Abstract
Clenbuterol is a β2-agonist used in the veterinary treatment of asthma in several countries. The drug is listed on the World Antidoping Agency’s prohibited list due to its effect on increased protein synthesis in the body. However, racemic clenbuterol has recently been shown to reduce the risk of Parkinson’s disease. In order to reveal which one (or both) of the enantiomers that cause this effect, pure enantiomers need to be separately studied. (R)-1-(4-Amino-3,5-dichlorophenyl)-2-bromoethan-1-ol has been synthesised in 93% enantiomeric excess (ee) by asymmetric reduction of the corresponding ketone catalysed by a ketoreductase and nicotinamide adenine dinucleotide phosphate (NADPH) as the cofactor in dimethyl sulfoxide (DMSO). (S)-N-(2,6-Dichloro-4-(1-hydroxyethyl)phenyl)acetamide has been synthesised in >98% ee by the same system. Both synthons are potential precursors for clenbuterol enantiomers.
1. Introduction
The need for enantiopure compounds for the treatment of human diseases and combatting microbial attacks on, for example, crops can be explained by the fact that all living organisms in nature are chiral. Protein biosynthesis and most metabolic processes are mediated by enzymes that are specific for a particular isomeric form of a certain substrate, and this is essential for ensuring the high degree of three-dimensional organization that is found in structures within cells. It is presumably evolutionary chance that has determined that life is based on l- rather than d-amino acids [1].
When drug–receptor interactions are considered, it is postulated that the lower the effective dose of a drug, the greater the difference in the pharmacological effect of the optical isomers [2]. The ratio of the more active enantiomer (eutomer) compared to the less active enantiomer (distomer) is defined as the eudismic ratio; the higher the eudismic ratio is, the higher the effectiveness of the drug [3].
The β2-agonists clenbuterol and salbutamol are drugs used in the treatment of for instance angina pectoris, asthma, and other disorders related to the sympathetic nervous system [4,5]. β2-Receptors are mainly found in the bronchi, bronchioles, and gastrointestinal tract, and one of their main actions is the relaxation of smooth muscles (bronchodilation) [6]. Racemic clenbuterol and salbutamol are shown in Figure 1. Clenbuterol is marketed with a racemic active pharmaceutical ingredient (API), while salbutamol is marketed both as the racemate and as Xopenex®, with the R-enantiomer as the API. The (R)-1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-ol ((R)-2a) and S)-N-(2,6-dichloro-4-(1-hydroxyethyl)phenyl)acetamide ((S)-6a) alcohols are also shown in Figure 1.
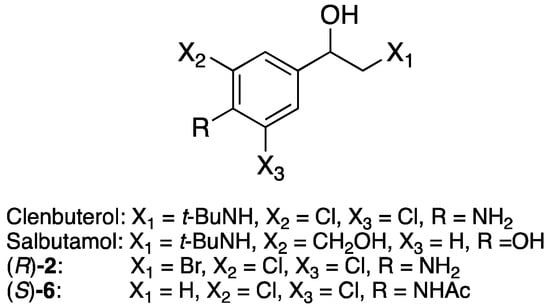
Figure 1.
Structures of racemic β2-agonists clenbuterol and salbutamol and (R)-1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-ol ((R)-2a) and (S)-N-(2,6-dichloro-4-(1-hydroxyethyl)phenyl)acetamide ((S)-6a).
Clenbuterol (Ventipulmin®, Boehringer Ingelheim Vetmedica, Inc., Duluth, GA, USA) is a long-acting, selective β2-agonist with anabolic properties used in the treatment of shock and airway-obstructing diseases in veterinary medicine [7,8]. It has not been approved by the U.S. Food and Drug Administration (FDA) for human or food-producing animals use, although it is illegally added to livestock feed to promote the growth of lean meat [9]. Clenbuterol can reduce body fat, and it is extensively used by athletes and bodybuilders even though it is listed on the World Antidoping Agency’s prohibited list [10]. In addition to the desired effects of smooth-muscle relaxation, clenbuterol can cause several side effects, such as heart palpitations, muscle tremors, and nervousness [8].
Previously, it was reported that (S)-clenbuterol had neuroprotective properties, reduced blood pressure, and enhanced blood-glucose levels in rats. While (R)-clenbuterol did not promote these properties, it was found that it caused decreased motor activity, head twitches, and tremors [11,12]. Upon examination of human urine of participants who ingested racemic clenbuterol, it was found that the S-enantiomer was retained for longer in the body [13]. Recently, Mittal et al. found that the brains of patients with Parkinson´s disease contained accumulations of α-synuclein protein, the so-called Lewy bodies. β2-agonists metaproterenol, clenbuterol, and salbutamol lowered the levels of α-synuclein gene mRNA and α-synuclein protein levels in human SK-N-MC neuroblastoma cells [14]. Only racemic compounds have been studied, but pure enantiomers of the drugs should be independently studied in order to reveal if enantiomers would show different effects.
Racemic clenbuterol has been synthesised through the incorporation of carbon monoxide in aryl iodides [15], while racemic 13C-labeled clenbuterol has been synthesised from acetanilide [16]. Enantiopure (R)-1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-ol, a precursor for (R)-clenbuterol, has been synthesized through an asymmetric reduction of the parent ketone, catalysed by an alcohol dehydrogenase from Rhodococcus erythropolis. No enantiomeric ratio was reported [17].
Our attempts to synthesize enantiopure precursors for (R)-clenbuterol include both lipase catalysed kinetic resolutions of racemates and asymmetrizations of substituted acetophenones catalysed by ketoreductases. Previously, we utilised chemoenzymatic methods in syntheses of enantiopure building blocks for compounds that are related to clenbuterol [18,19].
2. Results
In order to obtain a precursor for enantiopure (R)-clenbuterol, bromo alcohol 1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-ol (3) was synthesised in a 45% yield (Scheme 1) for the purpose of performing lipase-catalyzed kinetic resolution with lipases suitable for industrial-scale production. Bromination of substrate ketone 1 produced bromoketone 2 in an 86% yield. Various methods for the α-bromination of ketone 1 were performed: Addition of N-bromosuccinimide (NBS) (1.0 eq.) to 1 with para-toluenesulfonic acid (0.1 eq.) in methanol at 30 °C produced brominated ketone 2 in an 11% yield. Bromination of 1 with bromine (1.1 eq.) in acetic acid at 60 °C gave ketone 2 in a 27% yield, while reacting substrate 1 with NBS (1.3 eq.) catalysed by silica gel (10% w/w) in refluxing methanol produced 2 in a 30% yield. Finally, we were able to produce α-bromo ketone 2 in an 86% yield by the slow addition of Br2 (1.1 eq.) to 1 in dichloromethane and methanol (2:1 v/v) at room temperature. Subsequent reduction of 2 with sodium borohydride gave 3 in a 45% yield.
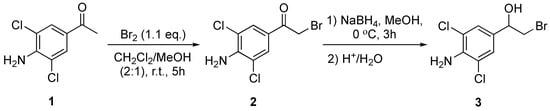
Scheme 1.
Synthesis of 1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-one (2) and 1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-ol (3) from commercial 1-(4-amino-3,5-dichlorophenyl)ethan-1-one (1).
2.1. Kinetic Resolutions
The kinetic resolutions of 3 were performed in dichloromethane with vinyl butanoate as the acyl donor catalysed by commercial immobilised lipases A and B from Candida antarctica (CALA and CALB, Sigma-Aldrich, Oslo, Norway). The reactions showed no conversion after five days. We also attempted to resolve racemic clenbuterol with CALA and CALB from Sigma-Aldrich and vinyl butanoate as the acyl donor in tetrahydrofuran, Scheme 2. However, these reactions resulted in several acylation products after five days and the enzyme showed low enantioselectivity.
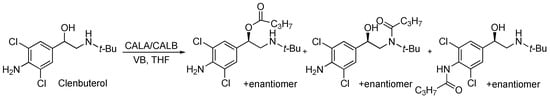
Scheme 2.
Kinetic resolution of clenbuterol catalysed by commercial immobilised lipases A and B from Candida antarctica (CALA and CALB) in dry tetrahydrofuran (THF) with vinyl butanoate (VB) as acyl donor, showed several acylation products. The E-value for the desired acylation was E = 1.3 after five days.
The electropherogram from the CALA-catalysed kinetic resolution of clenbuterol after 78 h (Figure 2) shows the peaks of (R)-clenbuterol at retention times (tR) 9.70 min and (S)-clenbuterol at tR 11.50 min, and the butanoic esters of (S)-clenbuterol at tR 12.52 min and ester of (R)-clenbuterol at tR 12.98 min. Elution orders are consistent with previously reported retention times [20]. The remaining peaks have not been identified, but are most likely the amide products from the acylation of the aromatic or aliphatic amines, which would produce two enantiomers each.
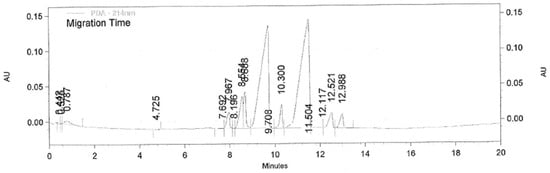
Figure 2.
Electropherogram from the kinetic resolution of clenbuterol, catalysed by CALA with vinyl butanoate in tetrahydrofuran. tR (R)-Clenbuterol 9.70 min, tR (S)-clenbuterol at 11.50 min, and the butanoic esters (R)- and (S)-clenbuterol at tR 12.52 min and tR 12.98 min, respectively.
We anticipate that the α-bromo substituent in 3 could be one reason for the low selectivity and low reaction rate in the transesterification. In order to reveal the influence of an electron-rich substituent next to the stereocenter compared to bromine, transesterification of the propargyl alcohol 8 was performed in dry hexane with vinyl butanoate as the acyl donor and CALB as the catalyst (Scheme 3). The catalyst showed excellent enantioselectivity towards 8 after 2.5 h with an E-value >200 resulting in the conversion of (R)-8 to (R)-9 with no sign of (R)-8 on the chromatogram, giving (S)-8 in 99% ee. The reaction progress is shown in Figure 3. From this test reaction, it seems that either the chlorosubstituents in meta-position or the amino substituent in para-position on the benzene ring on 3 must be the reason for the low selectivity and slow esterifications of 3 and clenbuterol with CALB.
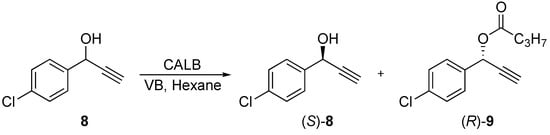
Scheme 3.
Kinetic resolution of 9 with CALB as the catalyst and vinyl butanoate in dry hexane.
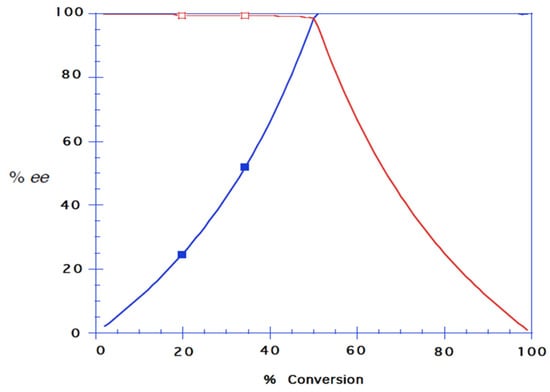
Figure 3.
Reaction progress of kinetic resolution of 8 with CALB in dry hexane with vinyl butanoate as acyl donor, E ≥ 200. Both the butanoate ester enantiomer of 8, (R)-9 and the remaining alcohol (S)-8 were obtained in 99% ee. Chiral GLC and HPLC analyses gave ees- and eep-values from which the degree of conversion was calculated according to c = ees/(ees + eep).
2.2. Asymmetrisations
Asymmetrisations of ketones with suitable enzymes is another way of obtaining enantiopure compounds. Depending on the catalysts stereoselectivity towards a chosen substrate, a theoretical yield of 100% might be obtained. Scheme 4 shows the asymmetrisation of ketones 1, 2, and 4–7 with ketoreductase KRED 228 (Syncozymes Co., Ltd., Shanghai, China). Several regeneration systems were tested in order to find a suitable system to regenerate nicotinamide adenine dinucleotide phosphate (NADP+), and we were successful with glucose-6-phosphate dehydrogenase with glucose-6-phosphate as the cosubstrate [21,22]. The cosolvent was DMSO in all asymmetrisation reactions.
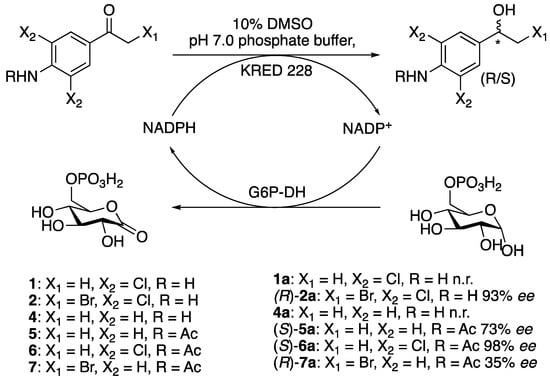
Scheme 4.
Reduction of ketones 1, 2, and 4–7 with ketoreductases (KRED 228 from Syncozymes Co., Ltd.) and glucose-6-phosphate dehydrogenase (G6P-DH) as the regeneration system for NADP+ with glucose-6-phosphate (G6P) as the cosubstrate in phosphate buffer with 10% dimethyl sulfoxide (DMSO).
Through the reduction of 1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-one (2) with KRED 228, (R)-2a was obtained in 93% ee. Reduction of N-(4-acetylphenyl)acetamide (5) by the same enzyme gave the S-alcohol (S)-N-(4(1-hydroxyethyl)phenyl)acetamide, (S)-5a, in 73% ee. Reduction of N-(4-acetyl-2,6-dichlorophenyl)acetamide, 6, produced the S-alcohol (S)-1-(4-amino-3,5-dichlorophenyl)ethan-1-ol, (S)-6a, in 98% ee. The reduction of N-(4-(2-bromoacetyl)phenyl)acetamide (7) gave an ee of 35% of the alcohol 7a, with R-configuration. The asymmetrisations of ketones 1 and 4 with the use of the same system were not successful. Asymmetrisations of the above-mentioned ketones were also performed with S. cereviciae and several alcohol dehydrogenases without any success. Valuable enantiopure synthons could be synthesised from (S)-6a (see suggestions in Scheme 5).
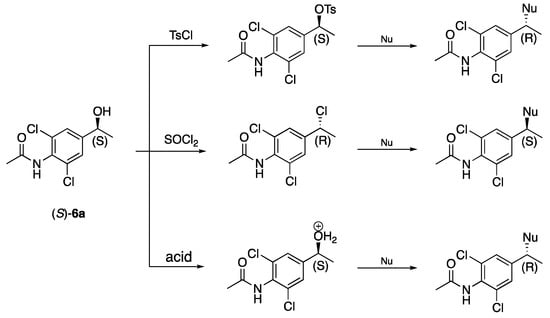
Scheme 5.
Stereoconfiguration of products from nucleophilic substitutions of enantiopure (S)-6a depends on the nucleophile (Nu).
3. Materials and Methods
Analytical-grade chemicals and solvents were purchased from Sigma-Aldrich (Oslo, Norway). HPLC-grade solvents were purchased from Sigma-Aldrich. Dry solvents (THF and CH2Cl2) were acquired from a MBraun MB-SPS-800 solvent purification system (MBraun, München, Germany). Candida antarctica lipase A (activity 1612 U/g; lot no. BCBR2259V), lipase B (activity 1800 U/g, lot no BCBP3525V) immobilised on Immobead 150, recombinant from Aspergillus oryzae and glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides (G6P-DH, ≥550 U/mg, lot no. 107K7700V) were also purchased from Sigma-Aldrich. Crystalline KRED 228 (331.9 U/g, lot no. 20160509) was from Syncozymes Co., Ltd., (Shanghai, China). Flash chromatography was performed with silica gel from Sigma-Aldrich (pore size 60 Å, 230–400 mesh, 40–63 m particle size). Ketones 1, 4, 7 and clenbuterol were bought from Sigma-Aldrich (Oslo, Norway).
The enzyme-catalysed kinetic resolutions were performed in a New Brunswick G24 Environmental Incubator Shaker (New Brunswick Co. Inc., Edison, NJ, USA) at 30 °C and 200 or 300 rpm. Optical rotations ([α]D) were determined at 20 °C using a Perkin-Elmer 343 instrument (Perkin Elmer, Waltham, MA, USA) concentrations are given in g/100 mL. NMR spectra were recorded with a Bruker Avance DPX 400 instrument (Bruker, Rheinstetten, Germany) operating at 400 MHz for 1H and 100 MHz for 13C, respectively. Chemical shifts are in ppm rel. to tetramethylsilane (TMS) and coupling constants are in Hertz (Hz). High-resolution MS was performed on a WatersSynapt G2-S Q-TOF instrument (Waters MS Technologies, Manchester, UK) with WatersTM Software (Masslynx V4.1 SCN871, (Waters MS Technologies, Manchester, UK). Sample ionization was performed with an ASAP probe (APCI).
3.1. Achiral Chromatographic Analyses
Achiral GLC analyses were performed on an Agilent 7890A gas chromatograph, with an Agilent 7890B autosampler, a split injector (280 °C), a 4 mm ID tap GW liner (Agilent Technologies, Santa Clara, CA, USA), a flame ionisation detector (FID, 280 °C). All analyses were performed on a Restek Rtx®-1701 column (Crossbond® 14% cyanopropylphenyl–86% dimethylpolysiloxane, 30 m × 0.32 mm ID, df 0.25 μm, (Restek Corporation, Bellefonte, PA, USA), carrier gas He 5.0, temp. prog.: 100–280 °C/10 °C min−1.
3.2. Chiral Analyses
3.2.1. Chiral GLC-Analyses
The chiral GLC analyses of 3 and 6a were performed on an Agilent 7890B gas chromatograph (Agilent Technologies, Palo Alto, CA, USA), with an Agilent 7890B autosampler, an Agilent G4513A–7693A autoinjector, a split injector (225–230 °C) and a flame ionization detector (FID, 275 °C). The enantiomers of alcohol 3 were separated on a CP-Chirasil-Dex CB column (24.3 × 0.25 mm ID, df 0.25 μm) from Agilent technologies (Santa Clara, CA, USA) with He 5.0 as the carrier gas, flow 81 min−1, split flow 75 mL min−1 (25:1), temp. prog.: 100–150 °C/10 °C min−1, 150–160 °C/3 °C min−1, 160–170 °C/1 °C min−1. Retention times: tR (R)-3 = 25.363 min, tR (S)-3, RS = 1.82.
Separation of enantiomers of 6a was performed on the same system: flow 75 mL min−1, split flow 1:30, temp. prog.: 100–160 °C/10 °C min−1, 160–170 °C/1 °C min−1, 170–185 °C/0.5 °C min−1. Retention times tR (S)-6a = 44.467 min, tR (R)-6a = 45.255 min, RS = 2.05.
The chiral GLC analyses of 8 were- performed on a Varian 3380 gas chromatograph (Varian Inc., Mississauga, ON, USA), with a Varian CP-8410 autosampler, a split injector (200 °C), and a flame ionization detector (FID, 200 °C). The enantiomers of alcohol 8 and butanoic ester 9 were separated on a CP-Chirasil-Dex CB column (25 × 0.25 mm ID, df 0.25 μm) from Agilent technologies. Carrier gas H2 5.0, gas pressure 8 psi, split flow 60 mL/min, temp prog 100–140 °C/10 °C min−1 (5), 140–180 °C/10 °C min−1 (7). Retention times tR (S)-8 = 12.690 min, tR (R)-8 = 12.901 min, RS = 3.5, tR (S)-9 = 13.162 min, tR (R)-9 = 14.359 min, RS > 1.5.
3.2.2. Chiral HPLC Analyses
HPLC analyses were performed on an Agilent HPLC 1100 system (Nacalai tesque, Japan) with a manual injector (Rheodyne 77245i/Agilent 20 μL loop), and a variable wavelength detector (VWD) set to 254 nm. Alcohol 5a was separated on a Chiralcel OD-H column (250 mm L × 4.6 mm ID, 5 μm particle size, Daicel, Chiral Technologies Europe, Illkirch-Graffenstaden, France). Eluent: 7% propan-2-ol/93% n-hexane; 1 mL min−1, 1.5 μL injected. Retention times: tR (R)-5a = 84.474 min, tR (S)-5a = 91.765 min, RS = 1.56.
Alcohol 7a was separated on the same column, eluent: 17% propan-2-ol/83% n-hexane; 1 mL min−1, 2 μL injected. Retention times: tR (R)-7a = 14.037 min, tR (S)-7a = 17.035 min, RS = 2.55.
3.2.3. Capillary Zone Electrophoresis
The chiral CZE analyses of clenbuterol and the corresponding butanoate were performed on a Beckman P/ACE MDQ capillary electrophoresis system (Beckman Coulter, Inc., Brea, CA, USA) in a fused silica capillary (31 cm × 50 μm, 21 cm to the detection window) with a diode array detector (DAD, 214 nm). Running buffer: pH 2.5 phosphate buffer (0.100 M), chiral selector: highly sulfonated β-cyclodextrin (5% w/v). Injection mode: hydrodynamic injection (0.5 psi for 5 s), separations voltage: −10 kV. Migration times: tM (R)-clenbuterol 9.708 min, tM (S)-clenbuterol at 11.504 min, tM butanoates: tM (R)-ester 12.521 min, tM (S)-ester 12.988 min.
3.3. Determination of Enantiomeric Excess (ee), Conversion (c), and Enantiomeric Ratio (E)
Chiral GLC and HPLC analyses gave ees- and eep-values from which the degree of conversion was calculated according to c = ees/(ees + eep). Enantiomeric ratios, E, were calculated based on ping-pong bi-bi kinetics using the computer program E and K Calculator 2.1b0 PPC (Department of chemistry, NTNU, Trondheim, Norway) [23]. Two or more replicates of the transesterification and asymmetrisation reactions were performed.
3.4. Absolute Configurations
The absolute configurations of (R)-2a, (S)-6a, and (R)-9 were determined by comparing the elution orders of the enantiomers with GLC elution orders of similar enantiopure compounds synthesised from (S)-epichlorohydrin. Elution orders on Daicel Chiralcel OD-H of the faster reacting enantiomers were the same [18,24,25]. Optical rotation values of (R)-2a, (S)-5, (S)-6a and (R)-7a have not been previously reported.
3.4.1. Synthesis of 1-(4-Amino-3,5-dichlorophenyl)-2-bromoethan-1-one (2)
To a solution of 1-(4-amino-3,5-dichlorophenyl)ethan-1-one (1) (1.10 g, 5.39 mmol) dissolved in CH2Cl2 (20 mL) and MeOH (10 mL), a solution of Br2 (0.167 mL, 3.23 mmol) in CH2Cl2 (6 mL) was added at a rate of 40 drops per minute under strong agitation at RT. After an observed color change from bromine red to pale yellow, two additional portions of Br2 (0.056 mL, 1.09 mmol) were added as described above. After the observed color change, the product was confirmed by TLC: Rf (2) = 0.58 (1:3 n-pentane: CH2Cl2). The reaction mixture was washed with satd. K2CO3 (2 × 40 mL) and brine (2 × 40 mL) before the organic layers were dried over MgSO4 and filtered before the solvents were removed under reduced pressure. The crude compound was stirred in EtOH (4.4 mL) at 50 °C for 30 min, and for an additional 60 min at RT. The solids were filtered and recrystallised from EtOAc. The crystals were dried under vacuum overnight to afford 2 in 86% yield (1.04 g, 4.22 mmol) with 98% purity (GLC). 1H NMR (400 MHz, CDCl3): δ 7.86 (s, 2H, Harom), 5.05 (s, 2H, NH2), 2.31 (s, 2H, CH2-Br). 13C NMR (600 MHz, CDCl3): δ 188.0, 144.9, 129.2, 124.0, 188.9, 29.8. MS (TOF-ASAP): [M + H]+ 281.9088 m/z.
3.4.2. Synthesis of 1-(4-Amino-3,5-dichlorophenyl)-2-bromoethan-1-ol (3)
1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-one (2) (0.50 g, 1.77 mmol) dissolved in EtOH (12 mL) at 0 °C was added NaBH4 (0.136 g, 3.60 mmol) in 4 portions over 20 min, under stirring. The reaction was slowly heated to RT, and the conversion was monitored by TLC: Rf (3) = 0.43 (1:3 n-pentane/CH2Cl2). After full conversion of the starting material, the reaction was quenched with dilute HCl until gas formation stopped. H2O and EtOH were removed through azeotropic distillation, and the solids were dissolved in EtOAc (50 mL), and extracted with satd. NaHCO3 (3 × 30 mL) and brine (3 × 30 mL). The aqueous layers were back extracted with EtOAc (2 × 50 mL), and the combined organic layers were dried over MgSO4. The solvent was removed under reduced pressure, and the crude product was purified by flash chromatography (1:3 n-pentane/CH2Cl2) to afford 3 as a light-yellow solid in 44% yield (0.22 g, 0.77 mmol) in 95% purity (GLC). 1H NMR (400 MHz, CDCl3): δ 7.22 (s, 2Harom), 4.80–4.76 (dd, 1H, CHOH, 3JHH = 3.4 Hz, 8.9 Hz), 4.49 (s, 2H, NH2), 3.59–3.55 (dd, 1H, CH-Br, 2JHH = 10.5 Hz, 3JHH = 3.4 Hz), 3.49–3.45 (dd, 1H, CH-Br, 3JHH = 8.9 Hz, 2JHH = 10.4 Hz). 13C NMR (400 MHz, CDCl3): δ 140.1, 130.4, 125.6, 119.6, 72.6, 39.8. MS (TOF-ASAP) [M + H]+ 283.9241 m/z.
3.4.3. Synthesis of N-(4-Acetyl-phenyl)acetamide (5)
To a stirred solution of 1-(4-aminophenyl)ethan-1-one (0.53 g, 3.92 mmol) in CH2Cl2 (15 mL), a solution of AcCl (0.56 mL, 7.84 mmol) in CH2Cl2 (2 mL) was added dropwise at RT. After 5 min, a white precipitate started to form, and a solution of Et3N (1.09 mL, 7.84 mmol) in CH2Cl2 (2 mL) was added dropwise, after which the solution cleared and turned a dark-yellow color. Full conversion was observed after 24 h by TLC: Rf (5) = 0.05 (1:4 EtOAc/n-pentane), and the solution was extracted with brine (3 × 15 mL). The aqueous layers were combined and extracted with EtOAc (2 × 15 mL), before the combined organic layers were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. The crude product was recrystallised from EtOAc to afford 5 in 53% yield (0.37 g, 2.09 mmol) and 98% purity (GLC). 1H NMR (600 MHz, CDCl3): δ 7.95–7.93 (m, 2Harom, 2JHH, ortho = 8.7 Hz, 3JHH, meta = 2.5 Hz), 7.62–7.61 (d, 2H, Harom, 2JHH = 8.3 Hz), 7.42 (s, 1H, NHR), 2.58 (s, 3H, CO-CH3), 2.22 (s, 3H, NCO-CH3). 13C NMR (600 MHz, CDCl3): δ 196.9, 168.4, 142.2, 132.9, 129.8, 118.8, 26.4, 24.8. MS (TOF-ASAP) [M + H]+ 178.0867 m/z.
3.4.4. Synthesis of N-(4-Acetyl-2,6-dichlorophenyl)acetamide (6)
To a stirred solution of 1-(4-amino-3,5-dichlorophenyl)ethan-1-one (1) (6.00 g, 29.40 mmol) in CH2Cl2 (150 mL) AcCl (10.49 mL, 147.01 mmol) in CH2Cl2 (15 mL) was added. To the stirred solution Et3N (4.29 mL, 30.78 mmol) in CH2Cl2 (15 mL) was added dropwise. The reaction was monitored by TLC. Rf (6) = 0.64 (1:4 EtOAc/n-pentane). After 48 h, full conversion was observed. The reaction mixture was washed with satd. K2CO3 (2 × 100 mL) and brine (2 × 100 mL), before the organic layers were collected and dried over anhydrous MgSO4, filtered, and the solvent removed under reduced pressure. The crude product was recrystallized from EtOAc, and the purified compound dried in vacuo overnight to afford 6 as white crystals in 58% yield (4.20 g, 17.07 mmol) in 99% purity. 1H NMR (400 MHz, CDCl3): δ 7.86 (s, 2H, Harom), 7.42 (s, 1H, NHR), 2.56 (s, 3H, CO-CH3), 2.22 (s, 3H, NCO-CH3). 13C NMR (400 MHz, CDCl3): δ 195.0, 168.4, 136.7, 136.4, 133.9, 128.3, 26.6, 23.2. MS (TOF-ASAP) [M + H]+ 246.0089 m/z.
3.4.5. Synthesis of 1-(4-Chlorophenyl)prop-2-yn-1-ol (8)
A solution of ethynylmagnesium bromide (0.50 M in THF; 18 mL, 9.0 mmol) was cooled to −20 °C. A solution of 4-chlorobenzaldehyde (1.0105 g, 7.19 mmol) in THF (2.5 mL) was added, and the flask washed out with THF (0.5 mL), which was then added to the reaction mixture. The cooling bath was removed, and the reaction mixture was stirred at room temperature (RT) for 5 min. The reaction was quenched by addition of satd. aq. NH4Cl (10 mL), and the mixture was extracted with Et2O (2 × 20 mL). The combined organic extracts were dried with Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (gradient 1:10–1:3 EtOAc/pentane) to give 8 in 90% yield (1.078 g, 6.47 mmol) as a pale yellow oil. Rf = 0.47 (1:3 EtOAc/pentane). 1H NMR (400 MHz, CDCl3): δ = 7.51–7.48 (m, 2H, Harom), 7.38–7.35 (m, 2H, Harom), 5.45 (dd, J = 6.1, 2.1 Hz, CHC≡), 2.68 (d, J = 2.2 Hz, ≡CH), 2.21 (d, J = 6.1 Hz, OH) ppm. 1H NMR spectroscopic data are in accordance with the literature data [26].
3.5. Kinetic Resolution of Secondary Alcohols with Lipases
3.5.1. Kinetic Resolution of Clenbuterol
1-(4-Amino-3,5-dichlorophenyl)-2-(tert-butylamino)ethan-1-ol (clenbuterol, 31.2 mg, 0.11 mmol) dissolved in dry THF (4.0 mL) was added 3 pellets of molecular sieve, and vinyl butanoate (0.058 mL, 0.55 mmol) before the mixture was incubated at 30 °C and 200 rpm and the reaction was started by addition of lipase A from Candida antarctica (CALA, 55.4 mg). Aliquots of 100 L were collected every 30 min for the first 5 h, then every hour for 3 h, and every 24 h for 5 days. The solvent in the aliquots was removed with N2 before 0.100 M pH 2.5 phosphate buffer (0.5 mL) was added, and the samples were analysed by chiral CE, from which the enantiomeric excess of (R)-clenbuterol was calculated to 11% ee.
3.5.2. Kinetic Resolution of 1-(4-Amino-3,5-dichlorophenyl)-2-bromoethan-1-ol (3)
To a solution of racemic 1-(4-amino-3,5-dichlorophenyl)-2-bromoethan-1-ol (3, 55 mg, 0.19 mmol) and vinyl butanoate (0.1225 mL, 0.97 mmol) in CH2Cl2 (3 mL), lipase B from Candida antarctica (CALB, 55 mg) was added before the solution was placed in an incubator set to 30 °C and 200 rpm. Aliquots of 100 L were collected every 30 min for the first 4 h, then at every 12 h for 3 days, and finally every 24 h for 4 days. The aliquots were analysed by chiral GLC, and no conversion was observed. A new reaction was performed with CALA (60 mg), and aliquots were collected as described above, then analysed by chiral GLC. No conversion was observed.
3.5.3. Kinetic Resolution of 1-(4-Chlorophenyl)prop-2-yn-1-ol (8)
To a solution of 1-(4-chlorophenyl)prop-2-yn-1-ol (8) (20.0 mg, 0.12 mmol), and vinyl butanoate (0.069 mL, 0.60 mmol) in dry n-hexane (3.0 mL), 5 pellets of molecular sieve were added before the mixture was incubated at 40 °C and 200 rpm. CALB (22.1 mg) was added to the mixture, and aliquots (0.100 mL) were collected every 30 min for the first 3 h, and finally after 19.5 h. The samples were analysed by chiral GLC: tR (S)-8 = 12.880 min, tR (R)-8 = 13.087 min, RS = 3.5. After 2.5 h, (R)-8 was not observed by GLC. The ester (R)-9 was produced in 99% ee.
3.6. Asymmetrisations of Ketones by Ketoreductases
3.6.1. General Procedure
The ketones 1, 2 and 4–7 (2.5–60.8 mg, 0.01–0.16 mmol) were dissolved in DMSO (0.10 mL) and transferred to a solution of ketoreductase 228 (KRED 228), NADPH, glucose-6-phosphate (G6P) and glucose-6-phosphate dehydrogenase (G6P-DH) in pH 7.0 phosphate buffer (0.9 mL). The reactions were incubated at 30 °C and 300 rpm for 1–28 days, and monitored by TLC (1:2 n-pentane/EtOAc) every 24 h. After full conversion of the starting material, the mixtures were extracted with EtOAc (4 × 10 mL) and the organic phases were combined, washed with water (2 × 20 mL), dried over MgSO4, and filtered before the organic solvent was removed under reduced pressure. The crude products were either purified by flash chromatography (silica, EtOAc/n-pentane) to afford the pure alcohols, or the crude product was analysed by chiral GLC or chiral HPLC.
3.6.2. Asymmetric Synthesis of (R)-1-(4-Amino-3,5-dichlorophenyl)-2-bromoethan-1-ol, (R)-2a
1-(4-Amino-3,5-dichlorophenyl)-2-bromoethan-1-one (2) (28.6 mg, 0.10 mmol) in DMSO (0.10 mL), KRED 228 (10.4 mg), G6P (77.7 mg), G6P-DH (0.1 mL, 20 U) and NADPH (0.3 mg) in pH 7.0 phosphate buffer (0.90 mL). Reaction time: 4 weeks, (R)-2a was obtained in 93% ee. GLC of rac-2: tR (R) = 25.363 min, tR (S) = 25.636 min, RS = 1.9.
3.6.3. Asymmetric Synthesis of (S)-N-(4-Acetyl-phenyl)acetamide, (S)-5a
N-(4-Acetylphenyl)acetamide (5) (20.3 mg, 0.11 mmol) in DMSO (0.10 mL), KRED 228 (7.2 mg), G6P (94.1 mg), G6P-DH (0.1 mL, 20 U) and NADPH (0.3 mg) in pH 7.0 phosphate buffer (0.90 mL) was incubated for 2 weeks. After flash chromatography, (100% EtOAc,), (S)-5a was obtained in a 45% isolated yield, (8.8 mg, 0.05 mmol), 96% purity, 73% ee. Rf (S)-5 = 0.42 (EtOAc). HPLC of rac-5: tR (R) = 84.474 min, tR (S) = 91.765 min, RS =1.56. = −0.11 (c 0.88, EtOH). 1H NMR (600 MHz, CD3OD): δ 7.50–7.49 (d, 2H, Harom, 3JHH = 8.4 Hz), 7.31–7.30 (d, 2H, Harom, 3JHH = 8.4 Hz), 4.80–4.77 (q, 1H, HO-CH, 3JHH = 6.5 Hz), 2.11 (s, 3H, CO-CH3), 1.42–1.41 (d, 3H, CH-CH3, 3JHH = 6.5 Hz) 13C NMR (600 MHz, MeOD): δ 171.6 (CO), 143.4 (C), 138.8 (C), 126.9 (CHarom), 121.1 (CHarom), 70.5 (CH), 25.4 (CH3), 23.7 (CH3). MS (TOF-ASAP): [M + H]+ 180.1025 m/z.
3.6.4. Asymmetric Synthesis of (S)-N-(2,6-dichloro-4-(1-hydroxyethyl)phenyl)acetamide, (S)-6a
N-(4-Acetyl-2,6-dichlorophenyl)acetamide (6) (32.1 mg, 0.13 mmol) in DMSO (0.10 mL), KRED 228 (8.1 mg), G6P (74.4 mg, 0.286 mmol), G6P-DH (0.1 mL, 20 U) and NADPH (0.2 mg) in phosphate buffer (pH 7.0, 0.90 mL). Reaction time 48 h, flash chromatography (1:2 n-pentane/EtOAc), isolated yield (S)-6a: 31% yield (10.1 mg, 0.04 mmol), 99% purity, >98% ee. GLC of rac-6: tR (S)-6a = 44.46 min, tR (R)-6a = 45.25 min, RS = 2.05. (S)-6a = −0.21 (c 1.01, EtOH). 1H NMR (600 MHz, MeOD,): δ 7.45 (s, 2H, Harom), 4.82–4.79 (q, 1H, CH-OH, 3JHH = 6.3 Hz), 2.17 (s, 3H, CO-CH3), 1.42–1.41 (d, 3H, CH(OH)-CH3, 3JHH = 6.3 Hz). 13C NMR (100 MHz MeOD,): δ 172 (CO), 150 (C), 135.2 (C), 132.2 (C), 126.5 (CHAr), 69.4 (CH), 25.4 (CH3), 22.3 (CH3). MS: (ASAP-TOF) [M + H]+ 248.0245 m/z.
3.6.5. Asymmetric Synthesis of (R)-N-(4-(2-Bromo-1-hydroxyethyl)phenyl)acetamide, (R)-7a
N-(4-(2-Bromoacetyl)phenyl)acetamide (7) (27.1 mg, 0.11 mmol) in DMSO (0.10 mL), KRED 228 (8.7 mg), G6P (82.9 mg, 0,319 mmol), G6P-DH (0.1 mL, 20 U) and NADPH (0.4 mg) in phosphate buffer (pH 7.0, 0.90 mL). Two days reaction time, flash chromatography (100% EtOAc), Rf (R)-7a = 0.57, isolated yield: 27% (7.3 mg, 0.03 mmol), 98% purity, 35% ee. HPLC of rac-7: tR (R) = 14.037 min, tR (S) = 17.035 min, RS = 2.55. = +0.07 (c 0.73, EtOH). 1H NMR (600 MHz, CD3OD): δ 7.56–7.55 (m, 2H, Harom, 3JHH = 8.7 Hz), 7.36–7.35 (m, 2H, Harom, 3JHH = 8.6 Hz), 4.85–4.83 (m, 1H, HO-CH, 3JHH = 4.6 Hz, 3JHH = 7.7 Hz), 3.62–3.60 (dd, 1H, CH-Br, 3JHH = 4.4 Hz, 2JHH = 10.5 Hz), 3.56–3.53 (dd, 1H, CH-Br, 3JHH = 7.7 Hz, 2JHH = 10.5 Hz), 2.13 (s, 3H, CO-CH3). 13C NMR (600 MHz, CD3OD): δ 170.2, 138.2, 137.6, 126.4, 119.6, 73.1, 37.8, 22.4. MS (TOF-ASAP): [M + H]+ 258.0130 m/z.
4. Conclusions
Asymmetrisation of ketones 2 and 6 with KRED 228 were the only asymmetrisations in the series of ketones 1, 2, and 4–7 that produced the respective alcohol with high enantiomeric excess, giving (R)-2a in a 93% ee (low yield) and (S)-6a in a 98% ee and 31% yield. (R)-2a may be reacted with t-butylamine, yielding (R)-clenbuterol in high enantiomeric excess. (S)-6a may be a precursor for important enantiopure compounds.
The absolute configuration of the alcohols was determined by comparing elution orders on GLC with similar compounds. KRED 228 from Syncozymes Co., Ltd. was found to be a suitable enzyme for the reduction of substitued arylketones with several substituents on the benzene ring, however when an α-halogen is present the enzyme is not fully efficient. In addition, protection of the amino group on the benzene ring seems to be cruicial.
Kinetic resolutions of the racemic bromo alcohol 3 with CALA and CALB were not successful. The transesterification reactions showed low reactivity and low stereoselectivity with both lipases. Due to the excellent selectivity of the CALB catalysed kinetic resolution of propargyl alcohol 8 with vinyl butanoate in hexane it was anticipated that both the amino and the chloro substituents in meta-positions of 3 cause the problem. Both steric hindrance and electronic effects of these substituents leading to an irreversible substrate-enzyme complex might be the reason for the low conversion of the two lipases.
Author Contributions
Investigation and writing—original draft preparation, F.H.B.; supervision and writing—review and editing, E.E.J.; investigation, M.B.H., S.E., and W.Z.
Acknowledgments
We thank EU COST ACTION CM1303 Systems Biocatalysis for the support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Palmer, T. Understanding Enzymes, 4th ed.; Prentice Hall/Ellis Horwood: Chichester, UK, 1995; p. 398. ISBN 9780131344709. [Google Scholar]
- Pfeiffer, C.C. Optical isomerism and pharmacological action, a generalization. Science 1956, 124, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, P.A; Rodriegues de Miranda, J.F.; Ariëns, E.J. Stereoselectivity and Affinity in Molecular Pharmacology. In Progress in Drug Research/Fortschritte der Arzneimittelforschung/Progrés des recherches pharmaceutiques; Birkhäuser: Basel, Switzerland, 1976; Volume 20, pp. 101–142. ISBN 978-3-0348-7094-8. [Google Scholar]
- Kearns, C.F.; McKeever, K.H. Clenbuterol and the horse revisited. Vet. J. 2009, 182, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Thomson, N.C. (R)-salbutamol in the treatment of asthma and chronic obstructive airways disease Expert Opin. Pharmacother. 2011, 12, 1133–1141. [Google Scholar] [CrossRef]
- Skachilova, S.Ya.; Shilova, E.V.; Chuchalin, A.G. Prospects for the development of bronchodilators. Russ. Chem. Bull. 2015, 64, 2022–2035. [Google Scholar] [CrossRef]
- Hieger, M.A.; Emswiler, M.P.; Maskell, K.F.; Sentz, J.T.; Miller, K.B.; Wolf, C.E.; Cumpston, K.L.; Wills, B.K. A case series of clenbuterol toxicity caused by adulterated heroin. J. Emerg. Med. 2016, 51, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Z.; Jiang, H.Y. Residues of β-andrenergic agonist in Animal Products and its Hazards. China Anim. Health Insp. 2011, 28, 27–28. [Google Scholar]
- Xiao, Y.-P.; Lu, H.-Y.; Lü, S.-J.; Xie, S.-X.; Wang, Z.-Z.; Chen, H.-W. Rapid Analysis of Trace Salbutamol and Clenbuterol in Pork Samples by Mass Spectrometry. Chin. J. Anal. Chem. 2016, 44, 1633–1638. [Google Scholar] [CrossRef]
- Prohibited List 2017. Available online: https://www.wada-ama.org/ (accessed on 18 October 2018).
- Culmsee, C.; Junker, V.; Thal, S.; Kremers, W.; Maier, S.; Schneider, H.J.; Plesnila, N.; Krieglstein, J. Enantio-selective effects of clenbuterol in cultured neurons and astrocytes, and in a mouse model of cerebral ischemia. Eur. J. Pharmacol. 2007, 575, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Puech, A.-J.; Brochet, D.; Soubrié, P.; Simon, P. Comparison of clenbuterol enantiomers using four psychopharmacological tests sensitive to β-agonists. Eur. J. Pharmacol. 1985, 117, 127–129. [Google Scholar] [CrossRef]
- Thevis, M.; Thomas, A.; Beuck, S.; Butch, A.; Dvorak, J.; Schänzer, W. Does the analysis of the enantiomeric composition of clenbuterol in human urine enable the differentiation of illicit clenbuterol administration from food contamination in sports drug testing? Rapid Commun. Mass Spectrom. 2013, 27, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.U.; Neumann, K.; Taaning, R.H.; Lindhardt, A.T.; Modvig, A.; Skrydstrup, T. Palladium-Catalyzed Double Carbonylation Using Near Stoichiometric Carbon Monoxide: Expedient Access to Substituted 13C2-Labeled Phenethylamines. J. Org. Chem. 2012, 77, 6155–6165. [Google Scholar] [CrossRef] [PubMed]
- González-Antuña, A.; Lavandera, I.; Rodríguez-González, P.; Rodríguez, J.; García Alonso, J.I.; Gotor, V. A straightforward route to obtain 13C1-labeled clenbuterol. Tetrahedron 2011, 67, 5577–5581. [Google Scholar] [CrossRef]
- Braun, M.; Leaux, B.; De Lacroix, B.F.; Na’Amnieh, S. Preparation of Optically Active 1-Hydroxy-2-alkylaminoethane Compounds, Useful as Bronchodilators for Treating e.g. Asthma, by Reacting an Amine with Reactive 2-Substituted Ethanol, also New Intermediates. DE10248277A1, 6 May 2004. [Google Scholar]
- Lund, I.T.; Bøckmann, P.L.; Jacobsen, E.E. Highly enantioselective CALB-catalyzed kinetic resolution of building blocks for β-blocker atenolol. Tetrahedron 2016, 72, 7288–7292. [Google Scholar] [CrossRef]
- Jacobsen, E.E.; Hoff, B.H.; Anthonsen, T. Enantiopure derivatives of 1,2-alkanediols: Substrate requirements of lipase B from Candida antarctica. Chirality 2000, 12, 654–659. [Google Scholar] [CrossRef]
- Chandkvetandze, B.; Lomsadze, K.; Bergenthal, D.; Breitkreudtz, J.; Bergander, K.; Blaschke, G. Mechanistic study on the opposite migration order of clenbuterol enantiomers in capillary electrophoresis with β-cyclodextrin and single-isomer heptakis(2,3-diacetyl-6-sulfo)-β-cyclodextrin. Electrophoresis 2001, 22, 3178–3184. [Google Scholar] [CrossRef]
- Matsuda, T.; Yamanaka, R.; Nakamura, K. Recent progress in biocatalysis for asymmetric oxidation and reduction. Tetrahedron Asymmetry 2009, 20, 513–557. [Google Scholar] [CrossRef]
- Au, S.K.; Hanefeld, U.; Pohl, M.; Bartsch, S.; Hussain, S.; Pressniitz, D.; Beecher, D.; Ilari, A.; Resch, V.; Boffi, D.; et al. Science of Synthesis: Biocatalysis in Organic Synthesis; Faber, K., Fessner, W-D., Turner, N.J., Eds.; Georg Thieme Verlag KG: Stuttgart, Germany, 2015; Volume 2, p. 672. ISBN 978-3-13-174161-5. [Google Scholar]
- Anthonsen, H.W.; Hoff, B.H.; Anthonsen, T. Calculation of enantiomer ratio and equilibrium constants in biocatalytic ping-pong bi-bi resolutions. Tetrahedron Asymmetry 1996, 7, 2633–2638. [Google Scholar] [CrossRef]
- Uray, G.; Stampfer, W.; Fabian, W.M.F. Comparison of Chirasil-DEX CB as gas chromatographic and ULMO as liquid chromatographic chiral stationary phase for enantioseparation of aryl- and heteroarylcarbinols. J. Chromatogr. Coruña 2003, 992, 151–157. [Google Scholar] [CrossRef]
- Lystvet, S.M.; Hoff, B.H.; Anthonsen, T.; Jacobsen, E.E. Chemoenzymatic synthesis of enantiopure 1-phenyl-2-haloethanols and their esters. Biocatal. Biotransform. 2010, 28, 272–278. [Google Scholar] [CrossRef]
- Bagley, M.C.; Glover, C. Bohlmann-Rahtz Cyclodehydration of Aminodienones to Pyridines Using N-Iodosuccinimide. Molecules 2010, 15, 3211–3227. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).