Hot and Dry Cleaning of Biomass-Gasified Gas Using Activated Carbons with Simultaneous Removal of Tar, Particles, and Sulfur Compounds

Abstract
:1. Introduction
2. Results and Discussion
2.1. Textural Parameters for Activated Carbons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET specific surface area | Pore volume | Average pore diameter |
|---|---|---|---|
| m2 g−1 | cm3(STP) g−1 | nm | |
| Fe0 | 1206.3 | 277.2 | 2.0 |
| Fe9 | 1126.0 | 258.7 | 2.0 |
| Fe17 | 1128.5 | 259.3 | 2.0 |

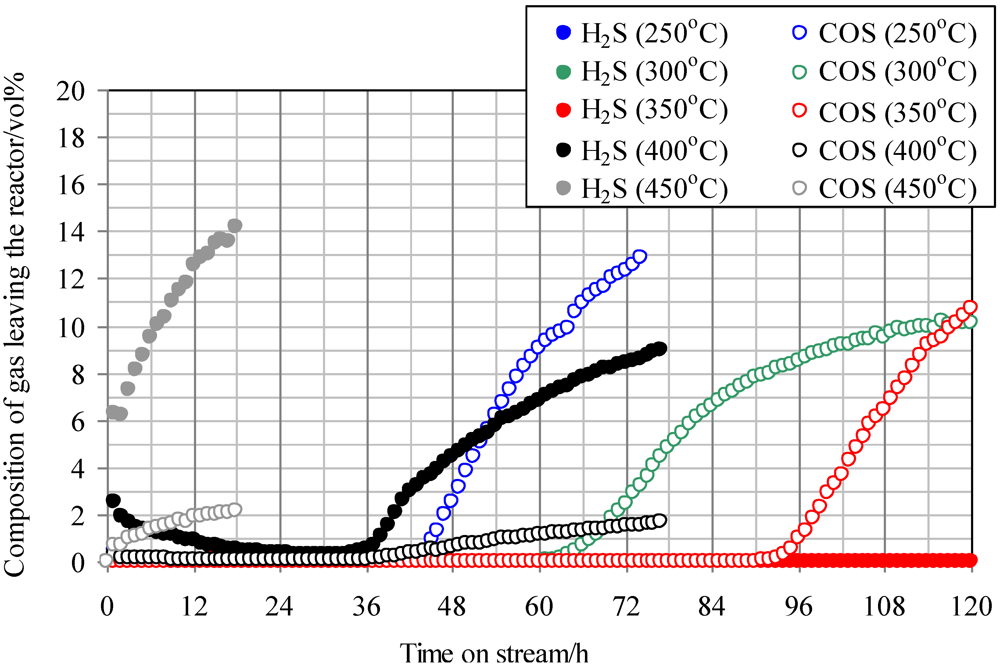
2.2. Effect of Temperature on COS Capture


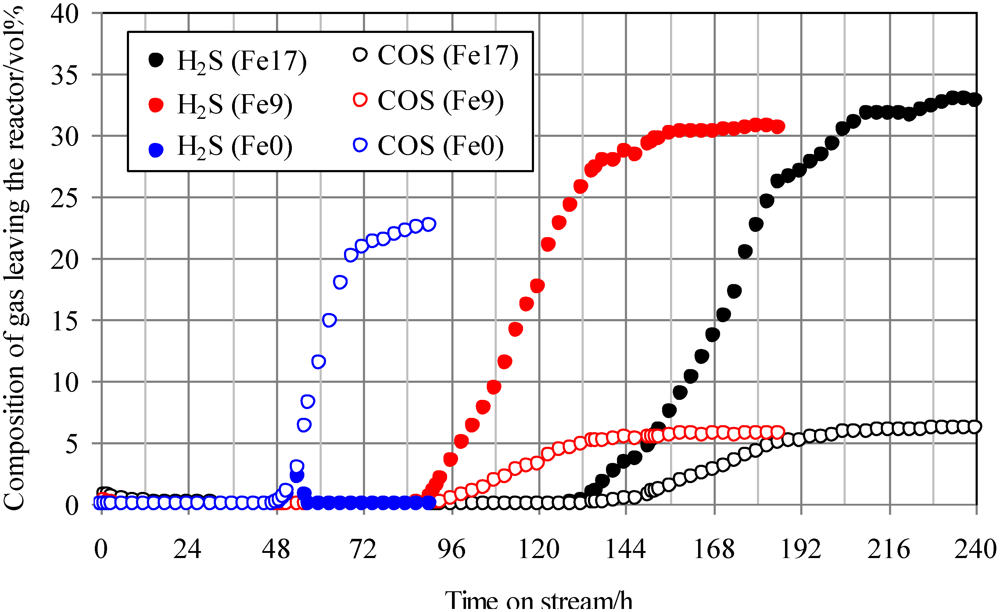
2.3. Effect of Addition of Fe to Activated Carbons

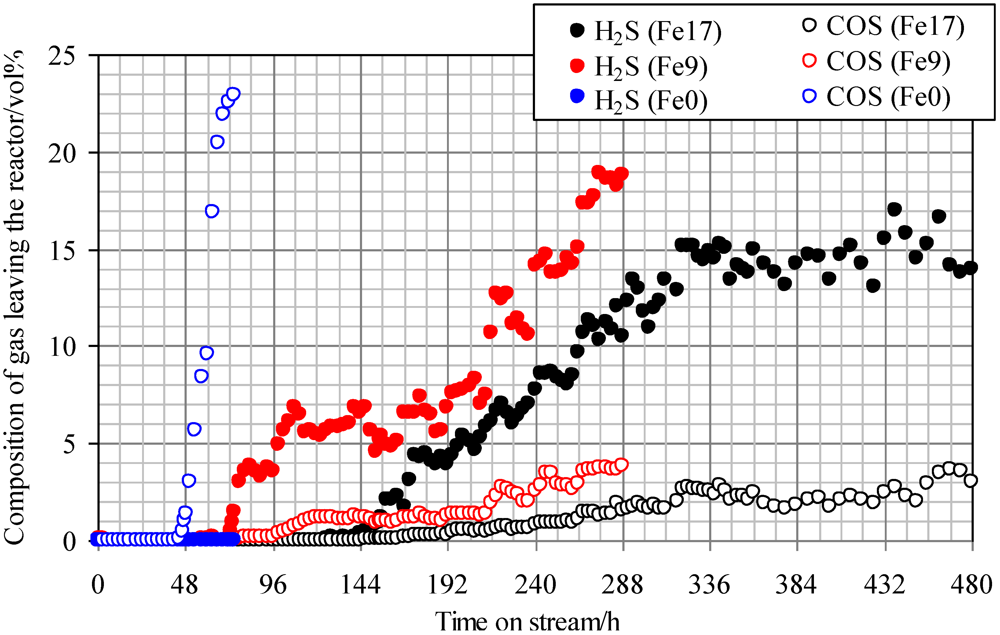
2.4. Effect of Steam on Simultaneous Removal of COS and H2S
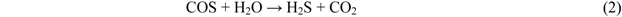

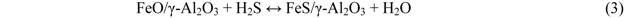
| Temperature | Gas composition/vol% on a dry basis | H2/CO | |||||
|---|---|---|---|---|---|---|---|
| °C | H2 | CO | CO2 | CH4 | C2H4 | N2 | - |
| 20 a | 28.1 | 41.0 | 0.4 | 4.2 | 0.1 | 26.3 | 0.69 |
| 250 | 27.1 | 39.8 | 0.6 | 4.0 | 0.1 | 28.3 | 0.68 |
| 300 | 27.3 | 39.2 | 1.2 | 4.0 | 0.1 | 28.2 | 0.70 |
| 350 | 27.4 | 38.7 | 1.7 | 4.0 | 0.1 | 28.1 | 0.71 |
| 400 | 29.2 | 34.8 | 4.4 | 3.9 | 0.1 | 27.6 | 0.84 |
| 450 | 30.5 | 30.8 | 7.5 | 3.9 | 0.1 | 27.2 | 0.99 |
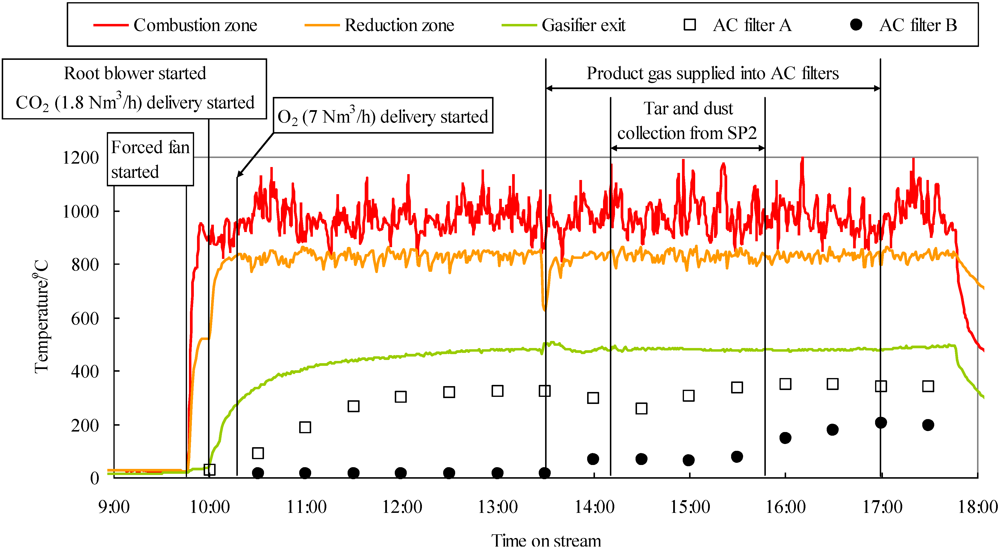
2.5. Simultaneous Removal of Impurities from Product Gas Derived from Woody Biomass Gasification Using Fe-Supported Activated Carbon

| Ultimate analysis/wt% daf basis | Proximate analysis/wt% | |||||||
|---|---|---|---|---|---|---|---|---|
| C | H | N | S a | O b | Moisture | Volatile matter | Fixed carbon | Ash |
| 50.7 | 5.9 | 0.2 | 0.015 | 43.2 | 13.1 | 70.9 | 15.4 | 0.6 |
2.5.1. Sulfur-Compound Removal and Water-Gas Shift Reaction

| SP | H2 | CO | CO2 | CH4 | C2H4 | C2H6 | N2 | H2S | COS | H2/CO |
|---|---|---|---|---|---|---|---|---|---|---|
| vol% | ppmv | - | ||||||||
| SP1 | 30.4 | 44.8 | 18.6 | 3.6 | 0.7 | 0.1 | 1.8 | 15.7 | 7.2 | 0.7 |
| SP2 | 35.6 | 28.6 | 27.9 | 3.9 | 0.8 | 0.2 | 3.0 | 0.1 | 0.0 | 1.2 |
| SP4 | 34.5 | 31.6 | 25.9 | 3.8 | 0.9 | 0.2 | 3.1 | 0.0 | 0.0 | 1.1 |
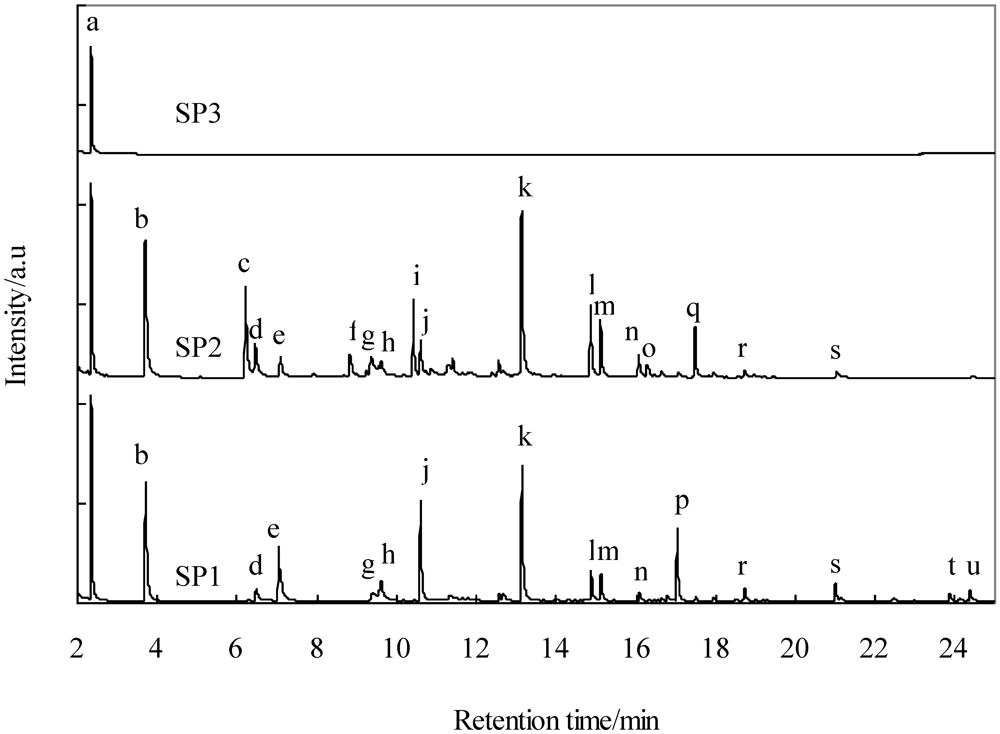
2.5.2. Removal of Tar and Particles
| SP | Tar | Particles |
|---|---|---|
| mg Nm−3 | mg Nm−3 | |
| SP1 | 2428 | 2244 |
| SP2 | 305 | 662 |
| SP3 | 102 | 181 |

| Symbol | Compound name |
|---|---|
| a | Benzene |
| b | Toluene |
| c | Ethylbenzene |
| d | o-Xylene |
| e | 1,3,5,7-Cyclooctatetraene |
| f | 1-Ethyl-3-methyl benzene |
| g | Phenol |
| h | Benzofuran |
| i | Indane |
| j | Indene |
| k | Naphthalene |
| l | 2-Methyl naphthalene |
| m | 1-Methyl naphthalene |
| n | Biphenyl |
| o | 1-Ethyl naphthalene |
| p | Biphenylene |
| q | Acenaphthene |
| r | Fluorene |
| s | Anthracene |
| t | Fluoranthene |
| u | Pyrene |
2.6. Improvement of Gas-Cleaning Process Using Fe-Supported Activated Carbons for BTL System
3. Experimental Section
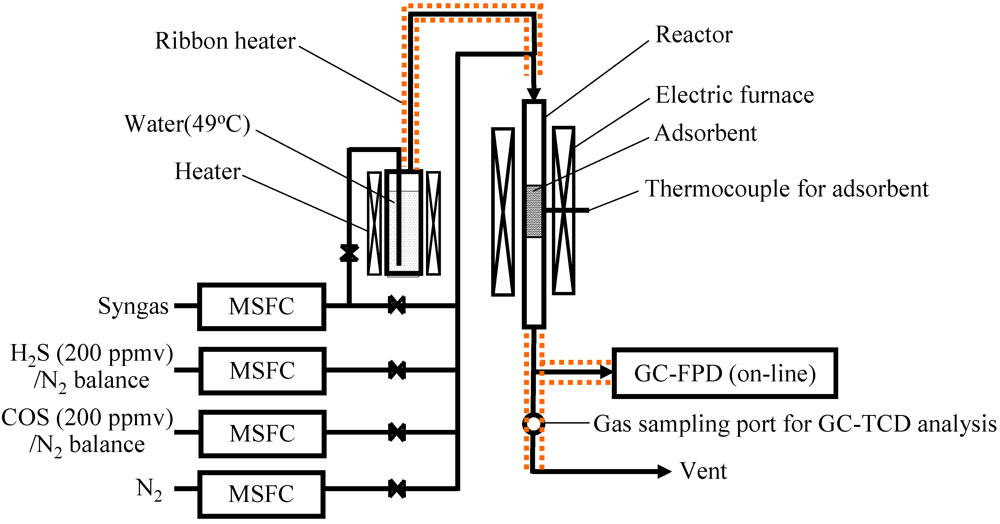
3.1. Desulfurization of Gas Using Activated Carbons on a Laboratory Scale

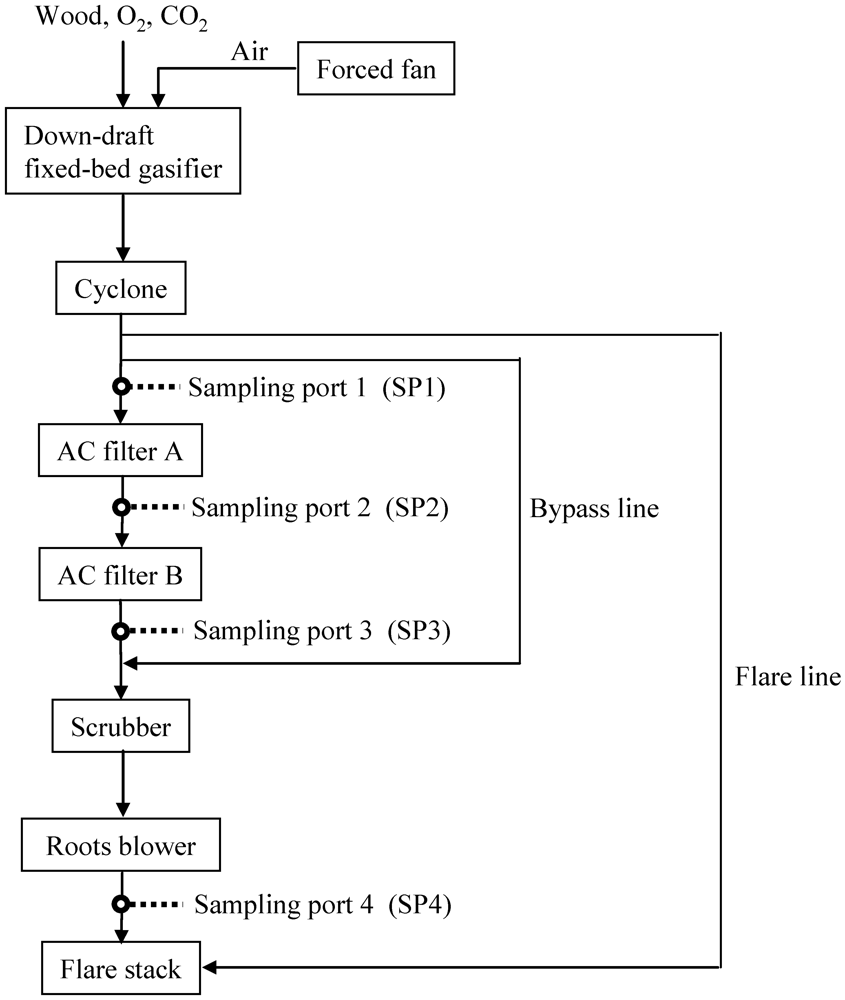
3.2. Removal of Impurities Using Fe-Supported Activated Carbons on a Bench Scale
4. Conclusions
Acknowledgments
References
- Minowa, T.; Hanaoka, T.; Yokoyama, S. Process evaluation of biomass to liquid fuel production system with gasification and liquid fuel synthesis. Stud. Surf. Sci. Catal. 2004, 153, 79–84. [Google Scholar]
- Sunde, K.; Brekke, A.; Solberg, B. Environment impacts and costs of woody Biomass-To-Liquid (BTL) production and use—A review. Forest Policy Econ. 2011, 13, 591–602. [Google Scholar]
- Devi, L.; Ptasinski, K.J.; Janssen, F.J.J.G. A review of the primary measures for tar elimination in biomass gasification processes. Biomass Bioenergy 2003, 24, 125–140. [Google Scholar]
- Brandt, P.; Larsen, E.; Henriksen, U. High tar reduction in a two-stage gasifier. Energy Fuels 2000, 14, 816–819. [Google Scholar]
- Gil, J.; Corella, J.; Aznar, M.P.; Caballero, M.A. Biomass gasification in atmospheric and bubbling fluidized bed. Effect of the type of gasifying agent on the product distribution. Biomass Bioenergy 1999, 17, 389–403. [Google Scholar] [CrossRef]
- Jansen, J.C.; Jonsson, K.; Hagman, M. Biological detoxification of tar-water. Water Sci. Technol. 2002, 46, 59–65. [Google Scholar]
- Nakamura, K. Status of EAGLE project: Multi-purpose coal gasification technology development (in Japanese). J. Jpn. Inst. Energy 2007, 86, 321–325. [Google Scholar]
- Gonzalez, J.M.; Roman, S.; Engo, G.; Encinar, J.M.; Martinez, G. Reduction of tars by dolomite cracking during two-stage gasification of olive cake. Biomass Bioenergy 2011, 35, 4324–4330. [Google Scholar]
- Andres, J.M.; Narros, A.; Rodriguez, M.E. Behaviour of dolomite, olivine and alumina as primary catalysts in air-steam gasification of sewage sludge. Fuel 2011, 90, 521–527. [Google Scholar]
- Corella, J.; Orio, A.; Toledo, J.M. Biomass gasification with air in a fluidized-bed: Exhaustive tar elimination with commercial steam reforming catalyst. Energy Fuels 1999, 13, 702–709. [Google Scholar]
- Nordgreen, T.; Liliedahl, T.; Sjostrom, K. Metallic iron as a tar breakdown catalyst related to atmospheric, fluidized bed gasification of biomass. Fuel 2006, 85, 689–694. [Google Scholar]
- Haibo, L.; Tianhu, C.; Xianlong, Z.; Jinhu, L.; Dongyin, C.; Lei, S. Effect of additives on catalytic cracking of biomass gasification tar over a nickel-based catalyst. Chin. J. Catal. 2010, 31, 409–414. [Google Scholar]
- Dou, B.; Gao, J.; Sha, X.; Baek, S.W. Catalytic cracking of tar component from high-temperature fuel gas. Appl. Therm. Eng. 2003, 23, 2229–2239. [Google Scholar]
- Lu, C.Y.; Wey, M.Y.; Fu, Y.H. The size, shape, and dispersion of active sites on AC-supported copper nanocatalysts with polyol process: The effect of precursors. Appl. Catal. A Gen. 2008, 344, 36–44. [Google Scholar] [CrossRef]
- Sakanishi, K.; Wu, Z.; Matsumura, A.; Saito, I.; Hanaoka, T.; Minowa, T.; Tada, M.; Iwasaki, T. Simultaneous removal of H2S and COS using activated carbons and their supported catalysts. Catal. Today 2005, 104, 94–100. [Google Scholar]
- Hanaoka, T.; Sakanishi, K.; Minowa, T. Hot gas cleaning of producer gas from biomass gasification using carbonaceous materials as a bed additive. J. Jpn. Inst. Energy 2004, 83, 828–831. [Google Scholar]
- Hu, X.; Hanaoka, T.; Sakanishi, K.; Shinagawa, T.; Matsui, S.; Tada, M.; Iwasaki, T. Removal of tar compounds produced from biomass gasification using activated carbons. J. Jpn. Inst. Energy 2007, 86, 707–711. [Google Scholar]
- Phuphuakrat, T.; Namioka, T.; Yoshikawa, K. Tar removal from biomass pyrolysis gas in two-step function of decomposition and adsorption. Appl. Energy 2010, 87, 2203–2211. [Google Scholar]
- Itaya, Y.; Kawahara, K.; Lee, C.W.; Kobayashi, J.; Kobayashi, N.; Hatano, S.; Mori, S. Dry gas cleaning process by adsorption of H2S into activated cokes in gasification of carbon resources. Fuel 2009, 88, 1665–1672. [Google Scholar]
- Adib, F.; Bagreev, A.; Bandosz, T.J. Analysis of the relationship between H2S removal capacity and surface properties of unimpregnated activated carbons. Environ. Sci. Technol. 2000, 34, 686–692. [Google Scholar]
- Xie, W.; Chang, L.; Wang, D.; Xie, K.; Wall, T.; Yu, J. Removal of sulfur at high temperatures using iron-based sorbents supported on fine coal ash. Fuel 2010, 89, 868–873. [Google Scholar]
- Wakker, J.P.; Gerritsen, A.W.; Moulijn, J.A. High temperature H2S and COS removal with MnO and FeO on γ-Al2O3 acceptors. Ind. Eng. Chem. Res. 1993, 32, 139–149. [Google Scholar]
- Encinar, J.M.; Gonzalez, J.F.; Rodriguez, J.J.; Ramiro, M.J. Catalysed and uncatalysed steam gasification of eucalyptus char: Influence of variables and kinetics study. Fuel 2001, 80, 2025–2036. [Google Scholar]
- Hanaoka, T.; Miyazawa, T.; Nurunnabi, M.; Hirata, S.; Sakanishi, K. Liquid fuel production from woody biomass via oxygen-enriched air/CO2 gasification on a bench scale. J. Jpn. Inst. Energy 2011, 90, 1072–1080. [Google Scholar]
- Stevens, D.J. Hot Gas Conditioning: Recent Progress with Larger-Scale Biomass Gasification Systems; NREL-SR-510-29952; National Renewable Research Laboratory: Golden, CO, USA, 2001. [Google Scholar]
- Baker, E.G.; Brown, M.D.; Moore, R.H.; Mudge, L.K.; Elliott, D.C. Engineering Analysis of Biomass Gasifier Product Gas Cleaning Technology; PNL-534; Biomass Energy Technology Division, Pacific Northwest Laboratory: Richland, WA, USA, 1986. [Google Scholar]
- Mastral, A.M.; Garcia, T.; Callen, M.S.; Navarro, M.V.; Galban, J. Removal of naphthalene, phenanthrene, and pyrene by sorbents from hot gas. Environ. Sci. Technol. 2001, 35, 2395–2400. [Google Scholar]
- Mastral, A.M.; Garcia, T.; Murillo, R.; Callen, M.S.; Lopez, J.M.; Navarro, M.V. Measurements of polycyclic aromatic hydrocarbon adsorption on activated carbons at very low concentrations. Ind. Eng. Chem. Res. 2003, 42, 155–161. [Google Scholar]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar]
- Mastral, A.M.; Garcia, T.; Murillo, R.; Callen, M.S.; Lopez, J.M.; Navarro, M.V. Moisture effects on the phenanthrene adsorption capacity by carbonaceous materials. Energy Fuels 2002, 16, 205–210. [Google Scholar]
- Milne, T.A.; Evans, R.J.; Abatzoglou, N. Biomass Gasifier “Tars”: Their Nature, Formation, and Conversion; NREL/TP-570-25357; National Renewable Energy Laboratory: Golden, CO, USA, 1998. [Google Scholar]
- Garcia, T.; Murillo, R.; Amoros, D.C.; Mastral, A.M.; Solano, A.L. Role of the activated carbon surface chemistry in the adsorption of phenanthrene. Carbon 2004, 42, 1683–1689. [Google Scholar]
- Li, Y.; Wang, T.; Yin, X.; Wu, C.; Ma, L.; Li, H.; Lv, Y.; Sun, L. 100 t/a-scale demonstration of direct dimethyl ether synthesis from corncob-derived syngas. Renew. Energy 2010, 35, 583–587. [Google Scholar]
- Tijmensen, M.J.A.; Faaij, A.P.C.; Hamelinck, C.N.; van Hardeveld, M.R.M. Exploration of the possibilities for production of Fischer Tropsch liquids and power via biomass gasification. Biomass Bioenergy 2002, 23, 129–152. [Google Scholar]
- Dry, M.E. High quality diesel via the Fischer-Tropsch process—A review. J. Chem. Technol. Biotechnol. 2001, 77, 43–50. [Google Scholar]
- Subramani, V.; Gangwal, S.K. A review of recent literature to search for an efficient catalytic process for the conversion of syngas to ethanol. Energy Fuels 2008, 22, 814–839. [Google Scholar]
- Boerrigter, H.; Calis, H.P.; Slort, D.J.; Bodenstaff, H. Gas Cleaning for Integrated Biomass Gasification (BG) and Fischer-Tropsch (FT) Systems; Experimental Demonstration of Two BG-FT Systems. In Proceedings of the 2nd World Conference and Technology Exhibition on Biomass for Energy, Industry and Climate Protection, Rome, Italy, 2004; pp. 51–56.
- Hanaoka, T.; Liu, Y.; Matsunaga, K.; Miyazawa, T.; Hirata, S.; Sakanishi, K. Bench-scale production of liquid fuel from woody biomass via gasification. Fuel Proc. Technol. 2010, 91, 859–865. [Google Scholar]
- Neeft, J.P.A.; Knoef, H.A.M.; Zielke, U.; Sjostrom, K.; Hasler, P.; Simell, P.A.; Dorrington, M.A.; Thomas, L.; Abatzoglou, N.; Deutch, S.; Greil, C.; Buffinga, G.J.; Brage, C.; Suomalainen, M. Guideline for Sampling and Analysis of Tar and Particles in Biomass Producer Gases; Energy project ERK6-CT1999-20002 (Tar protocol), Version 3.3; European Commission (DGXII),Netherlands Agency for Energy and the Environment (NOVEM),Swiss Federal Office of Education and Science, Danish Energy Agency (Energistyrelsen),US Department of Energy (DoE),National Resources Canada: Brussels, Belgium, 2002. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hanaoka, T.; Matsunaga, K.; Miyazawa, T.; Hirata, S.; Sakanishi, K. Hot and Dry Cleaning of Biomass-Gasified Gas Using Activated Carbons with Simultaneous Removal of Tar, Particles, and Sulfur Compounds. Catalysts 2012, 2, 281-298. https://doi.org/10.3390/catal2020281
Hanaoka T, Matsunaga K, Miyazawa T, Hirata S, Sakanishi K. Hot and Dry Cleaning of Biomass-Gasified Gas Using Activated Carbons with Simultaneous Removal of Tar, Particles, and Sulfur Compounds. Catalysts. 2012; 2(2):281-298. https://doi.org/10.3390/catal2020281
Chicago/Turabian StyleHanaoka, Toshiaki, Kotetsu Matsunaga, Tomohisa Miyazawa, Satoshi Hirata, and Kinya Sakanishi. 2012. "Hot and Dry Cleaning of Biomass-Gasified Gas Using Activated Carbons with Simultaneous Removal of Tar, Particles, and Sulfur Compounds" Catalysts 2, no. 2: 281-298. https://doi.org/10.3390/catal2020281
APA StyleHanaoka, T., Matsunaga, K., Miyazawa, T., Hirata, S., & Sakanishi, K. (2012). Hot and Dry Cleaning of Biomass-Gasified Gas Using Activated Carbons with Simultaneous Removal of Tar, Particles, and Sulfur Compounds. Catalysts, 2(2), 281-298. https://doi.org/10.3390/catal2020281