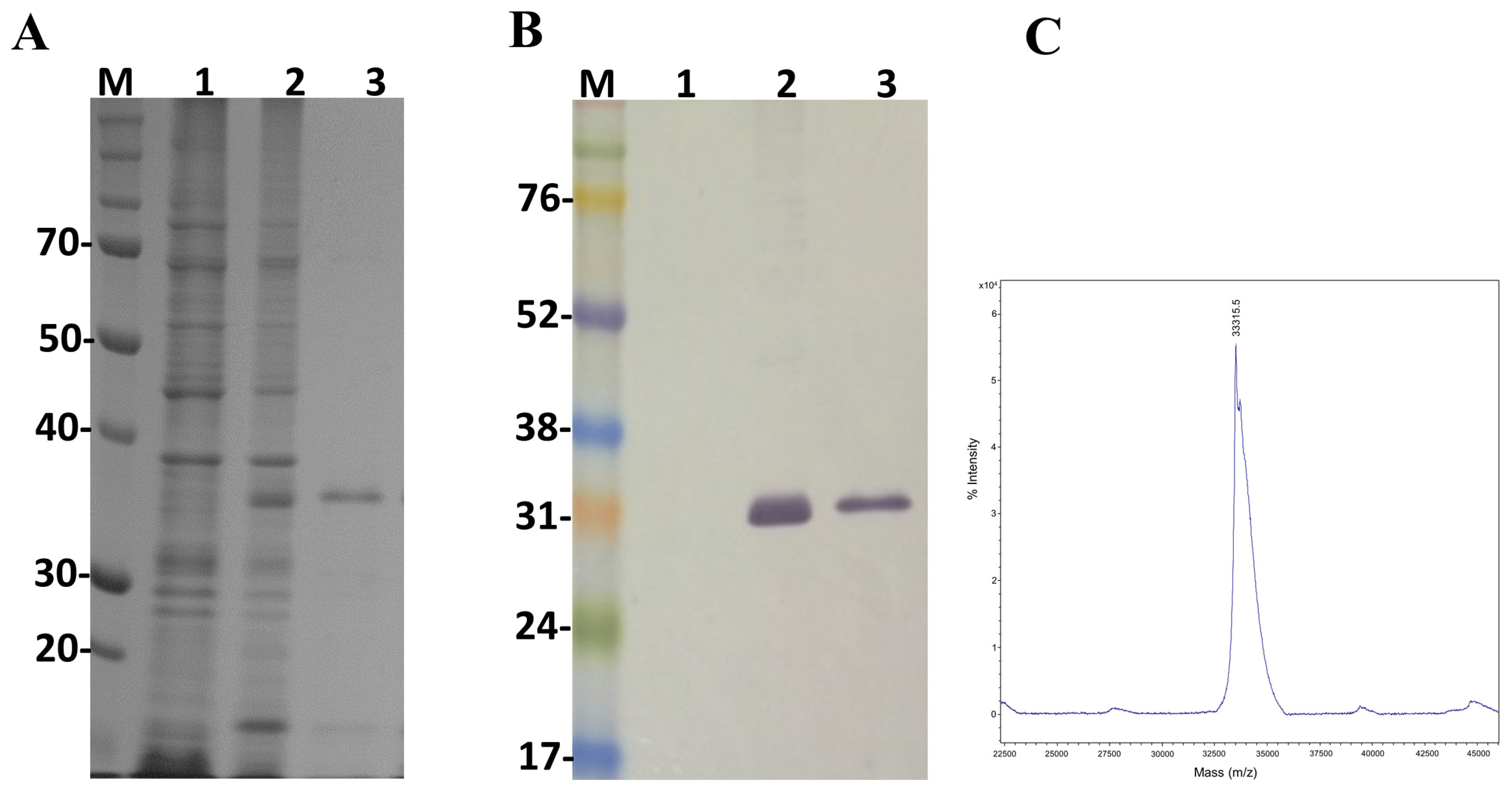
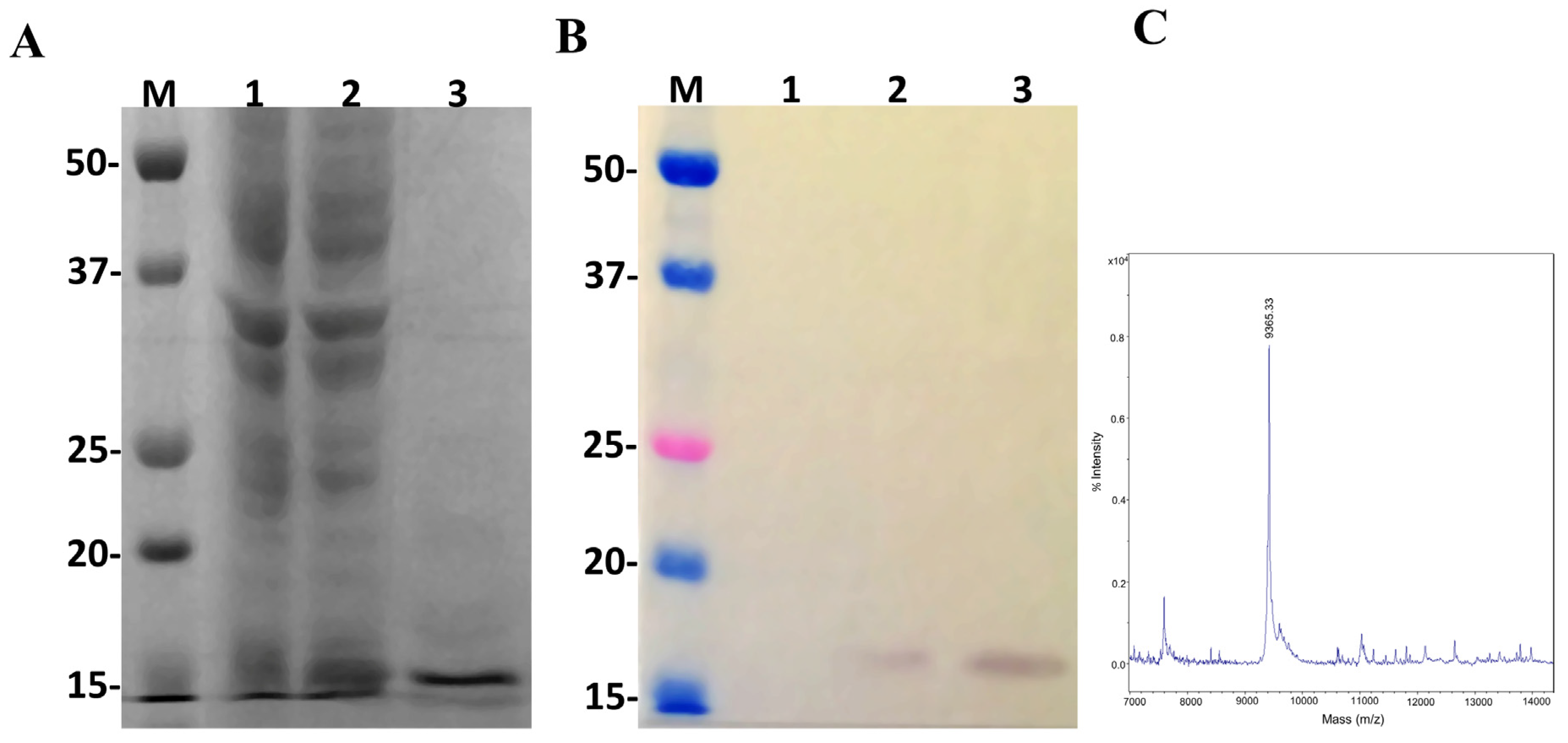
2.2. Evaluation of Ulp1 and SUMO Expression
2.3. Cloning of LgRec2 from L. gaucho Venom Gland and Alignment Analysis
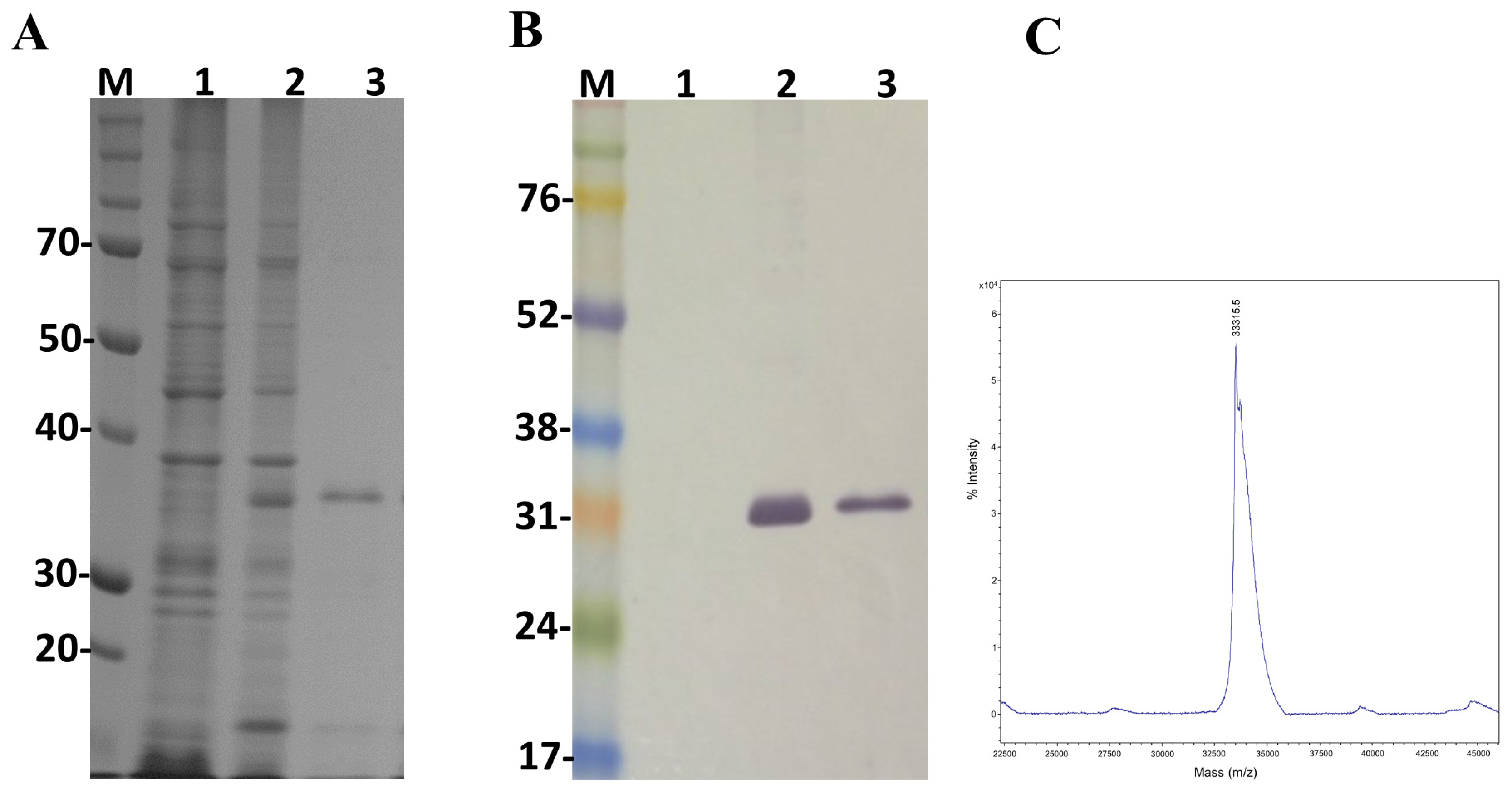
2.4. Expression of LgRec2 Using pSUMOUlp1 Vector
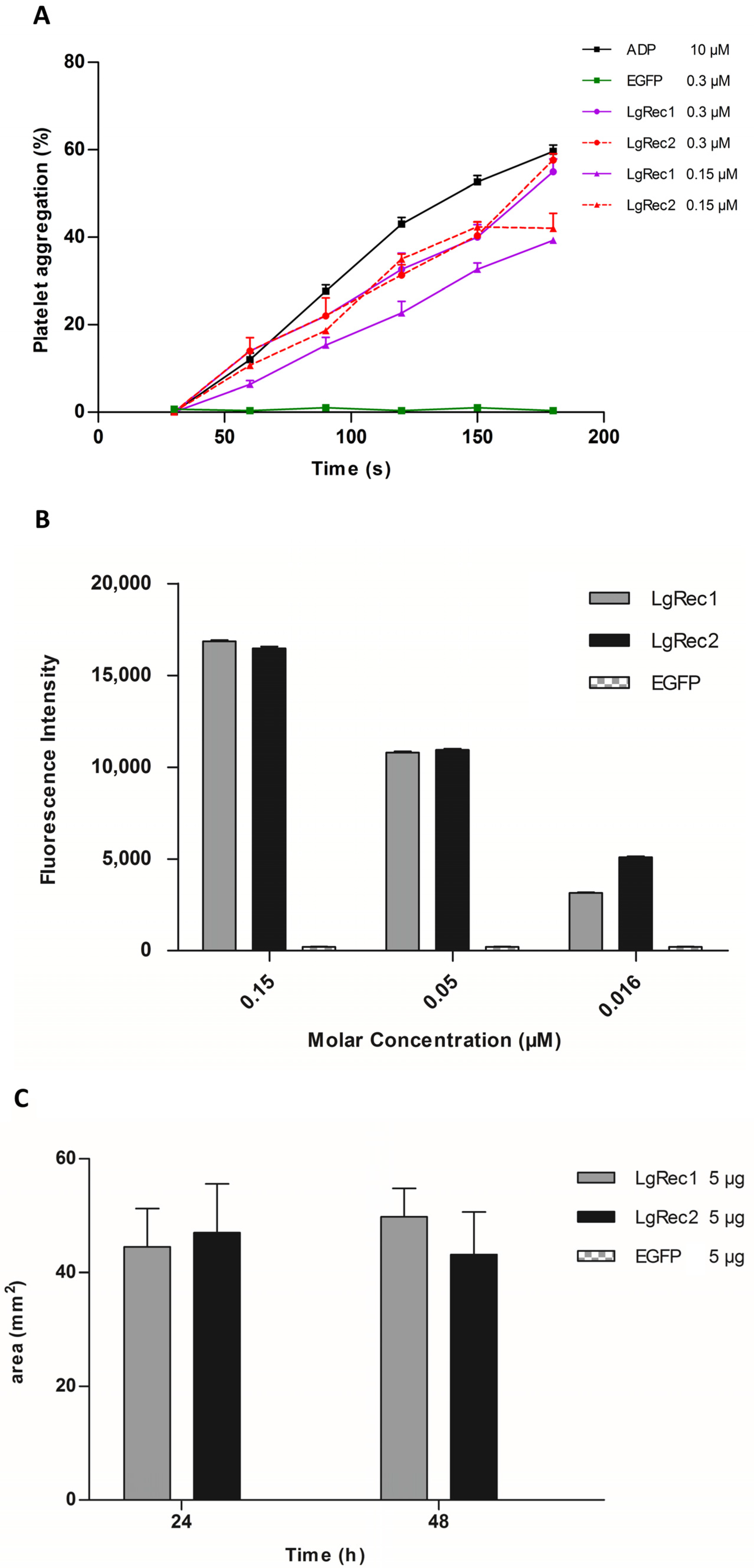
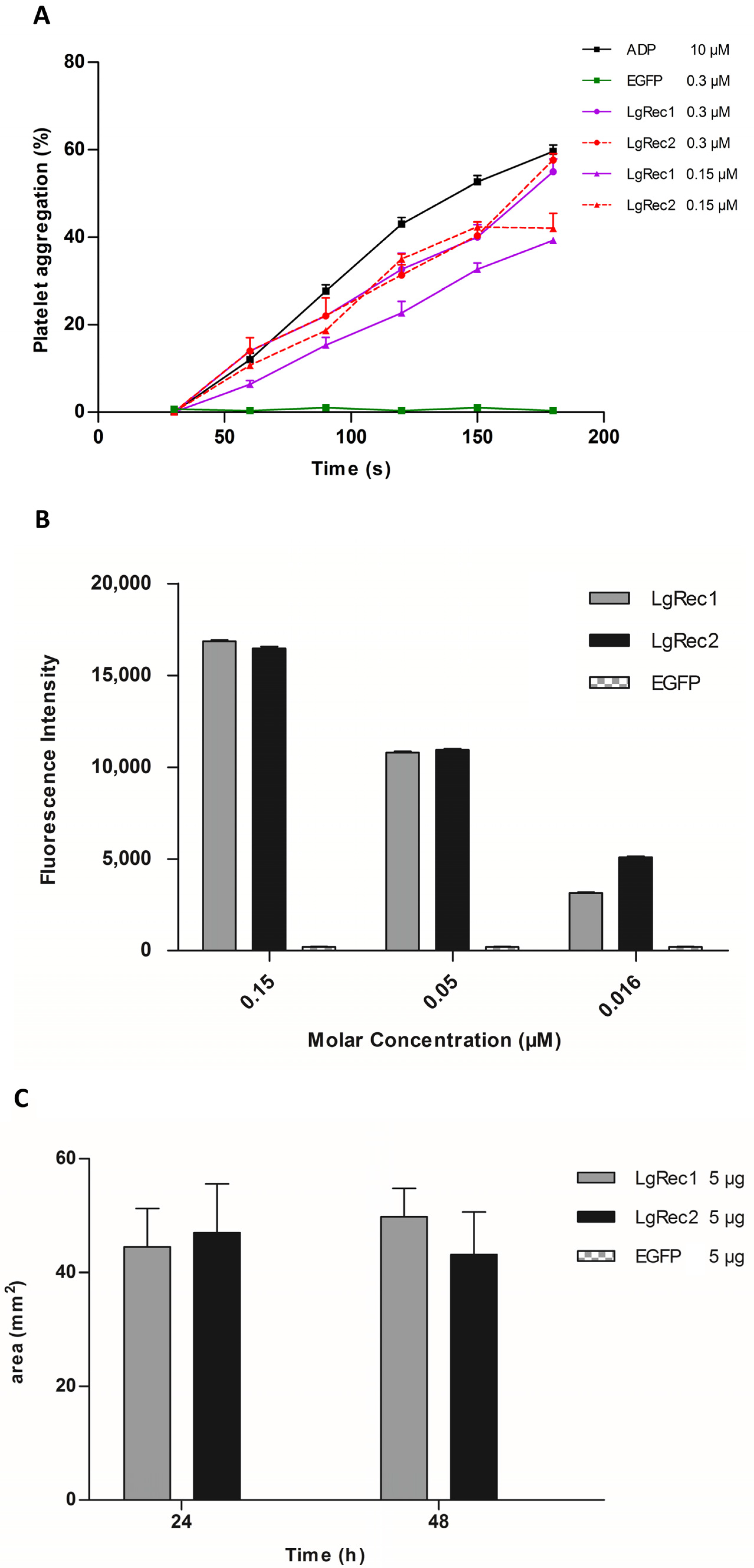
2.5. Analysis of Biological Activities of LgRec2
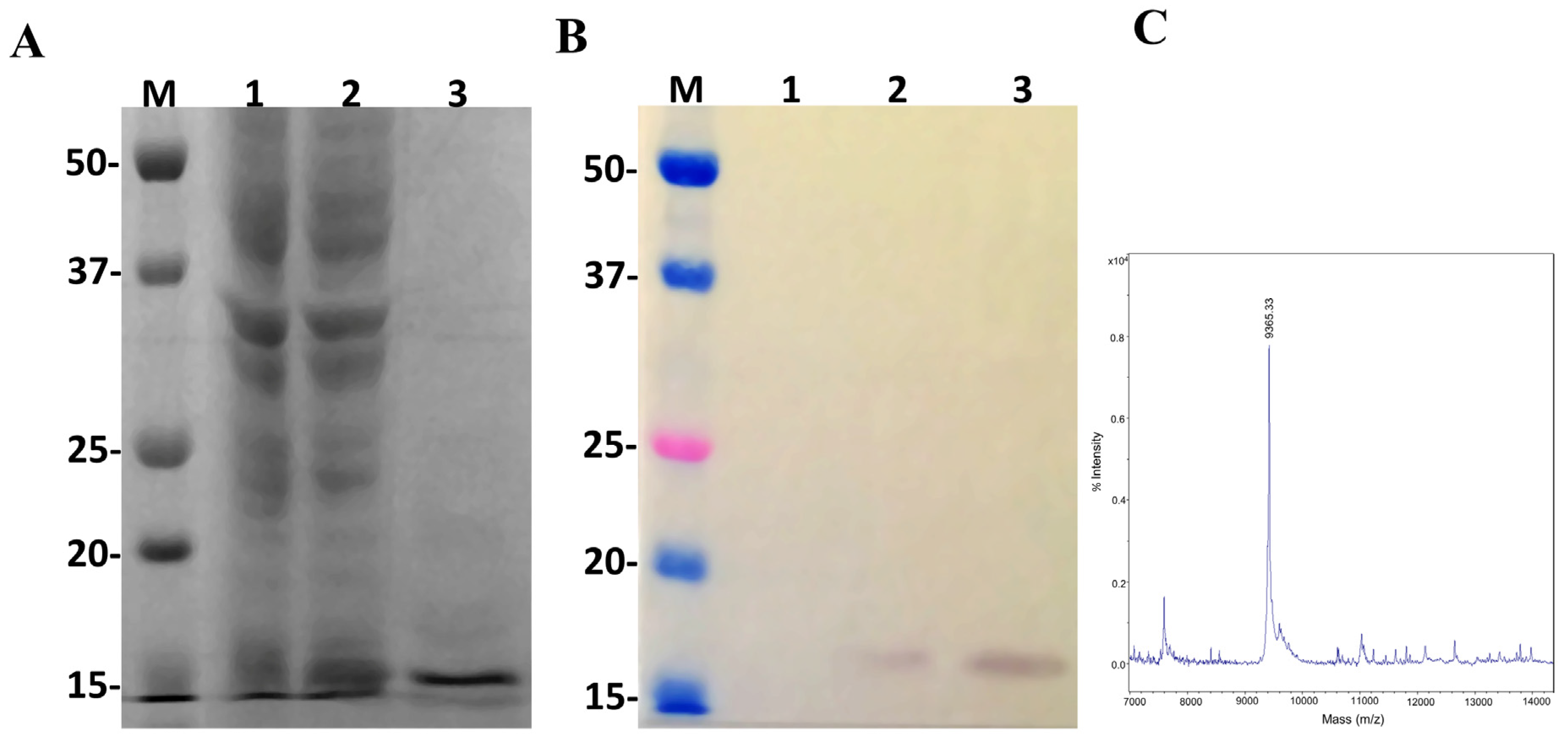
2.6. Expression of Insularin Using the pSUMOUlp1 Vector
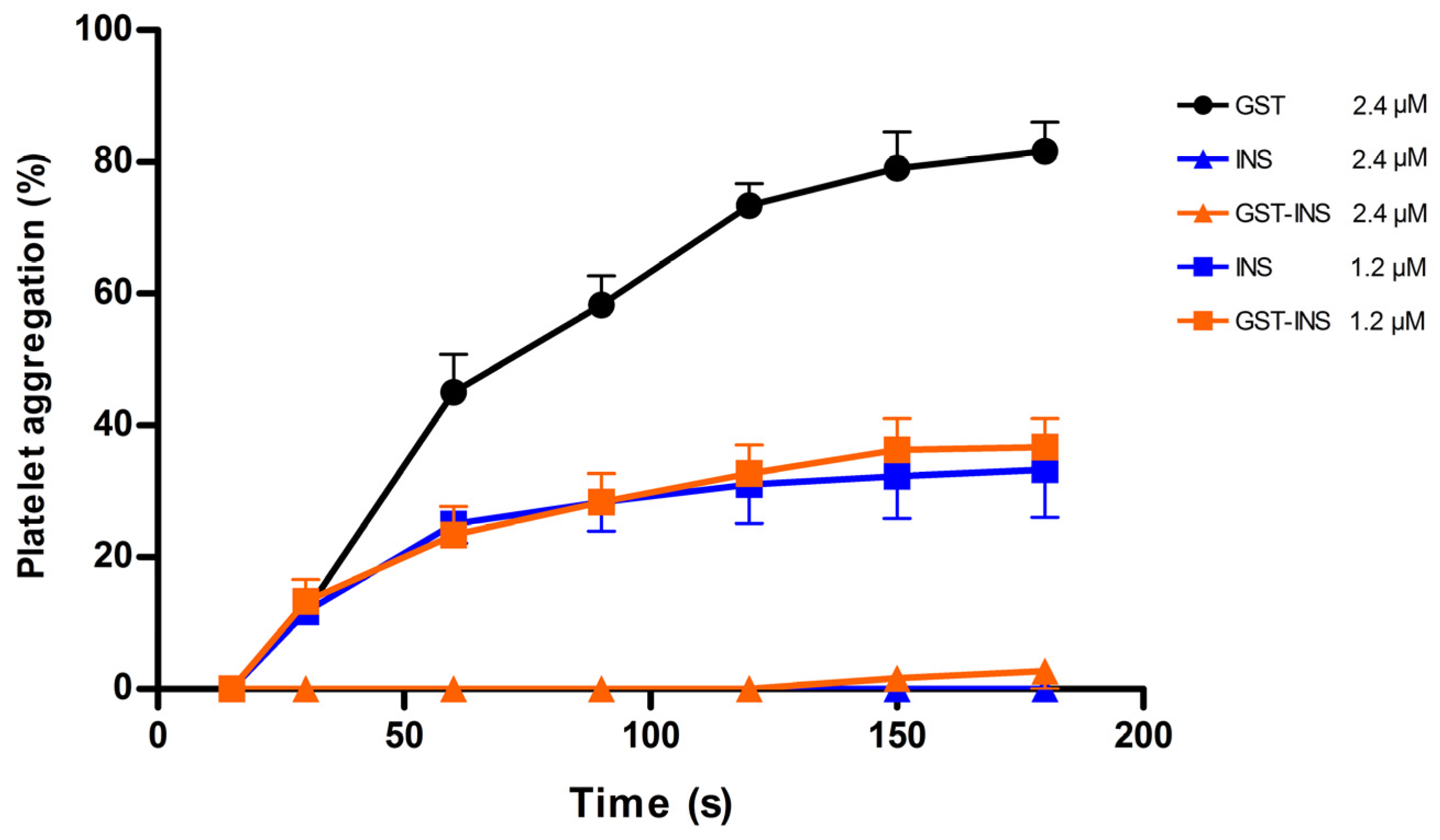
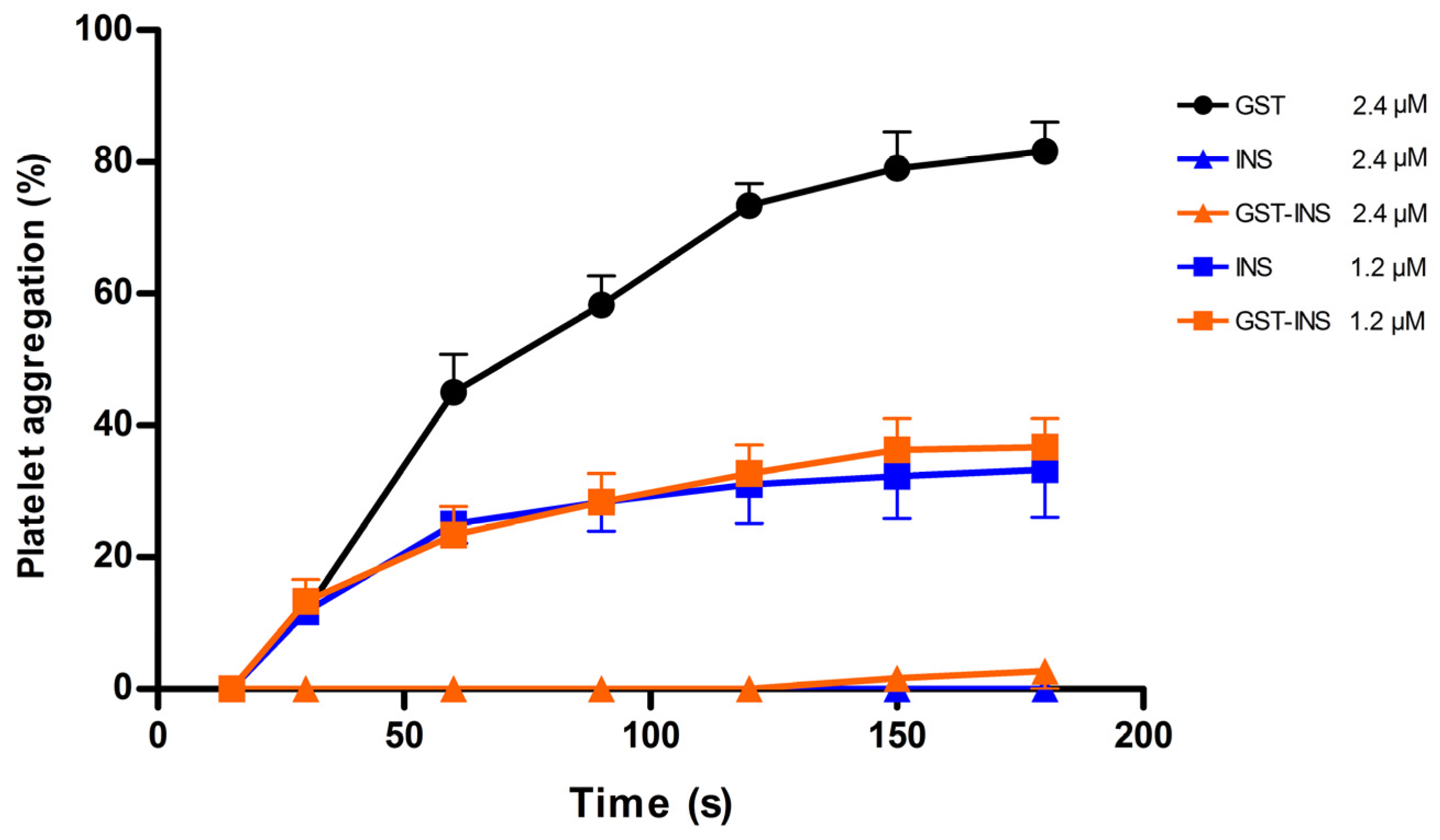
2.7. Analysis of Platelet Inhibition Activity of Insularin
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Construction of the pSUMOUlp1 Vector
5.2. Cloning of LgRec2 and INS into pSUMOUlp1 Vector
5.3. Protein Expression and Purification
5.4. Determination of Molecular Mass by Mass Spectrometry
5.5. SDS-Polyacrylamide Gel Electrophoresis and Western Blot Analysis
5.6. Sphingomyelinase Activity
5.7. Platelet Aggregation Assay
5.8. Dermonecrotic Activity
5.9. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chaisakul, J.; Hodgson, W.C.; Kuruppu, S.; Prasongsook, N. Effects of animal venoms and toxins on hallmarks of cancer. J. Cancer 2016, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- De Marco, V.; Stier, G.; Blandin, S.; de Marco, A. The solubility and stability of recombinant proteins are increased by their fusion to nusa. Biochem. Biophys. Res. Commun. 2004, 322, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Senff-Ribeiro, A.; da Silva, P.H.; Chaim, O.M.; Gremski, L.H.; Paludo, K.S.; da Silveira, R.B.; Gremski, W.; Mangili, O.C.; Veiga, S.S. Biotechnological applications of brown spider (loxosceles genus) venom toxins. Biotechnol. Adv. 2008, 26, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Menez, A. Functional architectures of animal toxins: A clue to drug design? Toxicon 1998, 36, 1557–1572. [Google Scholar] [CrossRef]
- Chaves-Moreira, D.; de Moraes, F.R.; Caruso, Í.P.; Chaim, O.M.; Senff-Ribeiro, A.; Ullah, A.; Da Silva, L.S.; Chahine, J.; Arni, R.K.; Veiga, S.S. Potential implications for designing drugs against the brown spider venom phospholipase-D. J. Cell. Biochem. 2016, 118, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Gopal, G.J.; Kumar, A. Strategies for the production of recombinant protein in Escherichia coli. Protein J. 2013, 32, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.P.; Andrade, A.B.; Ferreira, A.J.; Pires, A.C.G.; Damasceno, D.D.; Alves, M.N.M.; Gomes, E.R.M.; Kushmerick, C.; Lima, R.F.; Prado, M.A.M.; et al. Antiarrhythmogenic effects of a neurotoxin from the spider phoneutria nigriventer. Toxicon 2011, 57, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Shlyapnikov, Y.M.; Andreev, Y.A.; Kozlov, S.A.; Vassilevski, A.A.; Grishin, E.V. Bacterial production of latarcin 2a, a potent antimicrobial peptide from spider venom. Protein Expr. Purif. 2008, 60, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Sermadiras, I.; Revell, J.; Linley, J.E.; Sandercock, A.; Ravn, P. Recombinant expression and in vitro characterisation of active Huwentoxin-IV. PLoS ONE 2013, 8, e83202. [Google Scholar] [CrossRef] [PubMed]
- Bende, N.S.; Dziemborowicz, S.; Herzig, V.; Ramanujam, V.; Brown, G.W.; Bosmans, F.; Nicholson, G.M.; King, G.F.; Mobli, M. The insecticidal spider toxin SFI1 is a knottin peptide that blocks the pore of insect voltage-gated sodium channels via a large beta-hairpin loop. FEBS J. 2015, 282, 904–920. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Moreira, D.; Souza, F.N.; Fogaca, R.T.H.; Mangili, O.C.; Gremski, W.; Senff-Ribeiro, A.; Chaim, O.M.; Veiga, S.S. The relationship between calcium and the metabolism of plasma membrane phospholipids in hemolysis induced by brown spider venom phospholipase-D toxin. J. Cell. Biochem. 2011, 112, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.F.; Hsu, C.C.; Kuo, Y.J. Anti-thrombotic agents derived from snake venom proteins. Thromb. J. 2016, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Lyukmanova, E.N.; Shulepko, M.A.; Shenkarev, Z.O.; Kasheverov, I.E.; Chugunov, A.O.; Kulbatskii, D.S.; Myshkin, M.Y.; Utkin, Y.N.; Efremov, R.G.; Tsetlin, V.I.; et al. Central loop of non-conventional toxin WTX from Naja kaouthia is important for interaction with nicotinic acetylcholine receptors. Toxicon 2016, 119, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.S.; Ho, P.H.; Chang, H.H. Soluble P-selectin rescues viper venom-induced mortality through anti-inflammatory properties and PSGL-1 pathway-mediated correction of hemostasis. Sci. Rep. 2016, 6, 35868. [Google Scholar] [CrossRef] [PubMed]
- Yamane, E.S.; Bizerra, F.C.; Oliveira, E.B.; Moreira, J.T.; Rajabi, M.; Nunes, G.L.C.; de Souza, A.O.; da Silva, I.; Yamane, T.; Karpel, R.L.; et al. Unraveling the antifungal activity of a south american rattlesnake toxin crotamine. Biochimie 2013, 95, 231–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducancel, F.; Boulain, J.C.; Tremeau, O.; Menez, A. Direct expression in Escherichia coli of a functionally active protein A-snake toxin fusion protein. Protein Engineering 1989, 3, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Carrio, M.M.; Villaverde, A. Protein aggregation as bacterial inclusion bodies is reversible. FEBS Lett. 2001, 489, 29–33. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, P.F.; Meng, E.; Li, W.Y.; Zhou, L.; Zhu, L.Y.; Wu, L.; Li, M.J.; Liang, S.P.; Zhang, D.Y. An efficient strategy for heterologous expression and purification of active peptide Hainantoxin-IV. PLoS ONE 2015, 10, e0117099. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Xu, J.Q.; Yang, Q. Soluble expression, purification, and characterization of gloydius shedaoensis venom gloshedobin in Escherichia coli by using fusion partners. Appl. Microbiol. Biotechnol. 2010, 85, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.L.; Duan, H.Q.; Liu, C.J.; Liu, X.L.; Liu, T.T.; Tao, H.X.; Zhang, Z.S. The role of thioredoxin and disulfide isomerase in the expression of the snake venom thrombin-like enzyme calobin in Escherichia coli BL21 (DE3). Protein Expr. Purif. 2004, 38, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.T.; Jeong, B.; Yu, J.; Koo, B.K.; Jo, S.H.; Robinson, R.C.; Choe, H. Soluble prokaryotic expression and purification of crotamine using an n-terminal maltose-binding protein tag. Toxicon 2014, 92, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Cuebas, L.M.; White, M.M. Expression of a biologically-active conotoxin PrIIIE in Escherichia coli. Protein Expr. Purif. 2012, 82, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Marblestone, J.G.; Edavettal, S.C.; Lim, Y.; Lim, P.; Zuo, X.; Butt, T.R. Comparison of sumo fusion technology with traditional gene fusion systems: Enhanced expression and solubility with sumo. Protein Sci. 2006, 15, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Bird, L.E. High throughput construction and small scale expression screening of multi-tag vectors in Escherichia coli. Methods 2011, 55, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Malakhov, M.; Mattern, M.; Malakhova, O.; Drinker, M.; Weeks, S.; Butt, T. Sumo fusions and sumo-specific protease for efficient expression and purification of proteins. J. Struct. Funct. Genom. 2004, 5, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Gremski, L.H.; Trevisan-Silva, D.; Ferrer, V.P.; Matsubara, F.H.; Meissner, G.O.; Wille, A.C.M.; Vuitika, L.; Dias-Lopes, C.; Ullah, A.; de Moraes, F.R.; et al. Recent advances in the understanding of brown spider venoms: From the biology of spiders to the molecular mechanisms of toxins. Toxicon 2014, 83, 91–120. [Google Scholar] [CrossRef] [PubMed]
- Kalapothakis, E.; Araujo, S.C.; de Castro, C.S.; Mendes, T.M.; Gomez, M.V.; Mangili, O.C.; Gubert, I.C.; Chavez-Olortegui, C. Molecular cloning, expression and immunological properties of LiD1, a protein from the dermonecrotic family of, Loxosceles intermedia spider venom. Toxicon 2002, 40, 1691–1699. [Google Scholar] [CrossRef]
- Chaim, O.M.; Sade, Y.B.; da Silveira, R.B.; Toma, L.; Kalapothakis, E.; Chavez-Olortegui, C.; Mangili, O.C.; Gremski, W.; von Dietrich, C.P.; Nader, H.B.; et al. Brown spider dermonecrotic toxin directly induces nephrotoxicity. Toxicol. Appl. Pharmacol. 2006, 211, 64–77. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, R.B.; Pigozzo, R.B.; Chaim, O.M.; Appel, M.H.; Dreyfuss, J.L.; Toma, L.; Mangili, O.C.; Gremski, W.; Dietrich, C.P.; Nader, H.B.; et al. Molecular cloning and functional characterization of two isoforms of dermonecrotic toxin from Loxosceles intermedia (brown spider) venom gland. Biochimie 2006, 88, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, R.B.; Pigozzo, R.B.; Chaim, O.M.; Appel, M.H.; Silva, D.T.; Dreyfuss, J.L.; Toma, L.; Dietrich, C.P.; Nader, H.B.; Veiga, S.S.; et al. Two novel dermonecrotic toxins LiRecDT4 and LiRecDT 5 from brown spider (Loxosceles intermedia) venom: From cloning to functional characterization. Biochimie 2007, 89, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Appel, M.H.; da Silveira, R.B.; Chaim, O.M.; Paludo, K.S.; Silva, D.T.; Chaves, D.M.; da Silva, P.H.; Mangili, O.C.; Senff-Ribeiro, A.; Gremski, W.; et al. Identification, cloning and functional characterization of a novel dermonecrotic toxin (phospholipase D) from brown spider (Loxosceles intermedia) venom. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Vuitika, L.; Gremski, L.H.; Belisario-Ferrari, M.R.; Chaves-Moreira, D.; Ferrer, V.P.; Senff-Ribeiro, A.; Chaim, O.M.; Veiga, S.S. Brown spider phospholipase-D containing a conservative mutation (D233E) in the catalytic site: Identification and functional characterization. J. Cell. Biochem. 2013, 114, 2479–2492. [Google Scholar] [CrossRef] [PubMed]
- Catalan, A.; Cortes, W.; Sagua, H.; Gonzalez, J.; Araya, J.E. Two new phospholipase D isoforms of Loxosceles laeta: Cloning, heterologous expression, functional characterization, and potential biotechnological application. J. Biochem. Mol. Toxicol. 2011, 25, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.D.; de Azevedo, I.D.; Goncalves-de-Andrade, R.M.; van den Berg, C.W.; Ramos, C.R.R.; Ho, P.L.; Tambourgi, D.V. Molecular cloning and expression of a functional dermonecrotic and haemolytic factor from Loxosceles laeta venom. Biochem. Biophys. Res. Commun. 2002, 298, 638–645. [Google Scholar] [CrossRef]
- Tambourgi, D.V.; Pedrosa, M.D.F.; van den Berg, C.W.; Goncalves-de-Andrade, R.M.; Ferracini, M.; Paixao-Cavalcante, D.; Morgan, B.P.; Rushmere, N.K. Molecular cloning, expression, function and immunoreactivities of members of a gene family of sphingomyelinases from Loxosceles venom glands. Mol. Immunol. 2004, 41, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.I.D.; Fernandes-Pedrosa, M.D.; Junqueira-de-Azevedo, I.D.M.; Goncalves-de-Andrade, R.M.; Portaro, F.C.V.; Manzoni-de-Almeida, D.; Murakami, M.T.; Arni, R.K.; van den Berg, C.W.; Ho, P.L.; et al. Smase II, a new sphingomyelinase D from Loxosceles laeta venom gland: Molecular cloning, expression, function and structural analysis. Toxicon 2009, 53, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Della-Casa, M.S.; Junqueira-de-Azevedo, I.; Butera, D.; Clissa, P.B.; Lopes, D.S.; Serrano, S.M.T.; Pimenta, D.C.; Magalhães, G.S.; Ho, P.L.; Moura-da-Silva, A.M. “Insularin, a disintegrin from Bothrops insularis venom: Inhibition of platelet aggregation and endothelial cell adhesion by the native and recombinant gst-insularin proteins”. Toxicon 2011, 57, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Kuo, H.L.; Hsu, M.T.; Tseng, Y.J.; Lin, S.W.; Kuo, S.C.; Peng, H.C.; Lien, J.C.; Huang, T.F. A novel thromboxane receptor antagonist, nstpbp5185, inhibits platelet aggregation and thrombus formation in animal models. Thromb. Haemost. 2016, 116, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Macedo, J.K.A.; Fox, J.W.; Castro, M.D. Disintegrins from snake venoms and their applications in cancer research and therapy. Curr. Protein Pept. Sci. 2015, 16, 532–548. [Google Scholar] [CrossRef]
- Carey, C.M.; Bueno, R.; Gutierrez, D.A.; Petro, C.; Lucena, S.E.; Sanchez, E.E.; Soto, J.G. Recombinant rubistatin (r-Rub), an MVD disintegrin, inhibits cell migration and proliferation, and is a strong apoptotic inducer of the human melanoma cell line SK-Mel-28. Toxicon 2012, 59, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Suntravat, M.; Barret, H.S.; Jurica, C.A.; Lucena, S.E.; Perez, J.C.; Sánchez, E.E. Recombinant disintegrin (r-Cam-dis) from Crotalus adamanteus inhibits adhesion of human pancreatic cancer cell lines to laminin-1 and vitronectin. J. Venom Res. 2015, 6, 1–10. [Google Scholar] [PubMed]
- Magalhães, G.S.; Caporrino, M.C.; Della-Casa, M.S.; Kimura, L.F.; Prezotto-Neto, J.P.; Fukuda, D.A.; Portes, J.A.; Neves-Ferreira, A.G.C.; Santoro, M.L.; Barbaro, K.C. Cloning, expression and characterization of a phospholipase D from Loxosceles gaucho venom gland. Biochimie 2013, 95, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.T.; Fernandes-Pedrosa, M.F.; Tambourgi, D.V.; Arni, R.K. Structural basis for metal ion coordination and the catalytic mechanism of sphingomyelinases D. J. Biol. Chem. 2005, 280, 13658–13664. [Google Scholar] [CrossRef] [PubMed]
- de Giuseppe, P.O.; Ullah, A.; Silva, D.T.; Gremski, L.H.; Wille, A.C.M.; Moreira, D.C.; Ribeiro, A.S.; Chaim, O.M.; Murakami, M.T.; Veiga, S.S.; et al. Structure of a novel class II phospholipase D: Catalytic cleft is modified by a disulphide bridge. Biochem. Biophys. Res. Commun. 2011, 409, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Tavares, F.L.; Peichoto, M.E.; Rangel, D.D.; Barbaro, K.C.; Cirillo, M.C.; Santoro, M.L.; Sano-Martins, I.S. Loxosceles gaucho spider venom and its sphingomyelinase fraction trigger the main functions of human and rabbit platelets. Hum. Exp. Toxicol. 2011, 30, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, D.M.; Roberts, S.A.; Zobel-Thropp, P.A.; Delahaye, J.L.; Bandarian, V.; Binford, G.J.; Cordes, M.H.J. Variable substrate preference among phospholipase D toxins from Sicariid spiders. J. Biol. Chem. 2015, 290, 10994–11007. [Google Scholar] [CrossRef] [PubMed]
- Brahma, R.K.; McCleary, R.J.R.; Kini, R.M.; Doley, R. Venom gland transcriptomics for identifying, cataloging, and characterizing venom proteins in snakes. Toxicon 2015, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, D.; Gasparini, S.; Drevet, P.; Ducancel, F.; Bouet, F.; Boulain, J.C.; Harris, J.B.; Menez, A. Production of recombinant notechis 11’2l, an enzymatically active mutant of a phospholipase-a2 from notechis-scutatus scutatus venom, as directly generated by cleavage of a fusion protein produced in Escherichia coli. Eur. J. Biochem. 1993, 212, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Bouchier, C.; Ducancel, F.; Guignery-Frelat, G.; Bon, C.; Boulain, J.C.; Ménez, A. Cloning and sequencing of cDNAs encoding the two subunits of Crotoxin. Nucleic Acids Res. 1988, 16, 9050. [Google Scholar] [CrossRef] [PubMed]
- Ducancel, F.; Guigneryfrelat, G.; Boulain, J.C.; Menez, A. Nucleotide-sequence and structure-analysis of cDNAs encoding short-chain neurotoxins from venom glands of a sea-snake (Aipysurus laevis). Toxicon 1990, 28, 119–123. [Google Scholar] [CrossRef]
- Balduino, K.N.; Spencer, P.J.; Malavasi, N.V.; Chura-Chambi, R.M.; Lemke, L.S.; Morganti, L. Refolding by high pressure of a toxin containing seven disulfide bonds: Bothropstoxin-1 from Bothrops jararacussu. Mol. Biotechnol. 2011, 48, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Yin, S.J.; Li, T.; Cao, Z.J.; Ji, Y.H.; Wu, Y.L.; Li, W.X. Recombinant expression, purification, and characterization of scorpion toxin BmαTX14. Protein Expr. Purif. 2012, 82, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Estrada, G.; Silva, A.O.; Villegas, E.; Ortiz, E.; Beirao, P.S.L.; Corzo, G. Heterologous expression of five disulfide-bonded insecticidal spider peptides. Toxicon 2016, 119, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, V.P.; de Mari, T.L.; Gremski, L.H.; Silva, D.T.; da Silveira, R.B.; Gremski, W.; Chaim, O.M.; Senff-Ribeiro, A.; Nader, H.B.; Veiga, S.S. A novel hyaluronidase from brown spider (Loxosceles intermedia) venom (Dietrich’s hyaluronidase): From cloning to functional characterization. PLoS Negl. Trop. Dis. 2013, 7, e2206. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.M.A.; Oliveira, T.S.; Silveira, C.R.F.; Caporrino, M.C.; Rodriguez, D.; Moura-Da-Silva, A.M.; Ramos, O.H.P.; Rucavado, A.; Gutierrez, J.M.; Magalhães, G.S.; et al. A neutralizing recombinant single chain antibody, scFv, against BAP1, a P-I hemorrhagic metalloproteinase from Bothrops asper snake venom. Toxicon 2014, 87, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Lima-Dos-Santos, I.; Della-Casa, M.S.; Portes, J.A.; Calabria, P.A.L.; Magalhães, G.S.; Moura-da-Silva, A.M. Characterization of neuwiedin, a new disintegrin from Bothrops neuwiedi venom gland with distinct cysteine pattern. Toxicon 2015, 104, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Lima, C.D. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 2000, 5, 865–876. [Google Scholar] [CrossRef]
- Barbaro, K.C.; Sousa, M.V.; Morhy, L.; Eickstedt, V.R.D.; Mota, I. Compared chemical properties of dermonecrotic and lethal toxins from spiders of the genus Loxosceles (araneae). J. Protein Chem. 1996, 15, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lynch, K.R. Brown recluse spider (Loxosceles reclusa) venom phospholipase D (PLD) generates lysophosphatidic acid (LPA). Biochem. J. 2005, 391, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Idicula-Thomas, S.; Balaji, P.V. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli. Protein Sci. 2005, 14, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.O.S.; Chaim, O.M.; da Silveira, R.B.; Gremski, L.H.; Sade, Y.B.; Paludo, K.S.; Senff-Ribeiro, A.; de Moura, J.; Chavez-Olortegui, C.; Gremski, W.; et al. Biological and structural comparison of recombinant phospholipase D toxins from Loxosceles intermedia (brown spider) venom. Toxicon 2007, 50, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Derman, A.I.; Prinz, W.A.; Belin, D.; Beckwith, J. Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 1993, 262, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.H.; Silva, C.A.; Assakura, M.T.; Camargo, A.C.M.; Serrano, S.M.T. Molecular cloning, functional expression, and molecular modeling of Bothrostatin, a new highly active disintegrin from Bothrops jararaca venom. Biochem. Biophys. Res. Commun. 2005, 329, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Peng, H.C.; Yih, J.B.; Huang, T.F. A new short chain RGD-containing disintegrin, accutin, inhibits the common pathway of human platelet aggregation. Biochim. Biophys. Acta Gen. Subj. 1998, 1425, 493–504. [Google Scholar] [CrossRef]
- Halder, T.; Agarwal, T.; Ray, S. Isolation, cloning, and characterization of a novel Sorghum dehydrin (SbDhn2) protein. Protoplasma 2016, 253, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Glibowicka, M.; Nadeau, V.G.; Chen, G.; Deber, C.M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.L.; Sano-Martins, I.S. Platelet dysfunction during Bothrops jararaca snake envenomation in rabbits. Thromb. Haemost. 2004, 92, 369–383. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimokawa-Falcão, L.H.A.L.; Caporrino, M.C.; Barbaro, K.C.; Della-Casa, M.S.; Magalhães, G.S. Toxin Fused with SUMO Tag: A New Expression Vector Strategy to Obtain Recombinant Venom Toxins with Easy Tag Removal inside the Bacteria. Toxins 2017, 9, 82. https://doi.org/10.3390/toxins9030082
Shimokawa-Falcão LHAL, Caporrino MC, Barbaro KC, Della-Casa MS, Magalhães GS. Toxin Fused with SUMO Tag: A New Expression Vector Strategy to Obtain Recombinant Venom Toxins with Easy Tag Removal inside the Bacteria. Toxins. 2017; 9(3):82. https://doi.org/10.3390/toxins9030082
Chicago/Turabian StyleShimokawa-Falcão, Lhiri H. A. L., Maria C. Caporrino, Katia C. Barbaro, Maisa S. Della-Casa, and Geraldo S. Magalhães. 2017. "Toxin Fused with SUMO Tag: A New Expression Vector Strategy to Obtain Recombinant Venom Toxins with Easy Tag Removal inside the Bacteria" Toxins 9, no. 3: 82. https://doi.org/10.3390/toxins9030082