
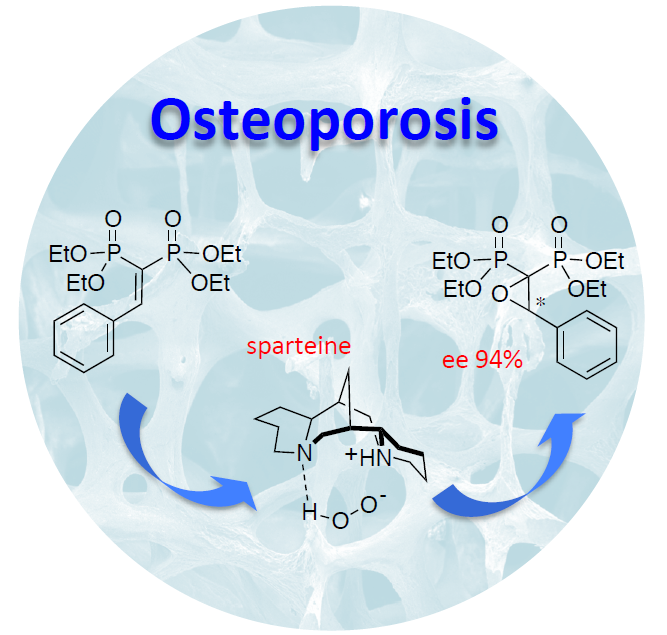
Organocatalytic Enantioselective Epoxidation of Some Aryl-Substituted Vinylidenebisphosphonate Esters: On the Way to Chiral Anti-Osteoporosis Drugs




Abstract
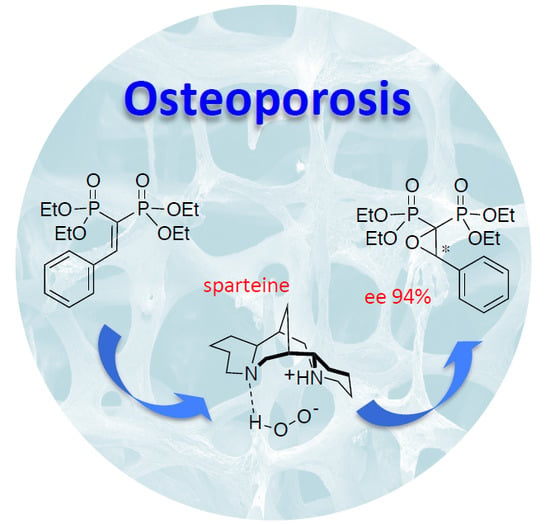
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of Methylenebisphosphonate Tetraethyl Ester (MBP) [38]
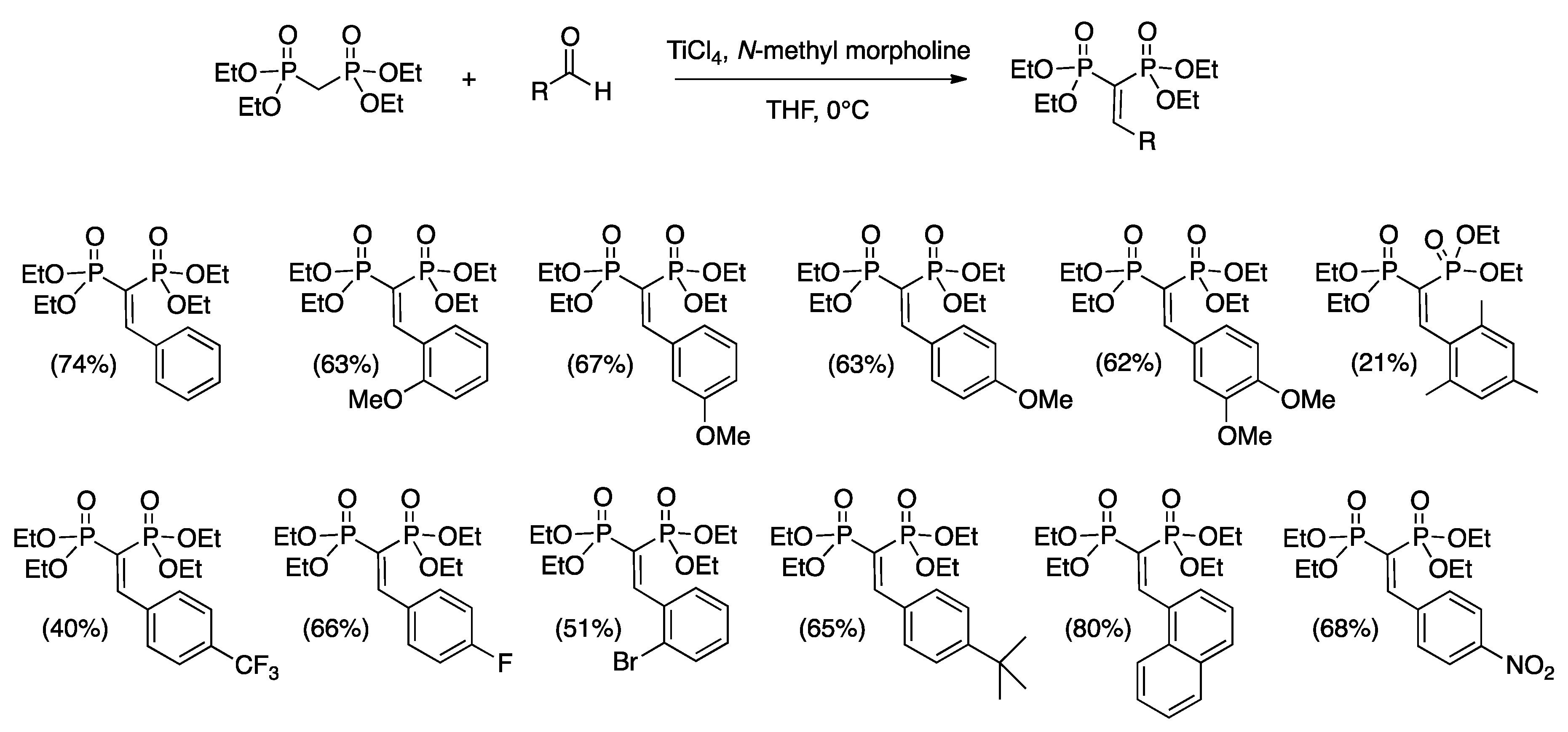
3.2. General Procedure for the Synthesis of the Mono-Substituted Alkylidene Bisphosphonates (VBPs)
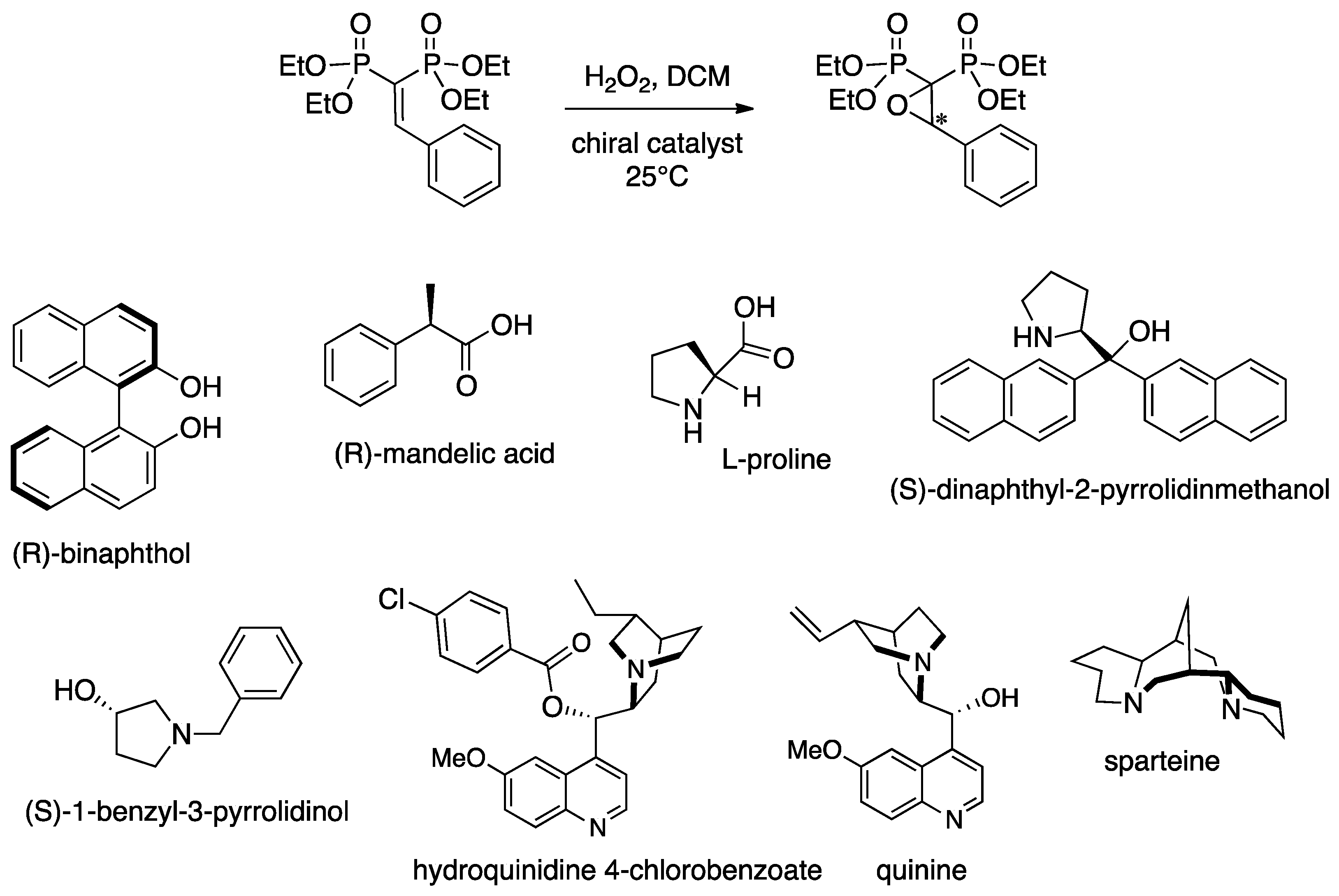
3.3. Epoxidation of Prochiral VBPs
3.4. Attempts of Oxirane Ring Opening with Organometals
3.5. Oxirane Ring Opening with S-Nucleophiles
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- NIH Consensus. Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. JAMA 2001, 285, 785–795. [Google Scholar]
- Randell, A.; Sambrook, P.N.; Nguyen, T.V.; Eisman, J.A. Direct clinical and welfare costs of osteoporotic fractures in elderly men and women. Osteoporos. Int. 1995, 5, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Melton, L.J. Hip fractures: A worldwide problem today and tomorrow. Bone 1993, 14 (Suppl. 1), S1–S8. [Google Scholar] [CrossRef]
- Zhang, S.; Gangal, G.; Uludağ, H. ‘Magic bullets’ for bone diseases: Progress in rational design of bone-seeking medicinal agents. Chem. Soc. Rev. 2007, 36, 507–531. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.C.; Monkkonen, J.; Blackburn, G.M. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5′-(beta, gamma-dichloromethylene) triphosphate, by mammalian cells in vitro. J. Bone Miner. Res. 1997, 12, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.P.; Hughes, D.E.; Coxon, F.P. Nitrogen-Containing Bisphosphonates Inhibit the Mevalonate Pathway and Prevent Post-Translational Prenylation of GTP-Binding Proteins, Including Ras. J. Bone Miner. Res. 1998, 13, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.; Pieterman, E.; Cohen, L. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem. Biophys. Res. Commun. 1999, 264, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Dunford, J.E.; Rogers, M.J.; Ebetino, F.H.; Phipps, R.J. Inhibition of Protein Prenylation by Bisphosphonates Causes Sustained Activation of Rac, Cdc42, and Rho GTPases. J. Bone Miner. Res. 2006, 21, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Gertz, B.J.; Holland, S.D.; Kline, W.F.; Quan, H. Studies of the oral bioavailability of alendronate. Clin. Pharmacol. Ther. 1995, 58, 288–298. [Google Scholar] [CrossRef]
- Capuzzi, M.; Perdicchia, D.; Jørgensen, K.A. Highly Enantioselective Approach to Geminal Bisphosphonates by Organocatalyzed Michael-Type Addition of β-Ketoesters. Chem. Eur. J. 2007, 14, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Sulzer-Mossè, S.; Tissot, M.; Alexakis, A. First Enantioselective Organocatalytic Conjugate Addition of Aldehydes to Vinyl Phosphonates. Org. Lett. 2007, 9, 3749–3752. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.T.; Faisca Philllips, A.M. Enamine Catalysis in the Synthesis of Chiral Structural Analogues of gem-Bisphosphonates Known To Be Biologically Active. Eur. J. Org. Chem. 2008, 15, 2525–2529. [Google Scholar] [CrossRef]
- Ebetino, F.H.; Dunford, J.E.; Lundy, M.W.; Pozzi, M.; Xia, Z.; Dobson, R. A mirror image pair of bisphosphonate analogs further demonstrates the mode of binding of the bisphosphonates in farnesyl pyrophosphate synthase. Bone 2008, 42, S36–S42. [Google Scholar] [CrossRef]
- Chiminazzo, A.; Sperni, L.; Damuzzo, M.; Strukul, G.; Scarso, A. Copper-mediated 1,4-Conjugate Addition of Boronic Acids and Indoles to Vinylidenebisphosphonate leading to gem-Bisphosphonates as Potential Antiresorption Bone Drugs. ChemCatChem 2014, 6, 2712–2718. [Google Scholar] [CrossRef]
- Ferrer-Casal, M.; Barboza, A.P.; Szajnman, S.H.; Rodriguez, J.B. 1,3-Dipolar Cycloadditions of the Versatile Intermediate Tetraethyl Vinylidenebisphosphonate. Synthesis 2013, 45, 2397–2404. [Google Scholar]
- Ruzziconi, R.; Rici, G.; Gioiello, A.; Couthon-Gourvès, H.; Gourvès, J.-P. First General Approach to Cyclohex-3-ene-1,1-bis(phosphonates) by Diels-Alder Cycloaddition of Tetraethyl Vinylidenebis(phosphonate) to 1,3-Dienes. J. Org. Chem. 2003, 68, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, O.; Mulani, I.; De Nino, A.; Maiuolo, L.; Nardi, M.; Russo, B.; Avnet, S. Efficient synthesis of isoxazolidine-substituted bisphosphonates by 1,3-dipolar cycloaddition reactions. Tetrahedron 2011, 67, 5635–5641. [Google Scholar] [CrossRef]
- Scarso, A.; Strukul, G. Transition-Metal-Catalyzed Stereoselective Oxidations in Drug and Natural Product Synthesis. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko, V., Andrushko, N., Eds.; Wiley-VCH: London, UK, 2013; pp. 1043–1070. [Google Scholar]
- Bianchini, G.; Scarso, A.; Chiminazzo, A.; Sperni, L.; Strukul, G. Water enhanced synthesis of gem-bisphosphonates via Rh(I) mediated 1,4-conjugate addition of aryl boronic acids to vinylidenebisphosphonate esters. Green Chem. 2013, 15, 656–662. [Google Scholar] [CrossRef]
- Granchi, D.; Scarso, A.; Bianchini, G.; Chiminazzo, A.; Minto, A.; Sgarbossa, P.; Michelin, R.A.; Di Pompo, G.; Avnet, S.; Strukul, G. Low toxicity and unprecedented anti-osteoclast activity of a simple sulfur-containing gem-bisphosphonate: A comparative study. Eur. J. Med. Chem. 2013, 65, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.; Löwik, C.; Que, I. Dissociation of binding and antiresorptive properties of hydroxybisphosphonates by substitution of the hydroxyl with an amino group. J. Bone Miner. Res. 1996, 11, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, W. Knoevenagel kondensationen mit TiCl4/base-IV: Umsetzungen von aldehyden und ketonen mit phosphonoessigester und methylendiphosphonsäureestern. Tetrahedron 1973, 30, 301–305. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, M.; Duan, W. Palladium-catalyzed asymmetric 1,6-addition of diphenylphosphine to (4-aryl-1,3-butadienylidene)bis(phosphonates) for the synthesis of chiral phosphines. Tetrahedron Lett. 2014, 55, 629–631. [Google Scholar]
- Xiang, H.; Qi, X.; Xie, Y.; Xub, G.; Yang, C. One-pot syntheses of novel pyrazole-containing bisphosphonate esters at room temperature. Org. Biomol. Chem. 2012, 10, 7730–7738. [Google Scholar]
- Ding, H.; Xu, G.; Wang, J.; Zhang, Y.; Wu, X.; Xie, Y. Catechol–bisphosphonate conjugates: New types of chelators for metal intoxication therapy. Heteroat. Chem. 2004, 15, 549–555. [Google Scholar] [CrossRef]
- Porter, M.G.; Skidmore, J. Asymmetric epoxidation of electron-deficient olefins. Chem. Commun. 2000, 14, 1215–1225. [Google Scholar] [CrossRef]
- Díez, D.; Núñez, M.G.; Antón, A.B.; García, P.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Basabe, P.; Urones, J.G. Asymmetric Epoxidation of Electron-Deficient Olefins. Curr. Org. Synth. 2008, 5, 186–216. [Google Scholar] [CrossRef]
- Weitz, E.; Scheffer, A. Über die Einwirkung von alkalischem Wasserstoffsuperoxyd auf ungesättigte Verbindungen. Chem. Ber. 1921, 54, 2327–2344. [Google Scholar] [CrossRef]
- Strukul, G.; Scarso, A. Environmentally Benign Oxidants. In Liquid Phase Oxidation via Heterogeneous Catalysis; Clerici, M.G., Kholdeeva, O., Eds.; Wiley: London, UK, 2013; pp. 1–20. [Google Scholar]
- Wynberg, H.; Greijdanus, B. Solvent effects in homogeneous asymmetric catalysis. Chem. Commun. 1978, 10, 427–428. [Google Scholar] [CrossRef]
- Colonna, S.; Molinari, H.; Banfi, S.; Julià, S.; Masana, J.; Alvarez, A. Synthetic enzymes—4: Highly enantioselective epoxidation by means of polyaminoacids in a triphase system: Influence of structural variations within the catalysts. Tetrahedron 1983, 39, 1635–1641. [Google Scholar] [CrossRef]
- Baccin, C.; Gusso, A.; Pinna, F.; Strukul, G. Platinum-Catalyzed Oxidations with Hydrogen Peroxide: The (Enantioselective) Epoxidation of alpha.,beta.-Unsaturated Ketones. Organometallics 1995, 14, 1161–1167. [Google Scholar] [CrossRef]
- Kakei, H.; Tsuji, R.; Ohshima, T.; Shibasaki, M. Catalytic Asymmetric Epoxidation of α,β-Unsaturated Esters Using an Yttrium-Biphenyldiol Complex. J. Am. Chem. Soc. 2005, 127, 8962–8963. [Google Scholar] [CrossRef] [PubMed]
- Sturtz, G.; Guervenou, J. Synthesis of Novel Functionalized gem-Bisphosphonates. Synthesis 1991, 661–662. [Google Scholar] [CrossRef]
- Johnson, J.B. Ring opening of epoxides, aziridines and cyclic anhydrides. In Science of Synthesis, Stereoselective Synthesis; Vries, J.G., Molander, G.A., Evans, P.A., Eds.; Thieme: Stuttgart, Germany, 2011; pp. 759–827. [Google Scholar]
- Szajnman, S.; Linares, G.; Moro, P.; Rodriguez, J. New Insights into the Chemistry of gem-Bis(phosphonates): Unexpected Rearrangement of Michael-Type Acceptors. Eur. J. Org. Chem. 2005, 3687–3696. [Google Scholar] [CrossRef]
- Nugent, R.A.; Murphy, M.; Schlatchter, S.T.; Dunn, C.J.; Smith, R.J.; Staite, N.D.; Galinet, L.A.; Shields, S.K.; Aspar, D.G.; Richard, K.A.; et al. Pyrazoline bisphosphonate esters as novel antiinflammatory and antiarthritic agents. J. Med. Chem. 1993, 36, 134–139. [Google Scholar] [CrossRef] [PubMed]
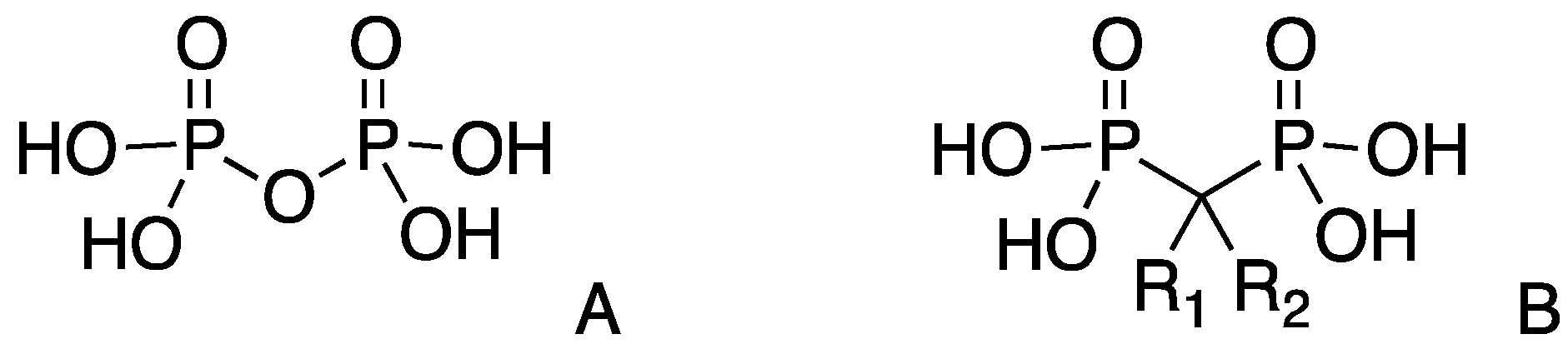
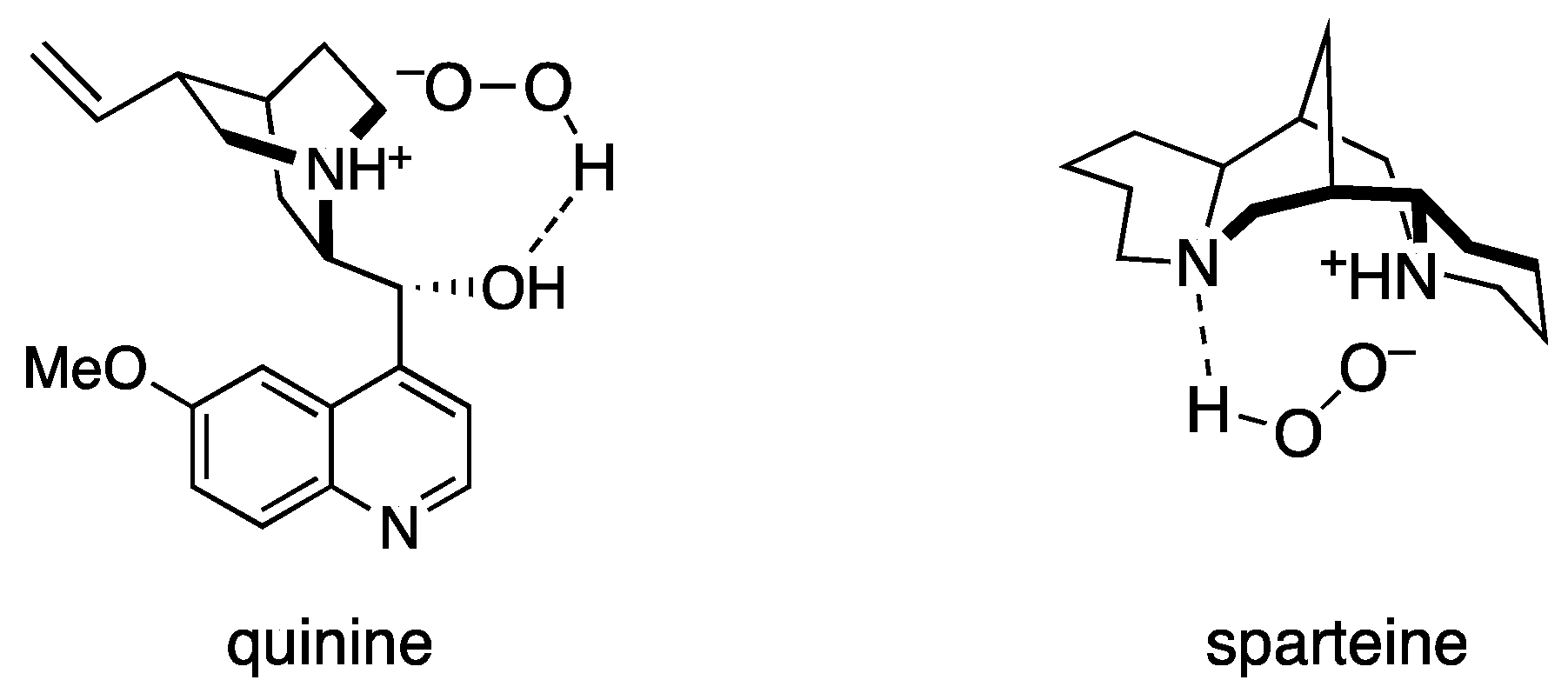

{kind=link}
{kind=link}
{kind=link}
{kind=link}
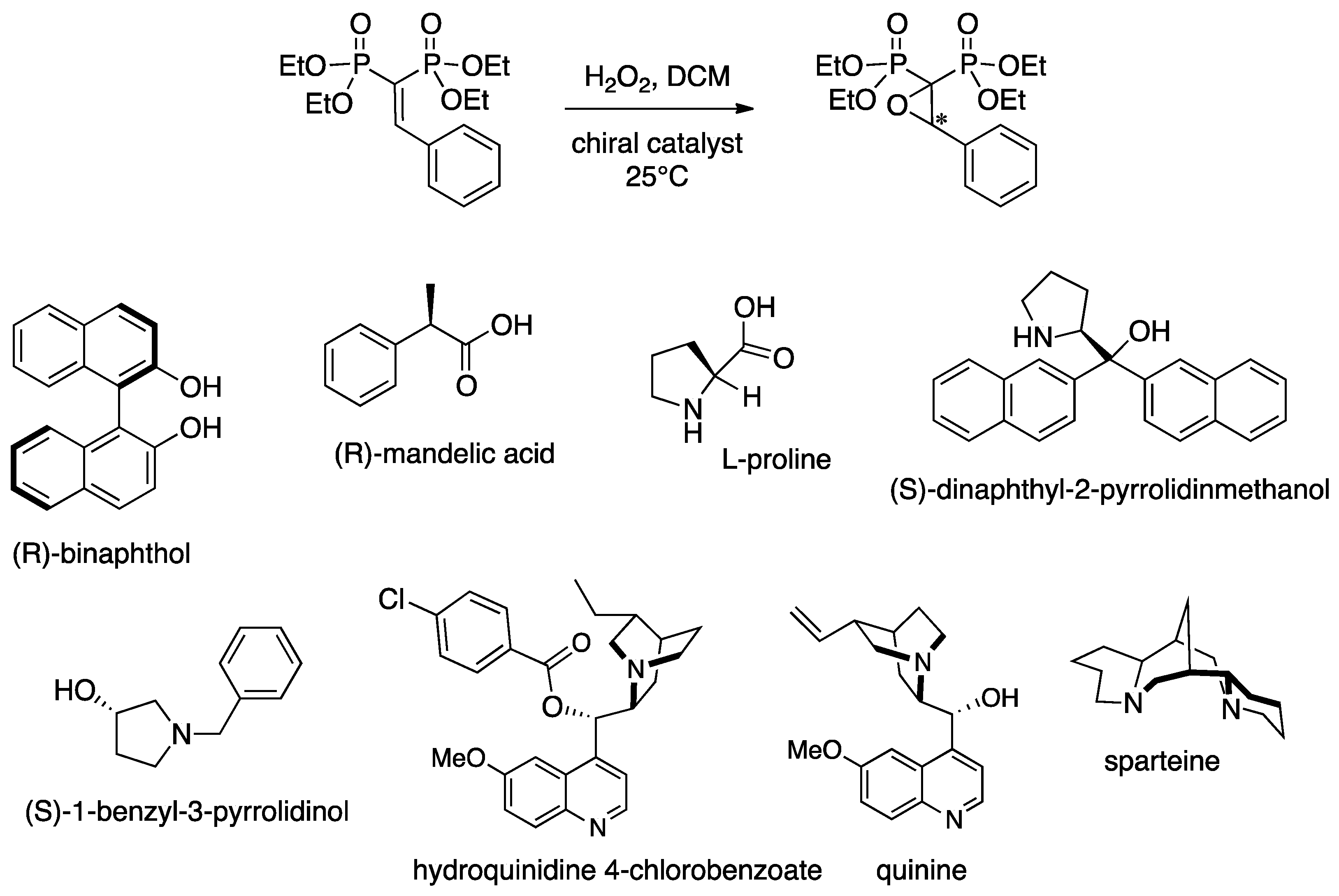{kind=link}
| Entry | Organocatalyst | Catalyst amt (mol %) | Oxidant | Yield a (%) | e.e. b (%) |
|---|---|---|---|---|---|
| 1 | (R)-1,1′-2-naphthol | 100 | H2O2 | 3 | = |
| 2 | (R)-mandelic acid | 100 | H2O2 | 0 | = |
| 3 | l-proline | 100 | H2O2 | 0 | = |
| 4 | (S)-dinaphthyl-2-pyrrolidinmethanol | 100 | H2O2 | 6 | = |
| 5 | (S)-1-benzyl-3-pyrrolidinol | 100 | H2O2 | 0 | = |
| 6 | hydroquinidine 4-chlorobenzoate | 100 | H2O2 | 2 | = |
| 7 | quinine | 100 | H2O2 | 73 | 68 |
| 8 | quinine | 50 | H2O2 | 42 | 67 |
| 9 | quinine | 10 | H2O2 | 4 | = |
| 10 | quinine | 100 | t-BuOOH | 0 | = |
| 11 | quinine | 100 | urea.H2O2 | >99 | 20 |
| 12 | sparteine | 100 | H2O2 | 90 | 94 |
| 13 | sparteine | 50 | H2O2 | 43 | 96 |
| 14 | sparteine | 20 | H2O2 | 36 | 94 |
| 15 | sparteine | 100 | t-BuOOH | 0 | = |
| 16 | sparteine | 100 | urea⋅H2O2 | >99 | 13 |
| Entry | Epoxide | Acronym | Yield a (%) | Ee b (%) |
|---|---|---|---|---|
| 1 | 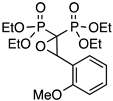 | PoOMeO | 63 | 4 |
| 2 | 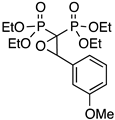 | PmOMeO | 74 | 81 |
| 3 |  | PpOMeO | 77 | 1 |
| 4 | 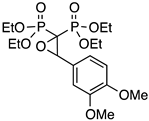 | PVERO | 71 | 9 |
| 5 |  | PMESO | 10 | = c |
| 6 |  | PptBuO | 0 | = |
| 7 |  | PpNO2O | 75 | = d |
| 8 |  | PpCF3O | 95 | 17 |
| 9 | 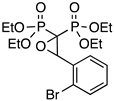 | PoBrO | 35 | 10 (17 e) |
| 10 | 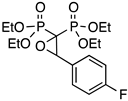 | PpFO | 71 | 5 (16 e) |
| 11 | 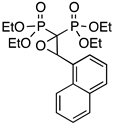 | P1NAPO | 65 | 3 (12 e) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiminazzo, A.; Sperni, L.; Scarso, A.; Strukul, G. Organocatalytic Enantioselective Epoxidation of Some Aryl-Substituted Vinylidenebisphosphonate Esters: On the Way to Chiral Anti-Osteoporosis Drugs. Catalysts 2017, 7, 90. https://doi.org/10.3390/catal7030090
Chiminazzo A, Sperni L, Scarso A, Strukul G. Organocatalytic Enantioselective Epoxidation of Some Aryl-Substituted Vinylidenebisphosphonate Esters: On the Way to Chiral Anti-Osteoporosis Drugs. Catalysts. 2017; 7(3):90. https://doi.org/10.3390/catal7030090
Chicago/Turabian StyleChiminazzo, Andrea, Laura Sperni, Alessandro Scarso, and Giorgio Strukul. 2017. "Organocatalytic Enantioselective Epoxidation of Some Aryl-Substituted Vinylidenebisphosphonate Esters: On the Way to Chiral Anti-Osteoporosis Drugs" Catalysts 7, no. 3: 90. https://doi.org/10.3390/catal7030090
APA StyleChiminazzo, A., Sperni, L., Scarso, A., & Strukul, G. (2017). Organocatalytic Enantioselective Epoxidation of Some Aryl-Substituted Vinylidenebisphosphonate Esters: On the Way to Chiral Anti-Osteoporosis Drugs. Catalysts, 7(3), 90. https://doi.org/10.3390/catal7030090