Polydimethylsiloxane (PDMS) Sub-Micron Traps for Single-Cell Analysis of Bacteria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (i)
- (ii)
2. Experimental
2.1. Soft Lithography
2.2. Sample Preparation
2.3. Experimental Procedure
3. Results and Discussion
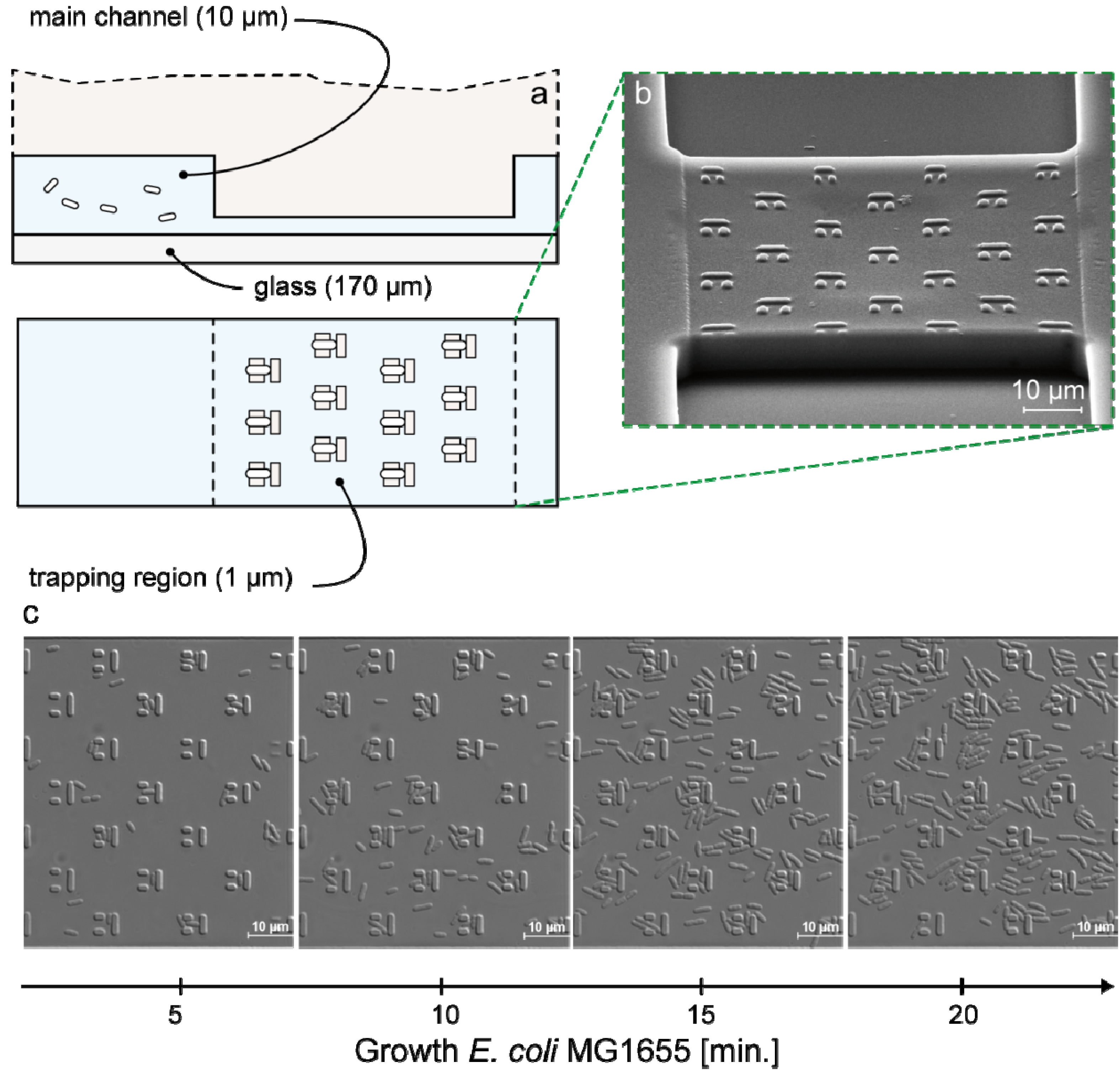
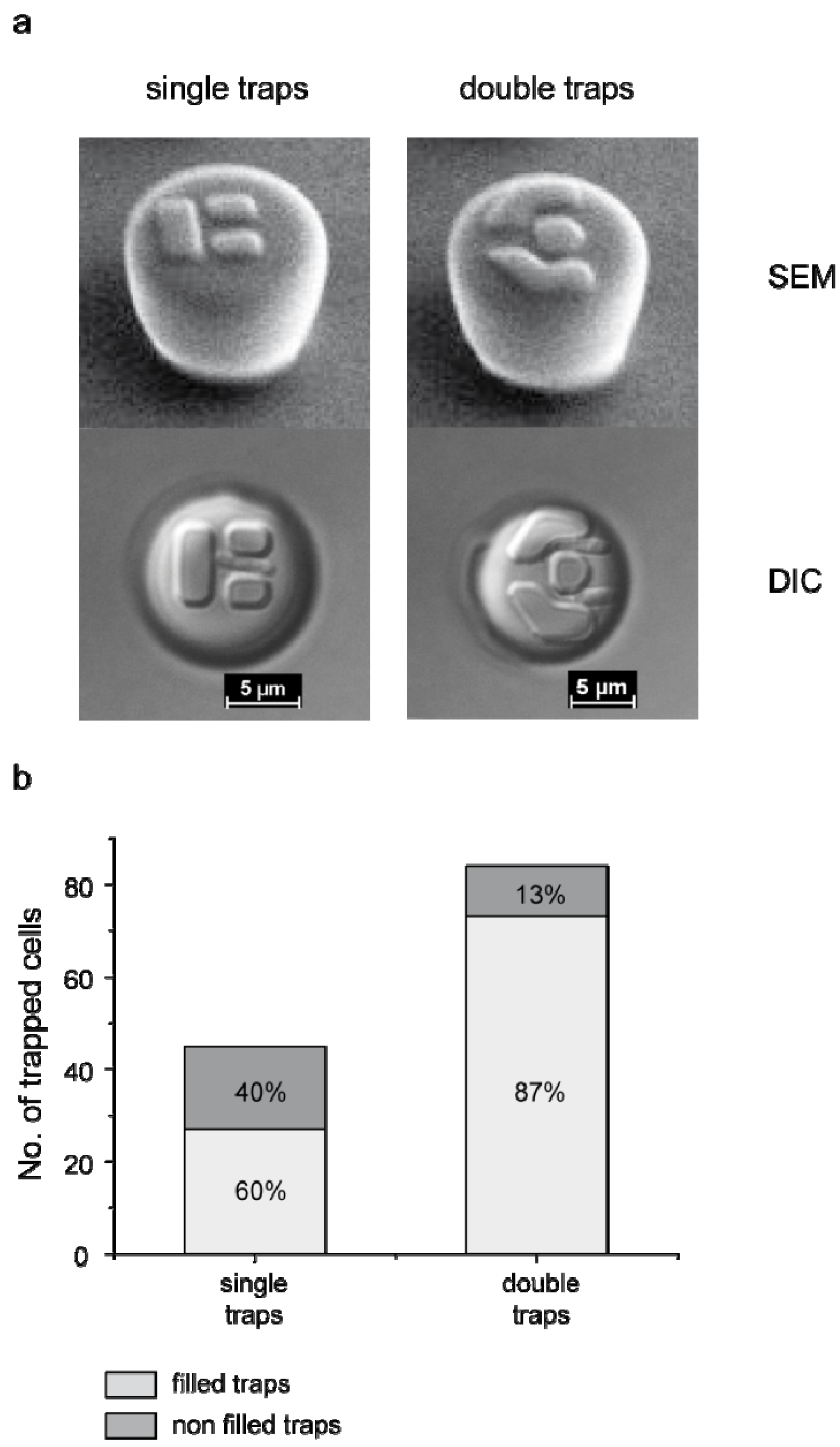
3.1. Trap Layout and Geometry


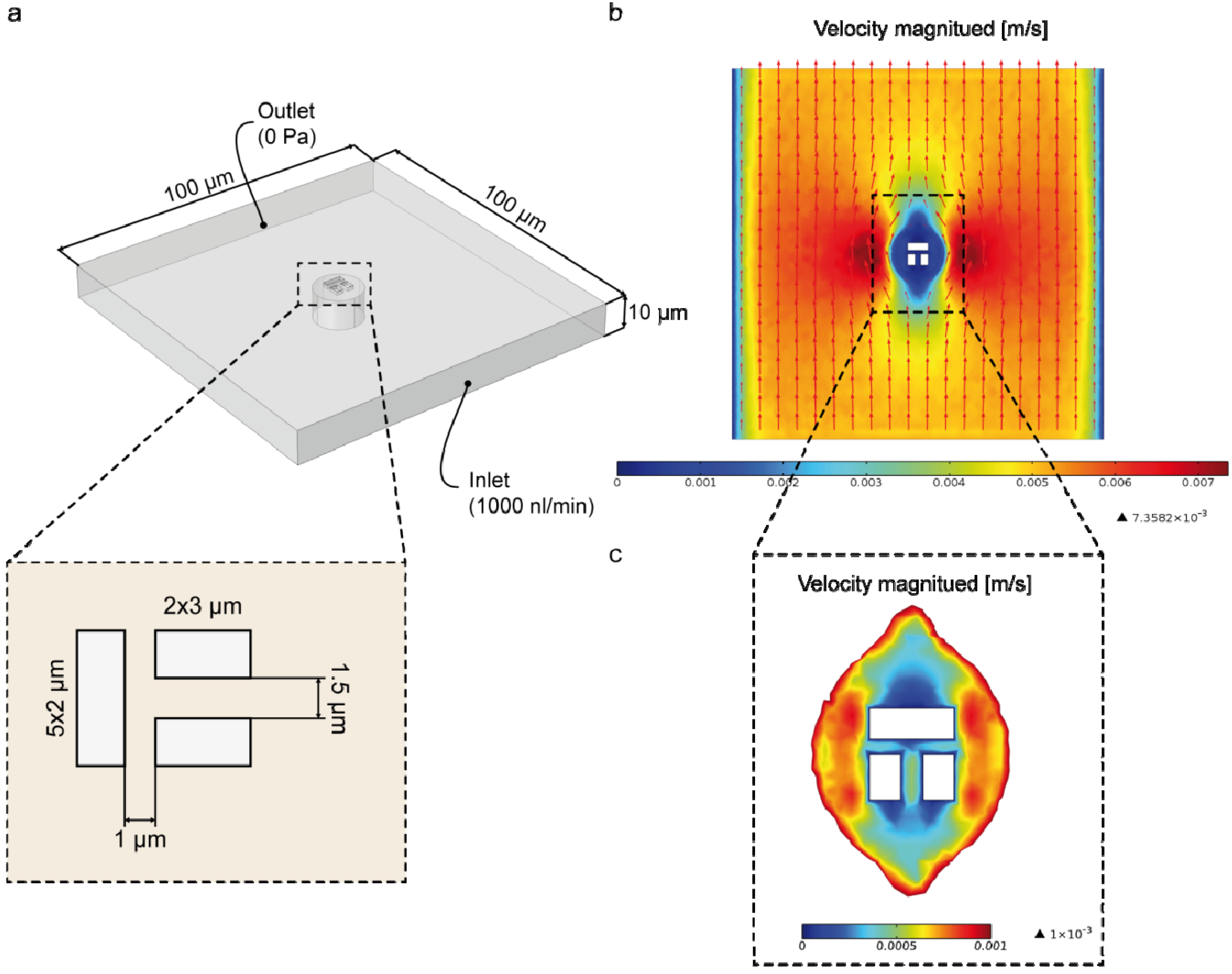
3.2. Numerical Simulation
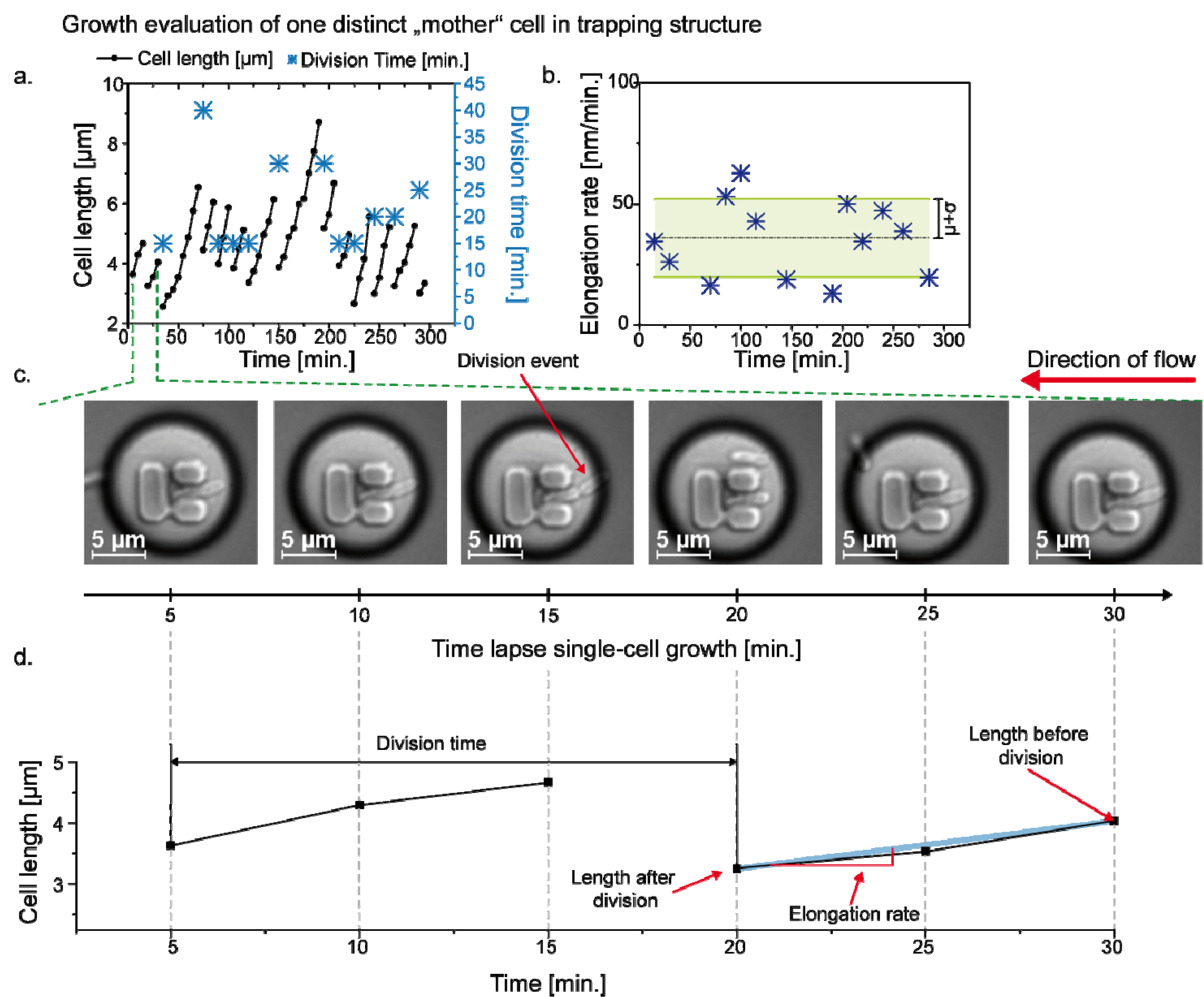
3.3. Single Cell Cultivation
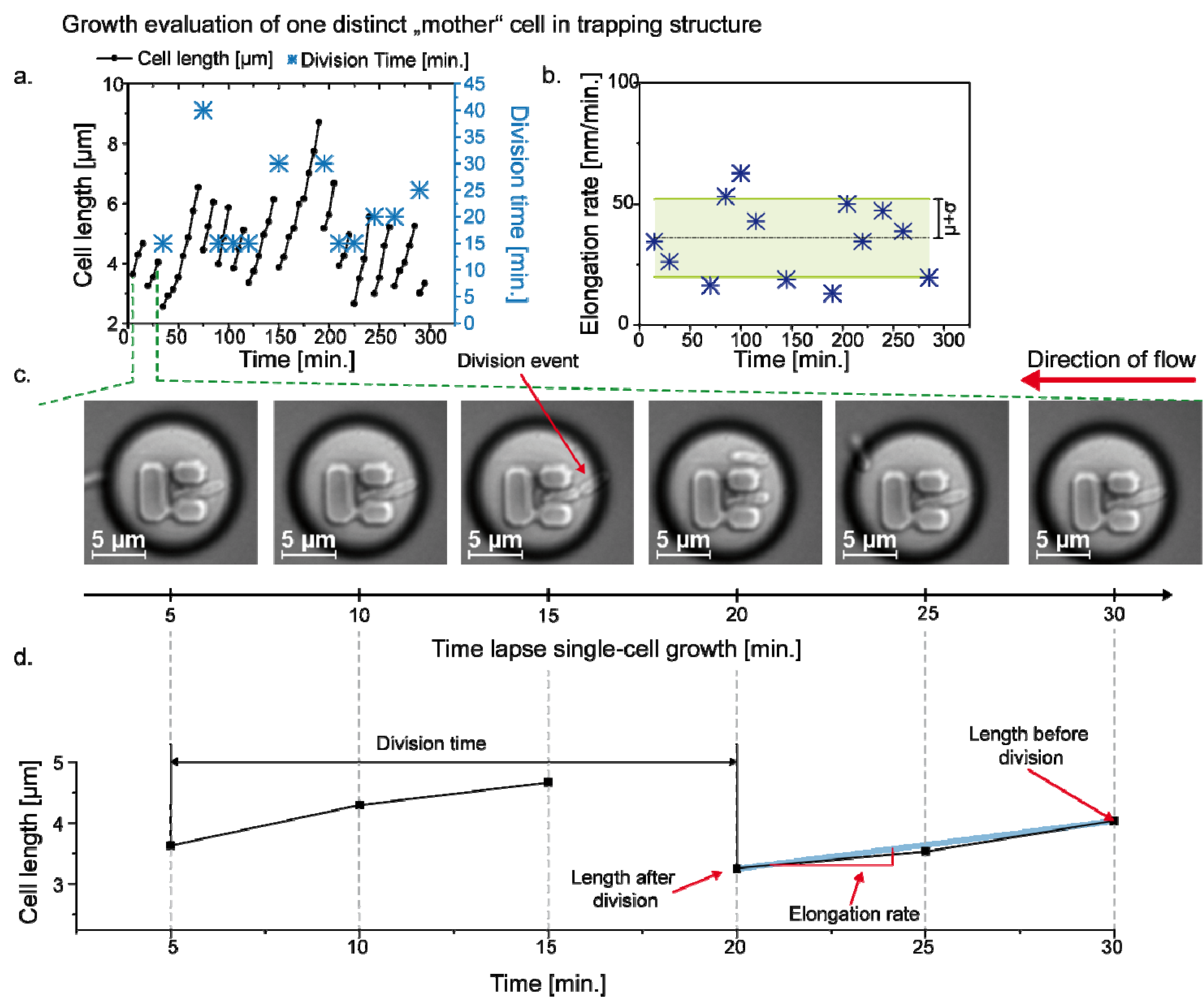
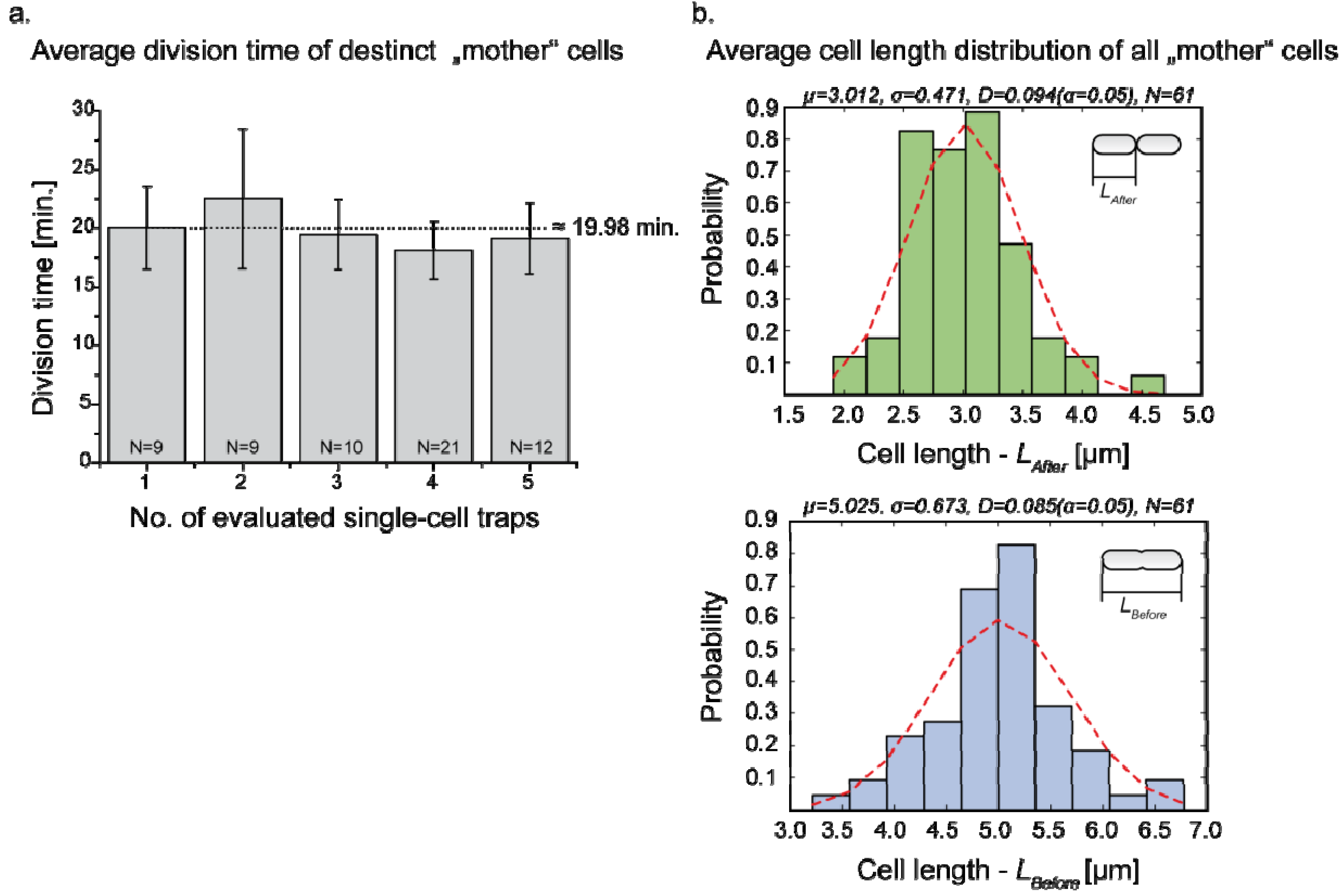
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Grünberger, A.; van Ooyen, J.; Paczia, N.; Rohe, P.; Schiendzielorz, G.; Eggeling, L.; Wiechert, W.; Kohlheyer, D.; Noack, S. Beyond growth rate 0.6: Corynebacterium glutamicum cultivated in highly diluted environments. Biotechnol. Bioeng. 2013, 110, 220–228. [Google Scholar] [CrossRef]
- Grunberger, A.; Paczia, N.; Probst, C.; Schendzielorz, G.; Eggeling, L.; Noack, S.; Wiechert, W.; Kohlheyer, D. A disposable picolitre bioreactor for cultivation and investigation of industrially relevant bacteria on the single cell level. Lab Chip 2012, 12, 2060–2068. [Google Scholar] [CrossRef]
- Mustafi, N.; Grünberger, A.; Kohlheyer, D.; Bott, M.; Frunzke, J. The development and application of a single-cell biosensor for the detection of l-methionine and branched-chain amino acids. Metab. Eng. 2012, 14, 449–457. [Google Scholar] [CrossRef]
- Schendzielorz, G.; Dippong, M.; Grünberger, A.; Kohlheyer, D.; Yoshida, A.; Binder, S.; Nishiyama, C.; Nishiyama, M.; Bott, M.; Eggeling, L. Taking control over control: Use of product sensing in single cells to remove flux control at key enzymes in biosynthesis pathways. ACS Synth. Biol. 2013. [Google Scholar] [CrossRef]
- Yin, H.; Marshall, D. Microfluidics for single cell analysis. Curr. Opin. Biotech. 2012, 23, 110–119. [Google Scholar] [CrossRef]
- Miesenbock, G.; de Angelis, D.A.; Rothman, J.E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 1998, 394, 192–195. [Google Scholar] [CrossRef]
- Prindle, A.; Samayoa, P.; Razinkov, I.; Danino, T.; Tsimring, L.S.; Hasty, J. A sensing array of radically coupled genetic “biopixels”. Nature 2012, 481, 39–44. [Google Scholar]
- Groisman, A.; Lobo, C.; Cho, H.; Campbell, J.K.; Dufour, Y.S.; Stevens, A.M.; Levchenko, A. A microfluidic chemostat for experiments with bacterial and yeast cells. Nat. Meth. 2005, 2, 685–689. [Google Scholar] [CrossRef]
- Di Carlo, D.; Aghdam, N.; Lee, L.P. Single-cell enzyme concentrations, kinetics, and inhibition analysis using high-density hydrodynamic cell isolation arrays. Anal. Chem. 2006, 78, 4925–4930. [Google Scholar] [CrossRef]
- Skelley, A.M.; Kirak, O.; Suh, H.; Jaenisch, R.; Voldman, J. Microfluidic control of cell pairing and fusion. Nat. Methods 2009, 6, 147–152. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Lee, J.B.; Cluzel, P. The single-cell chemostat: an agarose-based, microfluidic device for high-throughput, single-cell studies of bacteria and bacterial communities. Lab Chip 2012, 12, 1487–1494. [Google Scholar] [CrossRef]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. P. Natl. Acad. Sci. 2009, 106, 14195–14200. [Google Scholar] [CrossRef]
- Choi, K.; Ng, A.H.C.; Fobel, R.; Wheeler, A.R. Digital Microfluidics. Annu. Rev. Anal. Chem. 2012, 5, 413–440. [Google Scholar] [CrossRef]
- Ding, X.; Lin, S.-C.S.; Kiraly, B.; Yue, H.; Li, S.; Chiang, I.-K.; Shi, J.; Benkovic, S.J.; Huang, T.J. On-chip manipulation of single microparticles, cells, and organisms using surface acoustic waves. P. Natl. Acad. Sci. 2012. [Google Scholar]
- Chiou, P.Y.; Ohta, A.T.; Wu, M.C. Massively parallel manipulation of single cells and microparticles using optical images. Nature 2005, 436, 370–372. [Google Scholar] [CrossRef]
- Dusny, C.; Fritzsch, F.S.O.; Frick, O.; Schmid, A. Isolated microbial single cells and resulting micropopulations grow faster in controlled environments. Appl. Environ. Microbiol. 2012, 78, 7132–7136. [Google Scholar] [CrossRef]
- Fritzsch, F.S.O.; Rosenthal, K.; Kampert, A.; Howitz, S.; Dusny, C.; Blank, L.M.; Schmid, A. Picoliter nDEP traps enable time-resolved contactless single bacterial cell analysis in controlled microenvironments. Lab Chip 2013, 13, 397–408. [Google Scholar] [CrossRef]
- Hsu, H.-y.; Ohta, A.T.; Chiou, P.-Y.; Jamshidi, A.; Neale, S.L.; Wu, M.C. Phototransistor-based optoelectronic tweezers for dynamic cell manipulation in cell culture media. Lab Chip 2010, 10, 165–172. [Google Scholar] [CrossRef]
- Kortmann, H.; Chasanis, P.; Blank, L.M.; Franzke, J.; Kenig, E.Y.; Schmid, A. The envirostat—A new bioreactor concept. Lab Chip 2009, 9, 576–585. [Google Scholar] [CrossRef]
- Kim, S.H.; Yamamoto, T.; Fourmy, D.; Fujii, T. Electroactive microwell arrays for highly efficient single-cell trapping and analysis. Small 2011, 7, 3239–3247. [Google Scholar] [CrossRef]
- Ramser, K.; Hanstorp, D. Optical manipulation for single-cell studies. J. Biophotonics 2010, 3, 187–206. [Google Scholar] [CrossRef]
- Johann, R.M. Cell trapping in microfluidic chips. Anal. Bioanal Chem 2006, 385, 408–412. [Google Scholar] [CrossRef]
- Svoboda, K.; Block, S.M. Biological applications of optical forces. Annu. Rev. Biophys. Biomed. Struct. 1994, 23, 247–285. [Google Scholar] [CrossRef]
- Kobel, S.; Valero, A.; Latt, J.; Renaud, P.; Lutolf, M. Optimization of microfluidic single cell trapping for long-term on-chip culture. Lab Chip 2010, 10, 857–863. [Google Scholar] [CrossRef]
- Khine, M.; Lau, A.; Ionescu-Zanetti, C.; Seo, J.; Lee, L.P. A single cell electroporation chip. Lab Chip 2005, 5, 38–43. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Shaw, R.; Jan, L.; Lee, L.P. Microfluidic application-specific integrated device for monitoring direct cell–cell communication via gap junctions between individual cell pairs. Appl. Phys. Lett. 2005, 86, 223902. [Google Scholar] [CrossRef]
- Valero, A.; Post, J.N.; van Nieuwkasteele, J.W.; ter Braak, P.M.; Kruijer, W.; van den Berg, A. Gene transfer and protein dynamics in stem cells using single cell electroporation in a microfluidic device. Lab Chip 2008, 8, 62–67. [Google Scholar] [CrossRef]
- Zhu, Z.; Frey, O.; Ottoz, D.S.; Rudolf, F.; Hierlemann, A. Microfluidic single-cell cultivation chip with controllable immobilization and selective release of yeast cells. Lab Chip 2012, 12, 906–915. [Google Scholar] [CrossRef]
- Kim, M.-C.; Isenberg, B.C.; Sutin, J.; Meller, A.; Wong, J.Y.; Klapperich, C.M. Programmed trapping of individual bacteria using micrometre-size sieves. Lab Chip 2011, 11, 1089–1095. [Google Scholar] [CrossRef]
- Wang, P.; Robert, L.; Pelletier, J.; Dang, W.L.; Taddei, F.; Wright, A.; Jun, S. Robust growth of escherichia coli. Curr. Biol. 2010, 20, 1099–1103. [Google Scholar] [CrossRef]
- Long, Z.; Nugent, E.; Javer, A.; Cicuta, P.; Sclavi, B.; Cosentino Lagomarsino, M.; Dorfman, K.D. Microfluidic chemostat for measuring single cell dynamics in bacteria. Lab Chip 2013, 13, 947–954. [Google Scholar] [CrossRef]
- Gruenberger, A.; Probst, C.; Heyer, A.; Wiechert, W.; Frunzke, J.; Kohlheyer, D. Microfluidic picoliter bioreactor for microbial single-cell analysis: Fabrication, system setup and operation. J. Vis. Exp. 2013. [Google Scholar] [CrossRef]
- Männik, J.; Driessen, R.; Galajda, P.; Keymer, J.E.; Dekker, C. Bacterial growth and motility in sub-micron constrictions. P. Natl. Acad. Sci. 2009, 106, 14861–14866. [Google Scholar] [CrossRef]
- Lecault, V.; White, A.K.; Singhal, A.; Hansen, C.L. Microfluidic single cell analysis: From promise to practice. Curr. Opin. Chem. Biol. 2012, 16, 381–390. [Google Scholar] [CrossRef]
- Justice, S.S.; Hunstad, D.A.; Cegelski, L.; Hultgren, S.J. Morphological plasticity as a bacterial survival strategy. Nat. Rev. Micro. 2008, 6, 162–168. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Probst, C.; Grünberger, A.; Wiechert, W.; Kohlheyer, D. Polydimethylsiloxane (PDMS) Sub-Micron Traps for Single-Cell Analysis of Bacteria. Micromachines 2013, 4, 357-369. https://doi.org/10.3390/mi4040357
Probst C, Grünberger A, Wiechert W, Kohlheyer D. Polydimethylsiloxane (PDMS) Sub-Micron Traps for Single-Cell Analysis of Bacteria. Micromachines. 2013; 4(4):357-369. https://doi.org/10.3390/mi4040357
Chicago/Turabian StyleProbst, Christopher, Alexander Grünberger, Wolfgang Wiechert, and Dietrich Kohlheyer. 2013. "Polydimethylsiloxane (PDMS) Sub-Micron Traps for Single-Cell Analysis of Bacteria" Micromachines 4, no. 4: 357-369. https://doi.org/10.3390/mi4040357