Abstract
The current report contributes to the understanding of the stereoselectivity of chiral dirhodium(II) carboxylate catalysts carrying N-protected tert-leucine ligands. Investigating the possible effect of ligand stereo-purity on catalyst structure and enantioselectivity was carried out. This was justified through a new X-ray crystal structure for Rh2(S,S,S,R-PTTL)4 diastereomer.
1. Introduction
Chiral dirhodium(II) complexes are currently renowned as catalysts that can effectively drive a broad spectrum of reactions with superior levels of chemo-, regio- and stereo-selectivity [1,2,3,4,5,6,7,8,9,10,11,12,13,14]. Their remarkable selectivity has reached a stage where they can function as a powerful tool in building up value added molecules with complex structures [7,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34].
Out of the large dirhodium(II) catalyst family and by following different approaches for catalyst design, several highly enantioselective dirhodium(II) carboxylate catalysts have been reported to date [13,35,36,37,38,39,40,41,42,43,44]. Our research group, for example, has recently reported a novel approach for the design of dirhodium(II) tetracarboxylate complexes derived from S-amino acid ligands [35,36,45]. This approach is founded on tailoring the steric influences of the overall catalyst structure through reducing the local symmetry of the ligand’s N-heterocyclic tether. The application of this new approach led to the uncovering of Rh2(S-tertPTTL)4 (Figure 1) as a new member to the dirhodium(II) family with outstanding selectivity in asymmetric cyclopropanation reactions [35]. However, despite the number of successful approaches used for catalyst design, limited evidence is still available on how the four chiral carboxylate ligands within the dirhodium(II) carboxylate framework tailor the product chirality at the catalyst active center [46].
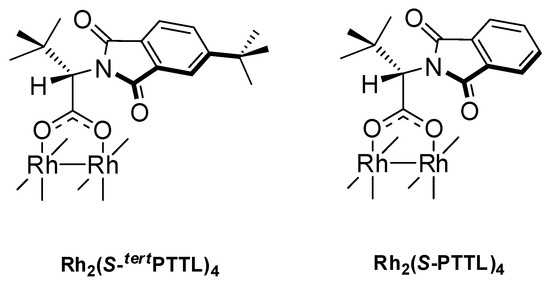
Figure 1.
Structure of Rh2(S-tertPTTL)4 and Rh2(S-PTTL)4.
At the heart of all enantioselectivities observed with chiral dirhodium(II) carboxylate complexes is, of course, the individual chirality of ligands. In the case of published X-ray crystal structures of various solvated adducts of Rh2(S-PTTL)4 [47,48], Rh2(S-tertPTTL)4 [35], Rh2(S-NTTL)4 [49], Rh2(S-PTAD)4 [35] and others [35,50,51], the incorporation of a ligand stereogenic center begets a “chiral cavity“ or a “chiral pocket”. Figure 2 [47,48] depicts the two most important features that are responsible for the establishment of the chiral binding pocket in Rh2(S-PTTL)4 and related complexes; C–C bond torsion (bond connecting carboxylate carbon to α-carbon) that is positioned so as to guide the C–N single bond leading to a clockwise twist of the chiral cavity walls (Figure 2a), and C–N bond torsion (bond connecting α-carbon to nitrogen) so as to permit docking of neighboring N-phthaloyl units (featuring alternating O...H–C closest contacts, Figure 2c). The overall visual result is that the eight oxygen atoms of the ligands’ N-phthaloyl units are placed at alternate high and low positions around the periphery of the carbene binding pocket (Figure 2b) [35]. Based on these observed features, the use of optically pure ligands in the process of catalyst preparation is essential for the construction of the symmetric chiral cavity and for achieving extremely reliable enantioselective catalysis [52]. This is because, the inclusion of a ligand with an opposite absolute configuration will result in the interruption of the symmetry of the chiral pocket.
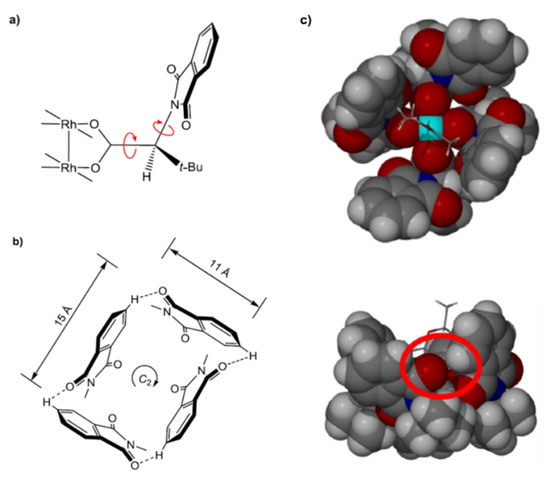
Figure 2.
(a) Features that contribute to the chirality nature of the carbene binding cavity of Rh2(S-PTTL)4 and associated catalysts; (b) Schematic presentation of the daisy chain manner by which the Rh2(S-PTTL)4 binding pocket is constructed (c) Space filling of mono-EtOAc adduct of Rh2(S-PTTL)4 (top and side views) [35,47,48]. (Adapted from Adly, F.G.; Gardiner, M.G.; Ghanem, A. Chem. Eur. J. 2016, 22, 3447, with permission from John Wiley and Sons, Hoboken, NJ, USA).
As a consequence, Hashimoto et al. explored the racemization free N-phthaloylation of tert-leucine [53]. They reported that minimal racemization can be achieved in refluxing toluene in the presence of triethylamine (TEA). Charette also reported that N-protection of different L-amino acids can be done in refluxing DMF to achieve the desired enantiomerically pure N-protected-L-amino acid ligands [41]. Likewise, N-1,8- [41,54] and N-2,3-naphthaloylation [55] of L-amino acids were also reported to take place under the same reaction conditions. In both latter cases, the prepared ligands were used in the preparation of the corresponding dirhodium(II) catalysts for enantioselective applications. Another report claimed that racemization free N-protection of amino acids was possible in refluxing acetic acid [56]. These later reports did not, however, support their claims by any quantitative information about the degree of racemization that accompanied the N-protection process under the reported reaction conditions and minimal/no racemization was assumed to take place.
Now the questions are, what is the effect if ligand enantio-impurity is present on the chiral cavity features explained earlier? and to what extent this impurity might affect the functionality of that cavity? To the best of our knowledge, there are no published reports to indicate the issues/benefits that might accompany the use of enantiomerically impure chiral ligand in the process of chiral dirhodium(II) catalyst preparation. Therefore, the trigger of the current research was to point out any aspects that can correlate the enantio-purity of the chiral carboxylate ligand used to the structure of the corresponding catalyst and to highlight any implications that this might have on catalyst enantioselectivity. We were inspired by the work done by Fox [40] and Charette [41], independently, as they both explored the interruption of the chiral cavity symmetry by substituting one of the four carboxylate ligands with an achiral ligand. Screening results revealed that interrupting the cavity framework has, indeed, a beneficial impact on the asymmetric induction of these complexes.
2. Results and Discussion
The current work started when PTTL ligand was synthesised three times using three different reaction solvents namely acetic acid, DMF and toluene/TEA, under refluxing conditions leading to chiral ligands with different degrees of enantiopurity (ee values of the obtained PTTL ligands from acetic acid, DMF and toluene/TEA solvents were found to be 78%, 86% and >99% ee, respectively). These prepared ligands were further used for the synthesis of three dirhodium(II) complexes, Rh2(PTTL78%ee)4, Rh2(PTTL86%ee)4 and Rh2(PTTL99%ee)4, respectively at which, the obtained complexes were expected to vary in their degree of stereo-purity.
When Rh2(PTTL78%ee)4 was crystallized from methanol/water in an open flask, two types of X-ray quality crystals were observed and isolated. The first were green prismatic crystals and, they were found to have the composition of [Rh2(S-PTTL)4.(H2O)][Rh2(S-PTTL)4.2(H2O)].5H2O by X-ray crystal structure determination (Figure 3a,b). Although the complex molecules are still retaining the chiral crown cavity of approximate C4 symmetry similar to the regular cavity structure of [Rh2(S-PTTL)4.(EtOAc)] [47,48], the inherent weakness of the cavity walls is apparent in the obtained aquated Rh2(S-PTTL)4 structure(s) (Figure 3a,b). This is owing to the obvious absence of strong interactions between adjacent N-phthaloyl units.
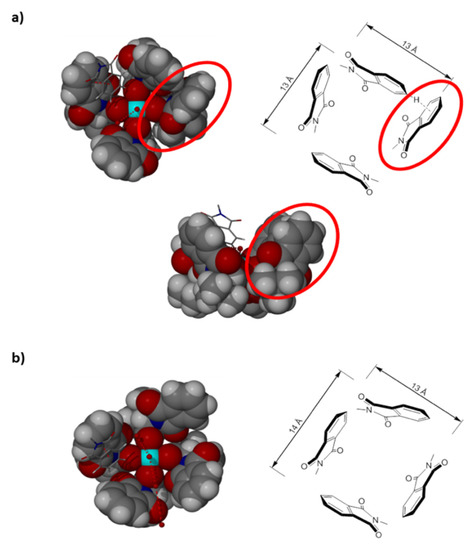
Figure 3.
Space filling and schematic representations of the irregularly shaped chiral crown cavities of; (a) [Rh2(S-PTTL)4.(H2O)] and (b) [Rh2(S-PTTL)4.2(H2O)]. Rh bound and lattice water molecules are depicted in stick form (hydrogen atoms of all water molecules could not be located and the extended hydrogen bonded structure is not shown beyond the asymmetric unit). Also shown in stick form are the endo-cavity residing N-phthaloyl units of the adjacent crystallographically distinct complex featuring face-to-face π-stacking interactions. A side view into the equatorial plane of the Rh centers is given in (a) to highlight the significantly different conformation of one of the ligands that results in “reversal” of the chirality of the crown cavity.
In fact, there are several structural features of major significance that comes to light through the detailed analysis of this newly determined X-ray structure of Rh2(PTTL)4 which can be correlated to the performance of this class of dirhodium(II) complexes in asymmetric catalysis. From the current structure and in light of the recent advances in the field, it is evident that the previously reported flattened rectangular shape structure of the crown cavity [47,48] is subject to substantial variations which appear to be influenced by the identity of the axial bound ligand and, at least in the solid state, by the intermolecular interactions including crystal packing forces. Whilst of irregular shape, the crown cavity of the current structure(s) (Figure 3a,b) can be described as being quite square shaped of ca. 13–14 Å width if compared to the rectangular shaped cavity reported for [Rh2(S-PTTL)4.(EtOAc)] (12.8 x 14.1 Å) [47,48].
Beyond the overall shape of the irregular crown cavities described above, the mono aquated complex, [Rh2(S-PTTL)4.(H2O)], features a partial “reversal” of its chiral nature (Figure 3a). Whilst all four ligands maintain their S-stereogenic carbon centres, the C–N bond from this carbon centre in one ligand is close to being aligned with the axial coordination site of the Rh centre (circled in Figure 3a). This is in contrast with the clockwise twist reported for [Rh2(S-PTTL)4.(EtOAc)] [47,48] and other related complexes [35,49,50,51]. As a result, the daisy-chain type docking of the N-phthaloyl units around the cavity seen in [Rh2(S-PTTL)4.(EtOAc)], which gives the cavity its main chiral feature (Figure 2c), is not only disrupted but is, in fact, partially reversed. This was most apparent in the formation of an edge (C–H)-face π-stacking of the involved N-phthaloyl unit with an adjacent ligand and a partial loss in regularity of the alternating locations of the oxygen atoms around the cavity compared to Rh2(S-PTTL)4.(EtOAc) structure (Figure 3a).
On the other hand, when Rh2(PTTL78%ee)4 was crystallized from anhydrous methanol under argon atmosphere, we were able to isolate crystals of a methanol solvated adduct and determine the X-ray crystal structure. The observed structure was different to the one crystallized from methanol/water. This showed the complex to be the di-solvated species [Rh2(S-PTTL)4.(MeOH)2].(MeOH)3/2. This complex exhibited a distorted crown cavity where one of the N-heterocycles features a reverse twist akin to the mono-aquated adduct [Rh2(S-PTTL)4.(H2O)] described above. In the solid state, there are two crystallographically independent molecules each having this structure, despite having different endo-cavity arrangements of hydrogen bonded MeOH molecules in addition to the axially coordinated solvent.
The ability of Rh2(S-PTTL)4 to have axial ligands bonded to the Rh centre shrouded by the crown cavity, as well as the more sterically encumbered Rh centre was, indeed, evident through the obtained di-aquated structure (Figure 3b). This fact was also previously observed through many other complexes from the same family [35,49,50,51] including Rh2(S-PTAD)4 which carry the bulkiest adamantyl groups at the ligands’ α-positions [35]. This observation can be also applied to carbene ligands during asymmetric catalysis which contradicts with the major assumption of Fox mechanistic study for Rh2(S-PTTL)4 in asymmetric cyclopropanations [47,48]. Fox assumed that only the Rh centre within the chiral crown cavity is accessible by the incoming substrate while the other is shielded by the four tert-butyl substituents of the PTTL ligands. This prior held assumption may not be valid and should be revised.
In addition to the aquated form of Rh2(S-PTTL)4 described above, we succeeded to identify another plate shaped green crystalline form from our methanol/water crystallized sample of Rh2(PTTL78%ee)4. These crystals were shown to be comprised of the catalyst diastereomeric form, [Rh2(S,S,S,R-PTTL)4(MeOH)2.].MeOH.H2O (Figure 4) by X-ray structure determination.
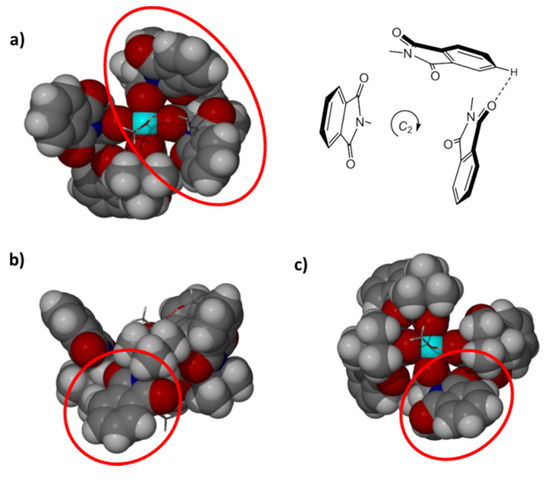
Figure 4.
Space filling and schematic representations of the partial crown shaped chiral cavity of [Rh2(S,S,S,R-PTTL)4(MeOH)2].MeOH.H2O viewed along the Rh-Rh bond (a) onto the cavity and (c) from the opposite side of the cavity. (b) A side view into the equatorial plane of the Rh centres to highlight the R-amino acid derived ligand which does not form part of the partial cavity. Rh bound MeOH and lattice MeOH/water molecules are depicted in stick form (hydroxyl groups of the Lewis bases were only located if Rh bound and the extended hydrogen bonded structure is not shown beyond the asymmetric unit).
In addition to identifying at least one of the forms in which the R-amino acid impurity manifests itself, an analysis of the structural details is also instructive here as it allows speculation as to the possible enantioselectivity that this impurity would achieve under catalytic conditions. This analysis is of major importance if the level of impurity is significant. The S,S,S,R- form features Rh bound methanol molecules at each axial coordination site and an α,α,α,β-conformation of the ligands, whereby the three N- phthaloyl units of the S-amino acid derived ligands form a partial crown cavity and the unique R-amino acid derived ligand faces the other axial binding site. Two of the N-phthaloyl groups forming the partial crown cavity exhibit conformations very similar to those in [Rh2(S-PTTL)4(EtOAc)] [47,48] and the predominant ligand types found in the aquated species described above (circled in Figure 4a). The third N-phthaloyl unit lacks the clockwise twist noted previously. This overall motif affords a partial cavity chirality (shown schematically in Figure 4a) of the same nature to the enantiomerically pure S,S,S,S- diastereomer of the complex (shown schematically in Figure 2). Based on previous results from Fox [40] and Charette [41] related to the functionality of the α,α,α,β-conformation, it can be hypothesized that the obtained S,S,S,R- form may still achieve a good degree of enantioselectivity and, importantly, towards the same product as obtained with the dominant S,S,S,S- form. Whilst we cannot disprove the presence of further diastereomeric forms existing in the same sample of Rh2(PTTL78%ee)4 at this stage, we have highlighted the potential advantage of the enantiomeric ligand purity assimilating as a minor component within the S,S,S,R- diastereomeric form rather than segregating as the R,R,R,R- enantiomer which will strictly afford the opposite enantioselectivity to the S,S,S,S- form.
At this stage and based on the obtained X-ray crystal structures, we decided to establish if the S,S,S,R- diastereomeric form of the catalyst is still functional and can lead in high enantioselectivity. we made use of the powerful method of asymmetric amplification by nonlinear effects. If the catalyst system contains a non-enantiopure chiral ligand with an enantiomeric excess eeligand, an enantiomerically enriched product with an enantiomeric excess of eeprod can be obtained. The calculation of the enantiomeric excess of the product (eemax) for an enantiopure ligand can easily be calculated if one assumes that the enantiomers of the ligand (in the catalyst) act independently. The proportionality between eeligand and eeprod in the below equation allows the eemax value to be calculated (ee values between 0 and 1). In other words, the expected enantiomeric excess of the reaction product should be proportional to the ee value of the chiral ligand in the catalyst (Linearity). However, in the case of multi-ligated catalysts (which is the case of chiral dirhodium(II) tetracarboxylate catalysts), the above equation generally is no longer obeyed. This is because diastereomeric species are produced which are impossible to generate from the enantiopure ligands. In fact, some deviation from linearity may be observed (non-Linear effect) [52].
In addition to the three prepared versions of Rh2(PTTL)4, five more dirhodium(II) complexes were prepared of which each complex was prepared three times using ligands obtained from acetic acid, DMF and toluene/TEA as reaction solvents (Scheme 1 and Scheme 2). Scheme 1 illustrates the extent of racemization in the prepared N-protected amino acid ligands by just changing the reaction solvent. It was obvious from HPLC trace that different degrees of racemization were introduced by changing the reaction conditions. It was clear that accessing ligands with minimal racemization can be readily accomplished by using toluene/TEA as a reaction solvent. This result is in agreement with the previous report by Hashimoto et al. for the preparation of the PTTL ligand [53]. By the application of this reaction conditions, the racemization degree in the prepared N-protected amino acids is restricted and kept to minimal. An exception to this was the case of N-1,2-naphthaloyl phenylalanine (1,2-NTPA), at which around 15% racemization took place (Scheme 1).
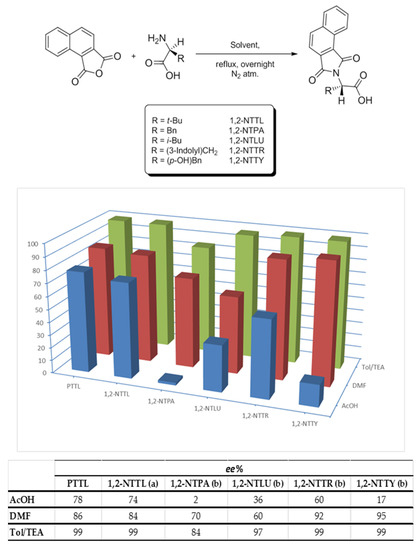
Scheme 1.
Effect of varying the reaction solvent on enantiomeric purity of the obtained N-protected amino acid ligands. Enantiomeric excess percentages (ee%) were determined by chiral HPLC using (a) Chiralpak® IB column, (b) Chiralpak® ID column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA, 254 nm, See experimental section for details.
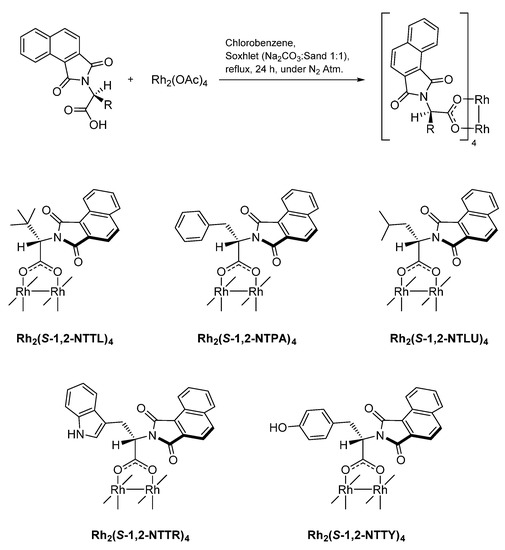
Scheme 2.
Preparation and structure of dirhodium(II) complexes.
At this point, all the prepared catalysts were subjected to catalysis and to facilitate the investigation, we employed our previously developed user-friendly one-pot cyclopropanation reaction of styrene in the presence of Meldrum’s acid. In this reaction, a phenyliodonium ylide is generated and used in situ [54,57]. All reactions were done at room temperature in DCM as reaction solvent.
In general, the cyclopropane product was generated in moderate isolated yields for all catalysts (45–60% yield) except for Rh2(S-1,2-NTTR)4 and Rh2(S-1,2-NTTY)4 (18% and 14%, respectively) and the obtained values were reproducible for all catalyst versions. The low yield observed with Rh2(S-1,2-NTTR)4 was returned to the poor solubility of the catalyst in reaction solvent. On the other hand, the low yield with Rh2(S-1,2-NTTY)4 was returned to the possible formation of a catalyst polymeric structure in solution at which some of the p-OH groups of the –CH2C6H4–OH substituents of one complex molecule might act as the axial ligands on each Rh centre of adjacent complex molecules to block both catalyst’s active sites and inhibits the catalyst from being functional [58]. In both cases, low conversion to product was observed.
In terms of catalyst enantioselectivity, results are illustrated in Figure 5. As expected, the obtained results for this study, graphically depicted in Figure 5, clearly revealed a modest but real, positive nonlinear effect for Rh2(PTTL)4, Rh2(1,2-NTTL)4, Rh2(1,2-NTPA)4 and Rh2(1,2-NTLU)4 catalysts. The results suggested that diastereomeric species with a partial crown structure similar to the S,S,S,R- diastereomer observed for Rh2(PTTL)4 might be still functional and can lead to enantioselectivity levels similar to the full crown structures.
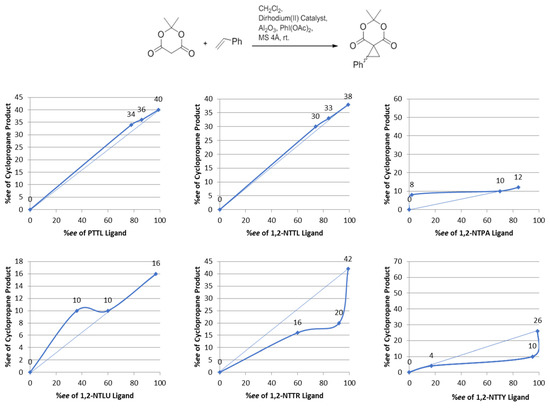
Figure 5.
Effect of ligand enantio-purity on the enantioselectivity of its corresponding dirhodium(II) catalyst.
On the other side, Rh2(1,2-NTTR)4 and Rh2(1,2-NTTY)4 showed negative deviation from linearity. In these particular cases, other solution species may have arisen and resulted in different influences on enantioselectivity levels. These other influences are untestable at this stage and beyond the scope of this report.
3. Materials and Methods
3.1. Chemicals
All starting materials and reagents were purchased from Sigma-Aldrich, Acros Organics and Tokyo Chemical Industry Co., Ltd. (TCI, Tokyo, Japan) and used without any further purification. All solvents were of HPLC grade and dried and distilled immediately prior to use. All reactions were performed using oven dried glassware and flame dried under vacuum prior to use. TLC was performed using Sigma-Aldrich pre-coated silica gel 60 F254 aluminium support (20 × 20 cm, layer thickness 0.2 mm) and spots were visualized by either using UV light (254 nm). Preparative TLC purifications were performed using Sigma-Aldrich pre-coated silica gel 60 F254 glass support (20 × 20 cm, 0.25 mm layer thickness). Column chromatography was carried out on silica gel 60 (130-270 mesh ASTM, Sigma-Aldrich, St. Louis, MO, USA) using the specified eluent compositions. Rh2(OAc)4 was purchased from Strem Chemicals (Newburyport, MA, USA).
3.2. Instruments
Melting points were measured on Stuart-SMP10 melting point apparatus and are uncorrected. Optical rotations were measured using Perkin–Elmer 341 polarimeter at the sodium D line (589 nm) and reported as [α]D25 in g/100 mL concentration (c) and in solvents indicated. IR spectroscopic measurements were carried out on PerkinElmer TravelIR FT-IR spectrometer and reported in units of cm−1. NMR spectra were recorded on Varian 400-MR and Varian Inova-500 spectrometers at room temperature. Mass Spectrometric analysis was recorded on Finnigen mat LCQ MS/MS ESI spectrometer and AB MDS Sciex 4800 MALDI-TOF-TOF mass analyser.
3.3. HPLC Analysis
The HPLC analysis was carried out at 25 °C using a Prominence Shimadzu System that consists of LC-20AD solvent delivery unit, SPD-M20A photodiode-array detector, SIL-20AHT auto sampler and CTO-20A column oven. For data processing, LabSolutions data managing software, version 5.54 SP2 was utilized. Chiralpak® IB (0.46 mm × 250 mm), Chiralpak® IG (0.46 mm × 250 mm) and Chiralpak® ID (0.46 mm × 250 mm) were obtained from Daicel Chiral Technologies. HPLC-grade n-hexane, ethyl acetate and 2-propanol were obtained from Scharlau Chemie S.A. (Sentmenat, Barcelona, Spain).
3.4. X-ray Crystallography for Dirhodium(II) Complexes
X-ray quality crystals of the dirhodium(II) complexes were obtained by dissolving the pure complex in the relevant solvent (for [Rh2(S-PTTL)4(H2O)][Rh2(S-PTTL)4(H2O)2].5H2O green prismatic crystals from MeOH/water, [Rh2(S,S,S,R-PTTL)4(MeOH)2].MeOH.H2O green plate shaped crystals from MeOH/water and for [Rh2(S-PTTL)4(MeOH)2].(MeOH)3/2 green prismatic crystals from anhydrous MeOH). The resulting solutions were subjected to sonication and Pasteur pipette filtration followed by slow evaporation of the solvent to yield crystals. Data were collected at −173 °C on crystals mounted on a Hampton Scientific cryoloop at the MX1 or MX2 beamlines, Australian Synchrotron, Victoria [59]. The structures were solved by direct methods with SHELXS-97, refined using full-matrix least-squares routines against F2 with SHELXL-97 [60], and visualised using X-SEED [61]. All non-hydrogen atoms were anisotropically refined, while, all hydrogen atoms were positioned in calculated locations and refined using a riding model with fixed C–H distances of 0.95 Å (sp2CH), 0.99 Å (CH2), 0.98 Å (CH3). The thermal parameters of all hydrogen atoms were estimated as Uiso(H) = 1.2Ueq(C) except for CH3 where Uiso(H) = 1.5Ueq(C).
CCDC-1834045-1834047 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures/.
3.5. General Procedure for Ligand Preparation
A mixture of 1,2-naphthalic anhydride (1.1 equiv.) and the L-amino acid (1 equiv.) in either anhydrous acetic acid, DMF or toluene/TEA (0.1 equiv.) was heated to reflux under nitrogen atmosphere overnight. After that time, the reaction was worked up according to one of the below procedures to afford the corresponding reaction product.
3.6. Reaction Workup Procedures
- (a)
- When using acetic acid as solventThe reaction solvent was evaporated in vacuo and the residue was directly purified on silica gel column chromatography using ethyl acetate: n-hexane as an eluent to afford the desired product.
- (b)
- When using DMF as solventThe reaction mixture was diluted in water and extracted with ethyl acetate twice. The organic layer was washed with water three times, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was then purified on silica gel column chromatography using ethyl acetate: n-hexane as an eluent to afford the desired product.
- (c)
- When using Toluene/TEA as solventThe mixture was diluted with ethyl acetate, washed twice with 0.1 M hydrochloric acid solution, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was then purified on silica gel column chromatography using ethyl acetate: n-hexane as an eluent to afford the desired product.
3.7. N-(Phthaloyl)-Tert-Leucine (PTTL)
Phthalic anhydride (0.5 g, 3.38 mmol), L-tert-Leucine (0.402 g, 3.07 mmol); white solid (0.746 g, 93%); mp 154 °C. Spectroscopic data were consistent with the previously published literature [53]. Conditions for chiral HPLC trace: Chiralpak® ID column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA, 0.25 mL/min, 254 nm, τ1 = 16.0 min, τ2 = 17.0 min.
3.8. N-(1,2-Naphthaloyl)-Tert-Leucine (1,2-NTTL)
1,2-Naphthalic anhydride (0.650 g, 3.28 mmol), L-tert-Leucine (0.391 g, 2.98 mmol); yellow oil (0.825 g, 89%). Spectroscopic data were consistent with the previously published literature [36]. Conditions for chiral HPLC trace: Chiralpak® IB column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA, 0.25 mL/min, 254 nm, τ1 = 22.5 min, τ2 = 24.5 min.
3.9. N-(1,2-Naphthaloyl)-Phenylalanine (1,2-NTPA)
1,2-Naphthalic anhydride (0.5 g, 2.52 mmol), L-Phenylalanine (0.35 g, 2.10 mmol); pale yellow solid (0.65 g, 89%); mp 182 °C. Spectroscopic data were consistent with the previously published literature [36]. Conditions for chiral HPLC trace: Chiralpak® ID column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA, 0.5 mL/min, 254 nm, τ1 = 40.0 min, τ2 = 44.0 min.
3.10. N-(1,2-Naphthaloyl)-Leucine (1,2-NTLU)
1,2-Naphthalic anhydride (0.5 g, 2.52 mmol), L-Leucine (0.3 g, 2.29 mmol); pale yellow solid (0.64 g, 90%); mp 138–139 °C. Spectroscopic data were consistent with the previously published literature [36]. Conditions for chiral HPLC trace: Chiralpak® ID column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA; 0.25 mL/min, 254 nm, τ1 = 56.0 min, τ2 = 61.0 min.
3.11. N-(1,2-Naphthaloyl)-Tryptophan (1,2-NTTR)
1,2-Naphthalic anhydride (0.5 g, 2.52 mmol), L-Tryptophan (0.47 g, 2.29 mmol); yellow solid (0.83 g, 85%); mp 238–239 °C. Spectroscopic data were consistent with the previously published literature [36]. Conditions for chiral HPLC trace: Chiralpak® ID column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA; 0.25 mL/min, 254 nm, τ1 = 110.0 min, τ2 = 131.0 min.
3.12. N-(1,2-Naphthaloyl)-Tyrosine (1,2-NTTY)
1,2-Naphthalic anhydride (0.5 g, 2.52 mmol), L-Tyrosine (0.42 g, 2.29 mmol); yellow solid (0.74 g, 90%); mp 248–249 °C. Spectroscopic data were consistent with the previously published literature [36]. Conditions for chiral HPLC trace: Chiralpak® ID column, 10% 2-propanol in n-hexane (v/v%) with 0.1% TFA, 0.5 mL/min, 254 nm, τ1 = 52.5 min, τ2 = 51.0 min.
3.13. General Procedure for Ligand Exchange
A mixture of the prepared carboxylic acid ligand (6 equiv.) and dirhodium(II) tetraacetate (Rh2(OAc)4, 1 equiv.) in dry chlorobenzene was refluxed for 24 h under nitrogen atmosphere using a Soxhlet extractor fitted with a thimble containing dry mixture of Na2CO3 and sand (1:1) for trapping the eliminated acetic acid molecules. After that time, the solvent was evaporated in vacuo and the residue was re-dissolved in DCM, washed with saturated NaHCO3 solution, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The green residue was then purified by means of silica gel column chromatography using ethyl acetate: n-hexane as mobile phase. Spectroscopic data of all prepared complexes were consistent with the previously published literature [36,53].
3.14. General Procedure for Cyclopropanation
2,2-Dimethyl-1,3-dioxane-4,6-dione (1 equiv.) was dissolved in dry dichloromethane and stirred with (diacetoxy)iodobenzene (1.4 equiv.), aluminium oxide (2.3 equiv.), 4Å molecular sieves and dirhodium(II) catalyst (0.01 equiv.) under nitrogen atmosphere for 5 min. After that time, Styrene (10 equiv.) was added and the mixture was stirred for 2 h. The reaction was then terminated by filtration through Celite® and concentrated in vacuo. The obtained residue was then purified by means of preparative TLC. Spectroscopic data were consistent with the previously published literature [54,57]. Enantiomeric excess of the cyclopropane product was determined by chiral HPLC analysis. Conditions for chiral HPLC trace: Chiralpak® IG column, 10% ethyl acetate in n-hexane (v/v%); 1 mL/min, 254 nm, τ1 = 25 min, τ2 = 33 min. Product yields are illustrated in Table 1.

Table 1.
Yields for the obtained cyclopropane product.
4. Conclusions
The current report contributes to the structural understanding of the reactivity and selectivity of dirhodium(II) carboxylate complexes derived from N-protected amino acid ligands. This contribution was through investigating the effect of ligand stereo-purity on the catalyst enantioselectivity. In conclusion, the use of partially resolved chiral ligand can produce some perturbations generated by the formation of diastereomeric complexes and such perturbations cannot be deducted from the sole behaviour of the catalyst prepared from the enantio-impure ligand. This was confirmed by the observation of Rh2(S,S,S,R-PTTL)4 diastereomer structure. The complex showed that it adapts an α,α,α,β- structure in solid state with the three S-ligands pointing to one side while the R-ligand is alone on the other. By having a partial chiral cavity, the observed complex was anticipated to perform catalysis in a similar way to its diastereomer Rh2(S-PTTL)4. The obtained catalysis results did show deviation from linearity and did reflect the anticipated effect caused by the presence of this type of diastereomer on the enantioselectivity of some dirhodium(II) carboxylate catalysts. In this regard, further research is currently being carried out in our group aiming for a more systematic study and endeavouring to get further insights into this interesting topic. Results will be reported in due course.
This report has also heightened awareness of subtle conformational variations that these complexes can adapt during the various phases of their catalytic cycles. This awareness evolved through the discovery of new Rh2(S-PTTL)4 structures including its mono- and di-aquated adducts, as well as its di-methanol adduct.
Generally, by drawing attention to the added complexity of these important structural issues, we can acknowledge that dirhodium(II) carboxylate catalysts are highly fluxional structures and more than one catalyst conformation might be present in solution at the same time. The energetics of these different conformers can change throughout the catalytic process and different conformers may have different energy barriers in productive processes involved in the catalytic cycle. Hence, the different conformers of the catalyst will contribute to the overall catalytic process to different extents proportional to their relative abundance in solution.
Supplementary Materials
The following are available online at http://www.mdpi.com/2073-4344/8/7/268/s1, [Rh2(S-PTTL)4.(H2O)][Rh2(S-PTTL)4.2(H2O)].cif, [Rh2(S-PTTL)4.2(MeOH)].(MeOH)3/2.cif and [Rh2(S,S,S,R-PTTL)4.2(MeOH)].MeOH.H2O.cif.
Author Contributions
A.G., F.G.A. and M.G.G. conceived and designed the experiments; F.G.A. performed all the experiments and analyzed the data; H.B. examined the reproducibility of the cyclopropanation results as part of her honors degree project. M.G.G. performed all the work related to the X-ray crystallography part, commented on the obtained structures and wrote the initial paper which has been rewritten by F.G.A.
Funding
This research was funded by the University of Canberra, Centre for Research on Therapeutic Solutions (CRESTS) strategic fund.
Acknowledgments
Data for the structures of the catalysts were obtained on the MX1/MX2 beamlines at the Australian Synchrotron, Victoria, Australia.
Conflicts of Interest
The authors declare no conflict of interest.
References and Note
- Adly, F.G.; Ghanem, A. Enantiomerically pure compounds by enantioselective synthetic chiral metal complexes. In Asymmetric Synthesis of Drugs and Natural Products; Nag, A., Ed.; CRC Press: Raton, FL, USA, 2018. [Google Scholar]
- Adly, F.G.; Ghanem, A. Chiral dirhodium(II) carboxylates and carboxamidates as effective chemzymes in asymmetric synthesis of three-membered carbocycles. Chirality 2014, 26, 692–711. [Google Scholar] [CrossRef] [PubMed]
- El-Deftar, M.; Adly, F.G.; Gardiner, M.G.; Ghanem, A. Chiral dirhodium catalysts: A new era for asymmetric catalysis. Curr. Org. Chem. 2012, 16, 1808–1836. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Manning, J.R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Qiu, H.; D Srinivas, H.; P Doyle, M. Chiral dirhodium(II) catalysts for selective metal carbene reactions. Curr. Org. Chem. 2016, 20, 61–81. [Google Scholar] [CrossRef]
- Trindade, A.F.; Coelho, J.A.S.; Afonso, C.A.M.; Veiros, L.F.; Gois, P.M.P. Fine tuning of dirhodium(II): Expolring the axial modification. ACS Catal. 2012, 2, 370–383. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Morton, D. Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bois, J.D.; Yu, J.-Q. C-H functionalization in organic synthesis. Chem. Soc. Rev. 2011, 40, 1855–1856. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic carbene insertion into C–H bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Hedley, S.J. Intermolecular reactions of electron-rich heterocycles with copper and rhodium carbenoids. Chem. Soc. Rev. 2007, 36, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Merlic, C.A.; Zechman, A.L. Selectivity in rhodium(II) catalyzed reactions of diazo compounds: Effects of catalyst electophilicity, diazo substitution, and substrate substitution. From chemoselectivity to enantioselectivity. Synthesis 2003, 34, 1137–1156. [Google Scholar] [CrossRef]
- Colacot, T.J. An overview on the application of “doyle catalysts” in asymmetric cyclopropanation, cyclopropenation and C-H insertion reactions. Proc. Indian Acad. Sci. (J. Chem. Sci.) 2000, 11, 197–207. [Google Scholar] [CrossRef]
- Liao, K.; Negretti, S.; Musaev, D.G.; Bacsa, J.; Davies, H.M.L. Site-selective and stereoselective functionalization of unactivated C–H bonds. Nature 2016, 533, 230. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Yang, H.; Ellis-Guardiola, K.; Lewis, J.C. Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat. Commun. 2015, 6, 7789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Yan, M.; Huang, D. Catalyzed addition of diazoacetoacetates to imines: Synthesis of highly functionalized aziridines. Org. Biomol. Chem. 2009, 7, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, M.; Tanaka, M.; Abe, T.; Anada, M.; Hashimoto, S. Catalytic enantioselective aziridination of alkenes using chiral dirhodium(II) carboxylates. Heterocycles 2007, 72, 709–721. [Google Scholar] [CrossRef]
- Catino, A.J.; Nichols, J.M.; Forslund, R.E.; Doyle, M.P. Efficient aziridination of olefins catalyzed by mixed-valent dirhodium(II,III) caprolactamate. Org. Lett. 2005, 7, 2787–2790. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Denton, J.R. Application of donor/acceptor-carbenoids to the synthesis of natural products. Chem. Soc. Rev. 2009, 38, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Kubiak, R.W.; Mighion, J.D.; Wilkerson-Hill, S.M.; Alford, J.S.; Yoshidomi, T.; Davies, H.M. Enantioselective intermolecular C-H functionalization of allylic and benzylic sp3 C–H bonds using n-sulfonyl-1, 2, 3-triazoles. Org. Lett. 2016, 18, 3118–3121. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P. Perspective on dirhodium carboxamidates as catalysts. J. Org. Chem. 2006, 71, 9253–9260. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Hodgson, D.M.; Stupple, P.A.; Pierard, F.Y.T.M.; Labande, A.H.; Johnstone, C. Development of dirhodium(II)-catalyzed generation and enantioselective 1,3-dipolar cycloaddition of carbonyl ylides. Chem. Eur. J. 2001, 7, 4465–4476. [Google Scholar] [CrossRef]
- Doyle, M.P.; Forbes, D.C.; Vasbinder, M.M.; Peterson, C.S. Enantiocontrol in the generation and diastereoselective reactions of catalytically generated oxonium and iodonium ylides. Metal-stabilized ylides as reaction intermediates. J. Am. Chem. Soc. 1998, 120, 7653–7654. [Google Scholar] [CrossRef]
- Doyle, M.P.; Hu, W. Macrocycle formation from catalytic metal carbene transformations. Synlett 2001, 2001, 1364–1370. [Google Scholar] [CrossRef]
- Doyle, M.P.; Phillips, I.M.; Hu, W. A new class of chiral lewis acid catalysts for highly enantioselective hetero-diels-alder reactions: Exceptionally high turnover numbers from dirhodium(II) carboxamidates. J. Am. Chem. Soc. 2001, 123, 5366–5367. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Valenzuela, M.; Huang, P. Asymmetric hetero-diels-alder reaction catalyzed by dirhodium(II) carboxamidates. Proc. Natl. Acad. Sci. USA 2004, 101, 5391–5395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wolf, J.; Zavalij, P.; Doyle, M.P. Cationic chiral dirhodium carboxamidates are activated for lewis acid catalysis. Angew. Chem. Int. Ed. 2008, 47, 1439–1442. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Shimada, N.; Anada, M.; Hashimoto, S. Enantio- and diastereoselective hetero-diels-alder reactions between 4-methyl-substituted rawal’s diene and aldhydes catalyzed by chiral dirhodium(II) carboxamidates: Catalytic asymmetric synthesis of (-)-cis-aerrangis lactone. Tetrahedron: Asymmetry 2014, 25, 63–73. [Google Scholar] [CrossRef]
- Anada, M.; Washio, T.; Shimada, N.; Kitagaki, S.; Nakajima, M.; Shiro, M.; Hashimoto, S. A new dirhodium(II) carboxamidate complex as a chiral lewis acid catalyst for enantioselective heteo-diels-alder reactions. Angew. Chem. Int. Ed. 2004, 43, 2665–2668. [Google Scholar] [CrossRef]
- Hansen, J.H.; Parr, B.T.; Pelphrey, P.; Jin, Q.; Autschbach, J.; Davies, H.M.L. Rhodium(II)-catalyzed cross-coupling of diazo compounds. Angew. Chem. Int. Ed. 2011, 50, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Protopopova, M.N. New aspecs of catalytic asymmetric cyclopropanation. Tetrahedron 1998, 54, 7919–7946. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Antoulinakis, E.G. Intermolecular metal-catalyzed carbenoid cyclopropanations. Org. React. 2001, 57, 1–326. [Google Scholar]
- Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A.B. Stereoselective cyclopropanation reactions. Chem. Rev. 2003, 103, 977–1050. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J. Modern Rhodium-Catalyzed Organic Reactions; Wiley: Weinheim, Germany, 2005. [Google Scholar]
- Adly, F.G.; Gardiner, M.G.; Ghanem, A. Design and synthesis of novel chiral dirhodium(II) carboxylate complexes for asymmetric cyclopropanation reactions. Chem. Eur. J. 2016, 22, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Maddalena, J.; Ghanem, A. Rh2(S-1,2-NTTL)4: A novel Rh2(S-PTTL)4 analog with lower ligand symmetry for asymmetric synthesis of chiral cyclopropylphosphonates. Chirality 2014, 26, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.P.; Lee, G.H.; Davies, H.M.L. Dirhodium tetracarboxylate derived from adamantylglycine as chiral catalyst for carbenoid reactions. Org. Lett. 2006, 8, 3437–3440. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bruzinski, P.R.; Fall, M.J. Effect of diazoalkane structure on the stereoselectivity of rhodium(II) (S)-N-(arylsulfonyl)prolinate catalyzed cyclopropanations. Tetrahedron Lett. 1996, 37, 4133–4136. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Panaro, S.A. Novel dirhodium tetraprolinate catalysts containing bridging prolinate ligands for asymmetric carbenoid reactions. Tetrahedron Lett. 1999, 40, 5287–5290. [Google Scholar] [CrossRef]
- Boruta, D.T.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Rh2(s-PTTL)3TPA—A mixed-ligand dirhodium(II) catalyst for enantioselective reactions of α-alkyl-α-diazoesters. Chem. Sci. 2012, 3, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, V.N.G.; Charette, A.B. Design and synthesis of chiral heteroleptic rhodium(II) carboxylate catalysts: Experimental investigations of halogen bond rigidification effects in asymmetric cyclopropanation. ACS Catal. 2012, 2, 1221–1225. [Google Scholar] [CrossRef]
- Yamawaki, M.; Tsutsui, H.; Kitagaki, S.; Anada, M.; Hashimoto, S. Dirhodium(II) tetrakis[N-tetrachlorophthaloyl-(S)-tert-leucinate]: A new chiral Rh(II) catalyst for enantioselective amidation of C–H bonds. Tetrahedron Lett. 2002, 43, 9561–9564. [Google Scholar] [CrossRef]
- Hashimoto, S.; Watanabe, N.; Ikegami, S. Enantioselective intramolecular C-H insertion of α-diazo β-keto esters catalyzed by homochiral rhodium(II) carboxylates. Tetrahedron Lett. 1990, 31, 5173–5174. [Google Scholar] [CrossRef]
- Qin, C.; Boyarskikh, V.; Hansen, J.H.; Hardcastle, K.I.; Musaev, D.G.; Davies, H.M.L. D2-symmetric dirhodium catalyst derived from a 1,2,2-triarylcyclopropanecarboxylate ligand: Design, synthesis and application. J. Am. Chem. Soc. 2011, 133, 19198–19204. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Ghanem, A. Polymer monolith-supported dirhodium(II)-catalyzed continuous flow cyclopropanation in capillary format. Tetrahedron Lett. 2016, 57, 852–857. [Google Scholar] [CrossRef]
- Adly, F.G. On the structure of chiral dirhodium(II) carboxylate catalysts: Stereoselectivity relevance and insights. Catalysts 2017, 7, 347. [Google Scholar] [CrossRef]
- DeAngelis, A.; Boruta, D.T.; Lubin, J.-B.; Plampin, J.N.; Yap, G.P.A.; Fox, J.M. The chiral crown conformation in paddlewheel complexes. Chem. Commun. 2010, 46, 4541–4543. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, A.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Chiral crown conformation of Rh2(PTTL)4: Enantioselective cyclopropanation with α-alkyl-α-diazoesters. J. Am. Chem. Soc. 2009, 131, 7230–7231. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Gardiner, M.G.; Williamson, R.M.; Müller, P. First X-ray structure of a N-naphthaloyl-tethered chiral dirhodium(II) complex: Structural basis for tether substitution improving asymmetric control in olefin cyclopropanation. Chem. Eur. J. 2010, 16, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- Mattiza, J.T.; Fohrer, J.G.G.; Duddeck, H.; Gardiner, M.G.; Ghanem, A. Optimizing dirhodium(II) tetrakiscarboxylates as chiral nmr auxiliaries. Org. Biomol. Chem. 2011, 9, 6542–6550. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, V.N.G.; Lin, W.; Charette, A.B. Experimental evidence for the all-up reactive conformation of chiral rhodium(II) carboxylate catalysts: Enantioselective synthesis of cis-cyclopropane α-amino acids. J. Am. Chem. Soc. 2009, 131, 16383–16385. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Abe, T.; Nakamura, S.; Anada, M.; Hashimoto, S. Practical synthesis of dirhodium(II) tetrakis[N-phthaloyl-(S)-tert-leucinate]. Chem. Pharm. Bull. 2005, 53, 1366–1368. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Aboul-Enein, H.Y.; Müller, P. One-pot synthesis and chiral analysis of cyclopropane derivatives. Chirality 2005, 17, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, S.; Anada, M.; Kataoka, O.; Matsuno, K.; Umeda, C.; Watanabe, N.; Hashimoto, S. Enantiocontrol in tandem carbonyl ylide formation and intermolecular 1,3-dipolar cycloaddition of α-diazo ketones mediated by chiral dirhodium(II) carboxylate catalyst. J. Am. Chem. Soc. 1999, 121, 1417–1418. [Google Scholar] [CrossRef]
- Yao, G.; Dai, W.; Ye, M.; Huang, R.; Pan, Y.-M.; Liao, Z.-X.; Wang, H.-S. Synthesis and antitumor properties of novel alizarin analogs. Med. Chem. Res. 2014, 23, 5031–5042. [Google Scholar] [CrossRef]
- Ghanem, A.; Lacrampe, F.; Aboul-Enein, H.Y.; Schurig, V. Diazo compounds and phenyliodonium ylides in inter- and intramolecular cyclopropanations catalyzed by dirhodium(II). Synthesis and chiral resolution by GC versus HPLC. Monatsh. Chem. 2005, 136, 1205–1219. [Google Scholar] [CrossRef]
- Adly, F.G.; Gardiner, M.G.; Ghanem, A. Unpublished X-ray Crystal Structure of Rh2(NTTY)4. The obtained X-ray crystal structure of Rh2(NTTY)4 revealed a polymeric structure where some of the p-OH groups of the –CH2C6H4–OH substituents of one complex molecule acts as the axial ligands on each Rh centre of adjacent complex molecules, 2014.
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the Distributed Control System: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Shelx-97, Programs for Crystal Structure Analysis; Universität of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Barbour, L.J. X-seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).