Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material


Abstract
:1. Introduction
2. Results
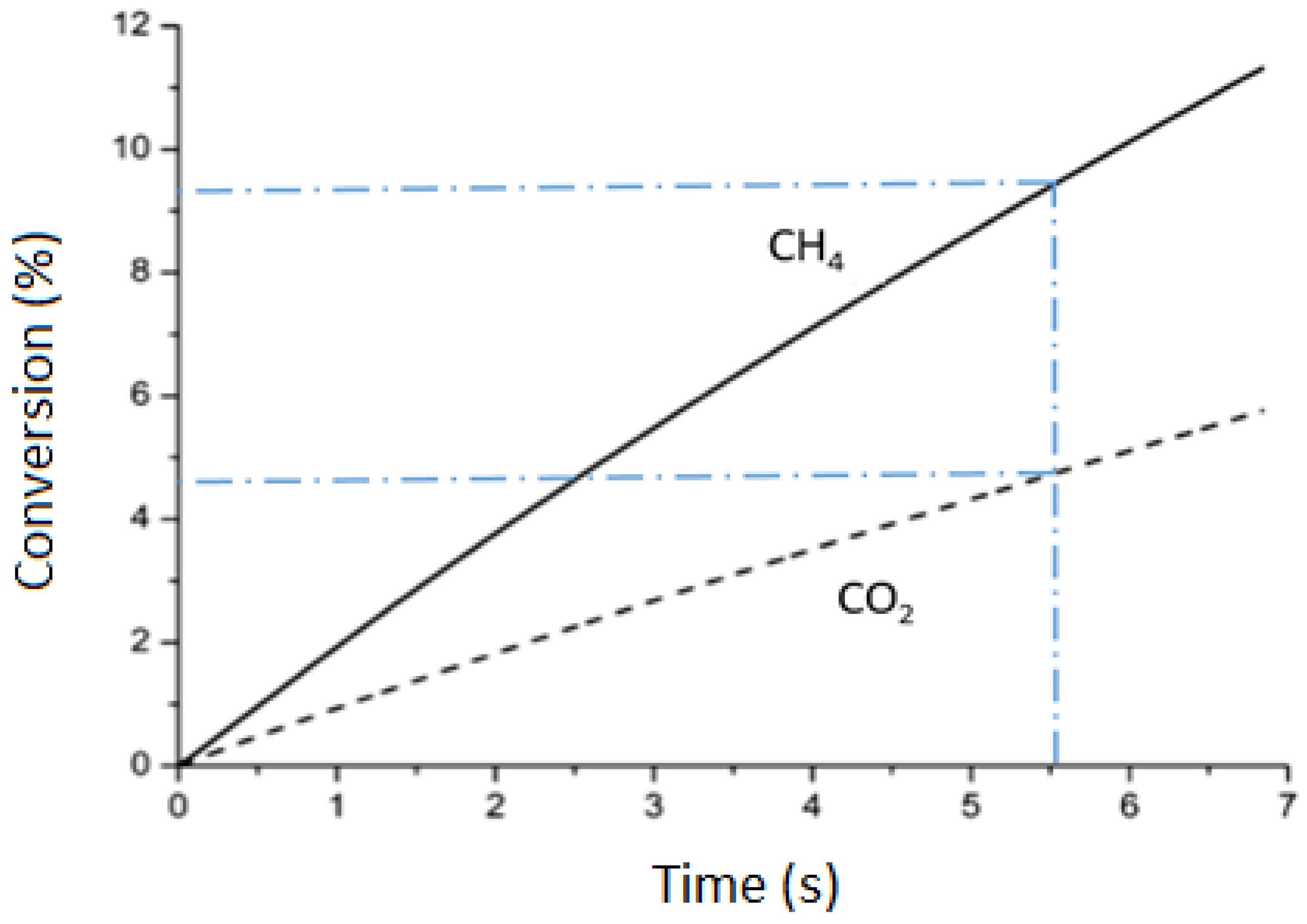
2.1. CO2 Conversion in DRM and Comparison with CO2 Splitting
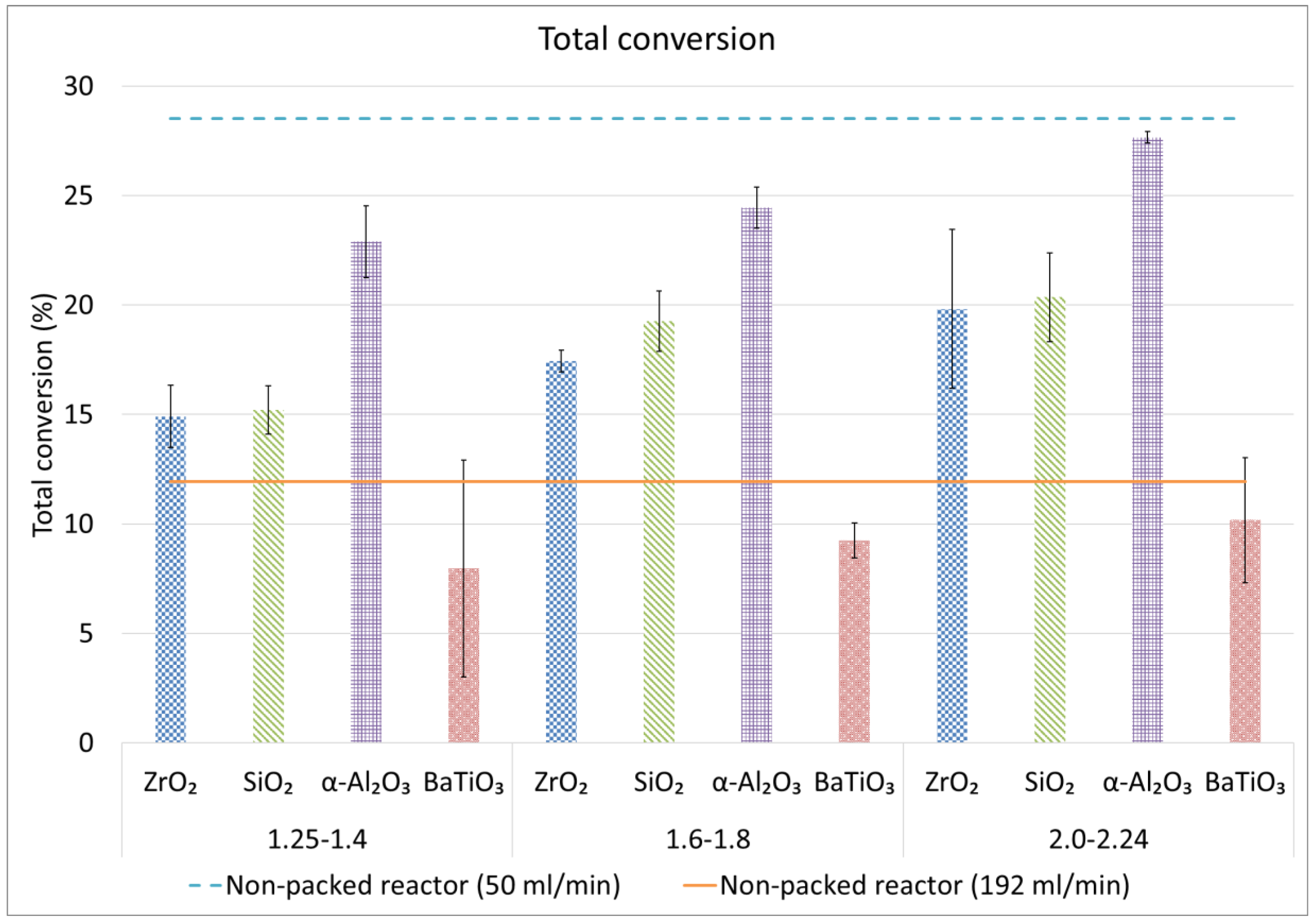
2.2. CH4 and Total Conversion
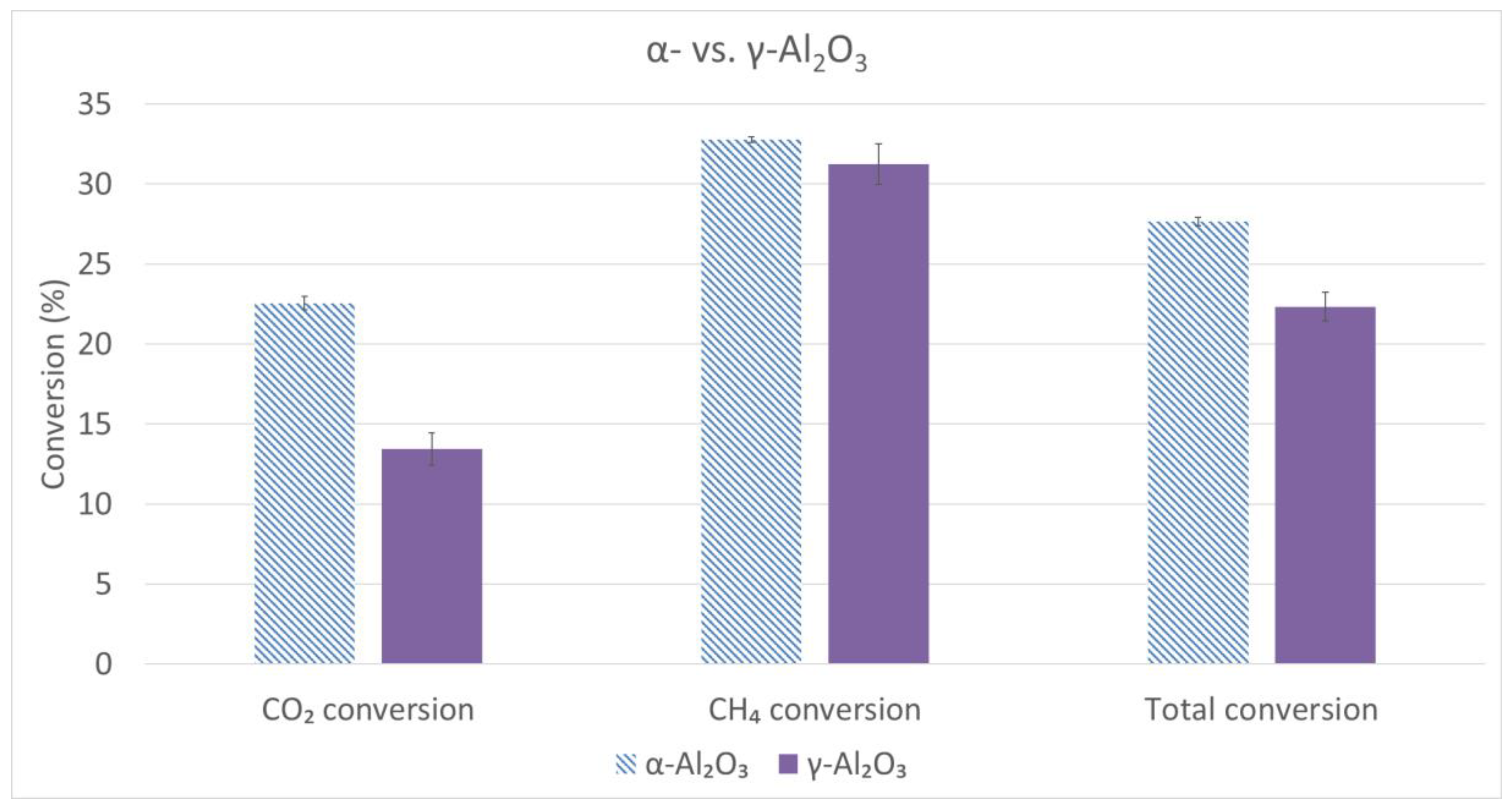
2.3. Comparison Studies α/γ-Al2O3
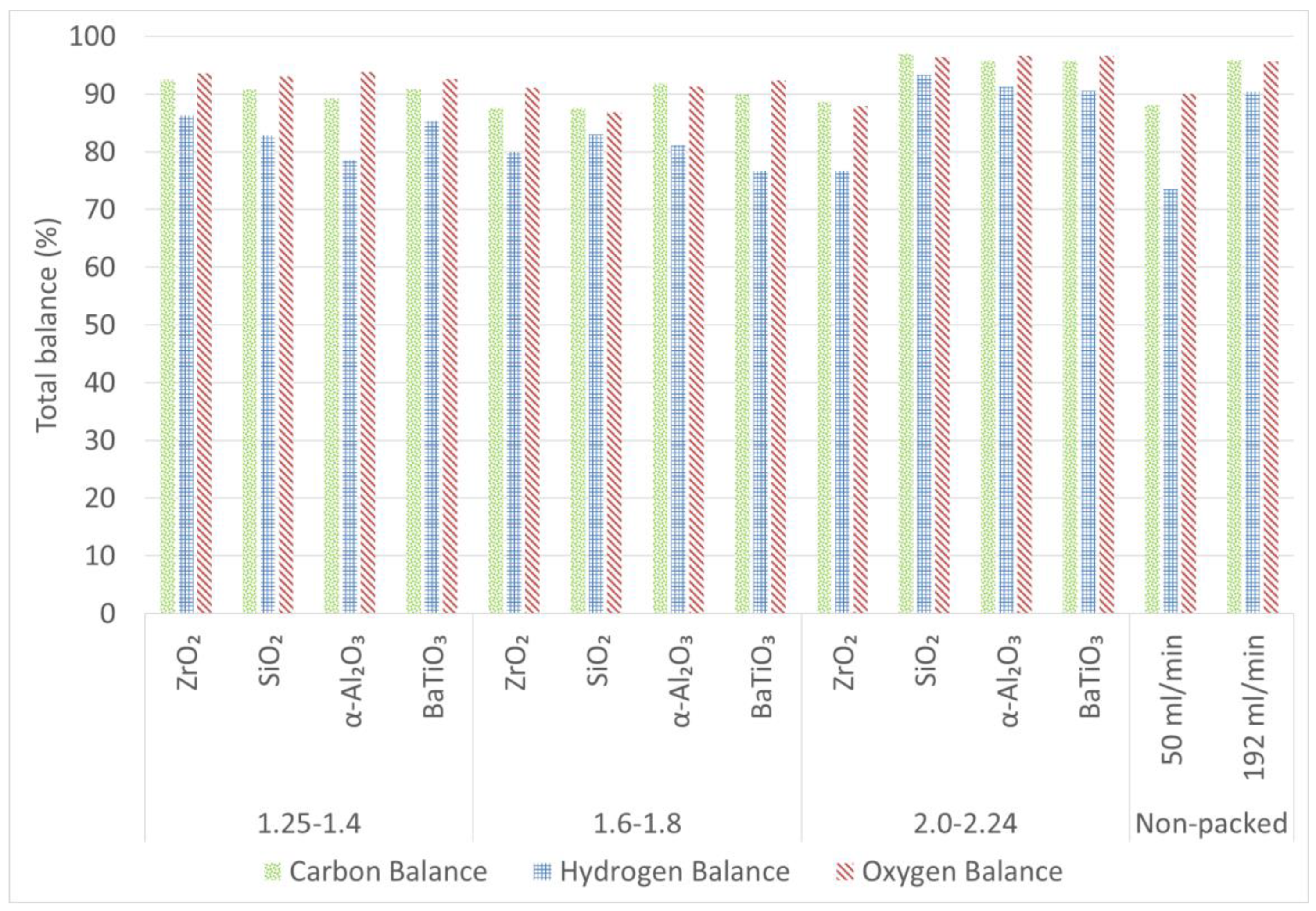
2.4. Carbon, Hydrogen and Oxygen Balances
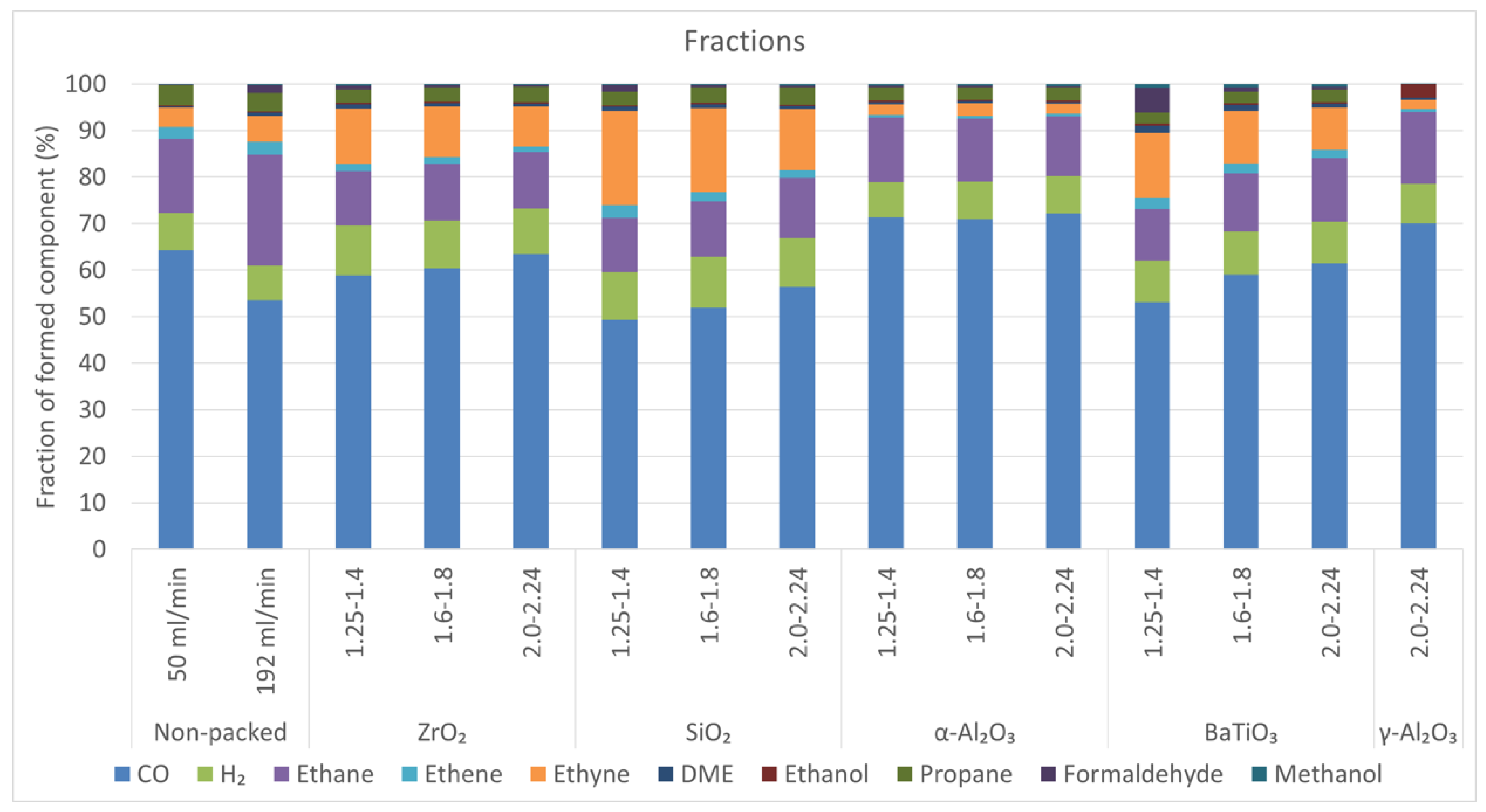
2.5. Product Fractions
- For the non-packed reactor at 50 mL/min and all α-Al2O3 spheres, the order is: ethane > H2 > propane > ethyne.
- For the non-packed reactor at 192 mL/min, the order is: ethane > H2 > ethyne > propane > ethene.
- For the smallest ZrO2 and BaTiO3 spheres and all SiO2 spheres, the order is: ethyne ≊ ethane > H2 > propane (formaldehyde in case of BaTiO3).
- For the two largest BaTiO3 spheres and the intermediate ZrO2 spheres, the order is: ethane > ethyne ≊ H2 > propane > ethene.
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snoeckx, R.; Bogaerts, A. Plasma technology—A novel solution for CO2 conversion? Chem. Soc. Rev. 2017, 46, 5805–5863. [Google Scholar] [CrossRef] [PubMed]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Chung, W.C.; Chang, M.B. Review of catalysis and plasma performance on dry reforming of CH4 and possible synergistic effects. Renew. Sustain. Energy Rev. 2016, 62, 13–31. [Google Scholar] [CrossRef]
- Usman, M.; Wan Daud, W.M.A.; Abbas, H.F. Dry reforming of methane: Influence of process parameters—A review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef]
- Jarvis, S.M.; Samsatli, S. Technologies and infrastructures underpinning future CO2 value chains: A comprehensive review and comparative analysis. Renew. Sustain. Energy Rev. 2018, 85, 46–68. [Google Scholar] [CrossRef]
- Chung, W.; Pan, K.; Lee, H.; Chang, M. Dry Reforming of Methane with Dielectric Barrier Discharge and Ferroelectric Packed-Bed Reactors. Energy Fuels 2014, 28, 7621–7631. [Google Scholar] [CrossRef]
- Arkatova, L.A. The deposition of coke during carbon dioxide reforming of methane over intermetallides. Catal. Today 2010, 157, 170–176. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Samukawa, S.; Hori, M.; Rauf, S.; Tachibana, K.; Bruggeman, P.; Kroesen, G.; Whitehead, J.C.; Murphy, A.B.; Gutsol, A.F.; Starikovskaia, S.; et al. The 2012 Plasma Roadmap. J. Phys. D Appl. Phys. 2012, 45, 253001. [Google Scholar] [CrossRef]
- Lavoie, J.-M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2, 81. [Google Scholar] [CrossRef] [PubMed]
- Kogelschatz, U. Dielectric-barrier discharges: Their history, discharge physics, and industrial applications. Plasma Chem. Plasma Process. 2003, 23, 1–46. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Wu, C.; Guo, H.; Tu, X. One-Step Reforming of CO2 and CH4 into High-Value Liquid Chemicals and Fuels at Room Temperature by Plasma-Driven Catalysis. Angew. Chem. Int. Ed. 2017, 56, 13679–13683. [Google Scholar] [CrossRef] [PubMed]
- Neyts, E.C.; Bogaerts, A. Understanding plasma catalysis through modelling and simulation—A review. J. Phys. D Appl. Phys. 2014, 47, 224010. [Google Scholar] [CrossRef]
- Aerts, R.; Somers, W.; Bogaerts, A. Carbon Dioxide Splitting in a Dielectric Barrier Discharge Plasma: A Combined Experimental and Computational Study. ChemSusChem 2015, 8, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, S.; Verheyde, B.; Tu, X.; De Bie, C.; Martens, T.; Petrovic, D.; Bogaerts, A.; Sels, B. Conversion of carbon dioxide to value-added chemicals in atmospheric pressure dielectric barrier discharges. Plasma Sources Sci. Technol. 2010, 19, 034015. [Google Scholar] [CrossRef]
- Ozkan, A.; Bogaerts, A.; Reniers, F. Routes to increase the conversion and the energy efficiency in the splitting of CO2 by a dielectric barrier discharge. J. Phys. D Appl. Phys. 2017, 50, 084004. [Google Scholar] [CrossRef]
- Zhang, A.-J.; Zhu, A.-M.; Guo, J.; Xu, Y.; Shi, C. Conversion of greenhouse gases into syngas via combined effects of discharge activation and catalysis. Chem. Eng. J. 2010, 156, 601–606. [Google Scholar] [CrossRef]
- Zheng, X.; Tan, S.; Dong, L.; Li, S.; Chen, H. Plasma-assisted catalytic dry reforming of methane: Highly catalytic performance of nickel ferrite nanoparticles embedded in silica. J. Power Sources 2015, 274, 286–294. [Google Scholar] [CrossRef]
- Zheng, X.; Tan, S.; Dong, L.; Li, S.; Chen, H. LaNiO3@SiO2 core–shell nano-particles for the dry reforming of CH4 in the dielectric barrier discharge plasma. Int. J. Hydrog. Energy 2014, 39, 11360–11367. [Google Scholar] [CrossRef]
- Karuppiah, J.; Manoj Kumar Reddy, P.; Linga Reddy, E.; Subrahmanyam, C. Catalytic non-thermal plasma reactor for decomposition of dilute chlorobenzene. Plasma Process. Polym. 2013, 10, 1074–1080. [Google Scholar] [CrossRef]
- Krawczyk, K.; Młotek, M.; Ulejczyk, B.; Schmidt-Szałowski, K. Methane conversion with carbon dioxide in plasma-catalytic system. Fuel 2014, 117, 608–617. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, Y.; Jin, Y. Dry reforming of methane in an atmospheric pressure plasma fluidized bed with Ni/γ-Al2O3 catalyst. Catal. Today 2009, 148, 275–282. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhu, X.; Mei, D.; Ashford, B.; Tu, X. Plasma-catalytic dry reforming of methane over-Al2O3 supported metal catalysts. Catal. Today 2015, 256, 80–87. [Google Scholar] [CrossRef]
- Zhang, K.; Mukhriza, T.; Liu, X.; Greco, P.P.; Chiremba, E. A study on CO2 and CH4 conversion to synthesis gas and higher hydrocarbons by the combination of catalysts and dielectric-barrier discharges. Appl. Catal. A Gen. 2015, 502, 138–149. [Google Scholar] [CrossRef]
- Tu, X.; Gallon, H.J.; Twigg, M.V.; Gorry, P.A.; Whitehead, J.C. Dry reforming of methane over a Ni/Al2O3 catalyst in a coaxial dielectric barrier discharge reactor. J. Phys. D Appl. Phys. 2011, 44, 274007. [Google Scholar] [CrossRef]
- Sentek, J.; Krawczyk, K.; Młotek, M.; Kalczewska, M.; Kroker, T.; Kolb, T.; Schenk, A.; Gericke, K.-H.; Schmidt-Szałowski, K. Plasma-catalytic methane conversion with carbon dioxide in dielectric barrier discharges. Appl. Catal. B Environ. 2010, 94, 19–26. [Google Scholar] [CrossRef]
- Pham, M.H.; Goujard, V.; Tatibouët, J.M.; Batiot-Dupeyrat, C. Activation of methane and carbon dioxide in a dielectric-barrier discharge-plasma reactor to produce hydrocarbons-Influence of La2O3/γ-Al2O3 catalyst. Catal. Today 2011, 171, 67–71. [Google Scholar] [CrossRef]
- Gallon, H.J.; Tu, X.; Whitehead, J.C. Effects of Reactor Packing Materials on H2 Production by CO2 Reforming of CH4 in a Dielectric Barrier Discharge. Plasma Process. Polym. 2012, 9, 90–97. [Google Scholar] [CrossRef]
- Song, H.K.; Choi, J.-W.; Yue, S.H.; Lee, H.; Na, B.-K.; Songu, H.K. Synthesis gas production via dielectric barrier discharge over Ni/γ-Al2O3 catalyst. Catal. Today 2004, 89, 27–33. [Google Scholar] [CrossRef]
- Tu, X.; Whitehead, J.C. Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: Understanding the synergistic effect at low temperature. Appl. Catal. B Environ. 2012, 125, 439–448. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, B.; Jin, Y.; Cheng, Y. Dry Reforming of Methane in a Dielectric Barrier Discharge Reactor with Ni/Al2O3 Catalyst: Interaction of Catalyst and Plasma. Energy Fuels 2009, 23, 4196–4201. [Google Scholar] [CrossRef]
- Michielsen, I.; Uytdenhouwen, Y.; Pype, J.; Michielsen, B.; Mertens, J.; Reniers, F.; Meynen, V.; Bogaerts, A. CO2 dissociation in a packed bed DBD reactor: First steps towards a better understanding of plasma catalysis. Chem. Eng. J. 2017, 326, 477–488. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Leys, C. Non-thermal plasmas for non-catalytic and catalytic VOC abatement. J. Hazard. Mater. 2011, 195, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Futamura, S.; Zhang, A.; Einaga, H.; Kabashima, H. Involvement of catalyst materials in nonthermal plasma chemical processing of hazardous air pollutants. Catal. Today 2002, 72, 259–265. [Google Scholar] [CrossRef]
- Kim, H.-H. Nonthermal Plasma Processing for Air-Pollution Control: A Historical Review, Current Issues, and Future Prospects. Plasma Process. Polym. 2004, 1, 91–110. [Google Scholar] [CrossRef]
- Subrahmanyam, C.; Magureanu, M.; Renken, A.; Kiwi-Minsker, L. Catalytic abatement of volatile organic compounds assisted by non-thermal plasma. Appl. Catal. B Environ. 2006, 65, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.H.; Ogata, A. Interaction of Nonthermal Plasma with Catalyst for the Air Pollution Control. Int. J. Plasma Environ. Sci. Technol. 2012, 6, 43–48. [Google Scholar]
- Francke, K.-P.; Miessner, H.; Rudolph, R. Plasmacatalytic processes for environmental problems. Catal. Today 2000, 59, 411–416. [Google Scholar] [CrossRef]
- Pasquiers, S. Removal of pollutants by plasma catalytic processes. Eur. Phys. J. Appl. Phys 2004, 28, 319–324. [Google Scholar] [CrossRef]
- Guaitella, O.; Thevenet, F.; Puzenat, E.; Guillard, C.; Rousseau, A. C2H2 oxidation by plasma/TiO2 combination: Influence of the porosity, and photocatalytic mechanisms under plasma exposure. Appl. Catal. B Environ. 2008, 80, 296–305. [Google Scholar] [CrossRef]
- Van Durme, J.; Dewulf, J.; Leys, C.; Van Langenhove, H. Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: A review. Appl. Catal. B Environ. 2008, 78, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Aerts, R.; Somers, W.; Bogaerts, A. A detailed description of the CO2 splitting by dielectric barrier discharges. ChemSusChem 2015, 8, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Kong, M.; Liu, T.; Fei, J.; Zheng, X. Characteristics of the Decomposition of CO2 in a Dielectric Packed-Bed Plasma Reactor. Plasma Chem. Plasma Process. 2012, 32, 153–163. [Google Scholar] [CrossRef]
- Mei, D.; Zhu, X.; He, Y.-L.Y.Y.; Yan, J.D.; Tu, X. Plasma-assisted conversion of CO2 in a dielectric barrier discharge reactor: Understanding the effect of packing materials. Plasma Sources Sci. Technol. 2015, 24, 15011. [Google Scholar] [CrossRef]
- Ramakers, M.; Michielsen, I.; Aerts, R.; Meynen, V.; Bogaerts, A. Effect of argon or helium on the CO2 conversion in a dielectric barrier discharge. Plasma Process. Polym. 2015, 12, 755–763. [Google Scholar] [CrossRef]
- Snoeckx, R.; Aerts, R.; Tu, X.; Bogaerts, A. Plasma-Based Dry Reforming: A Computational Study Ranging From Nanoseconds to Seconds Timescale. J. Phys. Chem. 2013, 117, 4957–4970. [Google Scholar] [CrossRef]
- Atkins, P.; Jones, L. Chemical Principles: The Quest for Insight, 4th ed.; Craig Bleyer: New York, NY, USA, 2008. [Google Scholar]
- Wang, W.; Berthelot, A.; Zhang, Q.; Bogaerts, A. Modelling of plasma-based dry reforming: How do uncertainties in the input data affect the calculation results? J. Phys. D Appl. Phys. 2018, 51, 204003. [Google Scholar] [CrossRef]
- Berthelot, A.; Bogaerts, A. Modeling of CO2 plasma: Effect of uncertainties in the plasma chemistry. Plasma Sources Sci. Technol. 2017, 26, 115002. [Google Scholar] [CrossRef]
- De Bie, C.; Van Dijk, J.; Bogaerts, A. The Dominant Pathways for the Conversion of Methane into Oxygenates and Syngas in an Atmospheric Pressure Dielectric Barrier Discharge. J. Phys. Chem. C 2015, 119, 22331–22350. [Google Scholar] [CrossRef]
- Wang, W.; Kim, H.H.; Van Laer, K.; Bogaerts, A. Streamer propagation in a packed bed plasma reactor for plasma catalysis applications. Chem. Eng. J. 2018, 334, 2467–2479. [Google Scholar] [CrossRef]
- Van Laer, K.; Bogaerts, A. Influence of Gap Size and Dielectric Constant of the Packing Material on the Plasma Behaviour in a Packed Bed DBD Reactor: A Fluid Modelling Study. Plasma Process. Polym. 2017, 14, e1600129. [Google Scholar] [CrossRef]
- Van Laer, K.; Bogaerts, A. How bead size and dielectric constant affect the plasma behaviour in a packed bed plasma reactor: A modelling study. Plasma Sources Sci. Technol. 2017, 26, 085007. [Google Scholar] [CrossRef]
- Butterworth, T.D. The Effects of Particle Size on CO2 reduction in Packed Bed Dielectric Barrier Discharge Plasma Reactors. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2015. [Google Scholar]
- Aerts, R.; Snoeckx, R.; Bogaerts, A. In-Situ Chemical Trapping of Oxygen in the Splitting of Carbon Dioxide by Plasma. Plasma Process. Polym. 2014, 11, 985–992. [Google Scholar] [CrossRef]
- Snoeckx, R.; Ozkan, A.; Reniers, F.; Bogaerts, A. The Quest for Value-Added Products from Carbon Dioxide and Water in a Dielectric Barrier Discharge: A Chemical Kinetics Study. ChemSusChem 2017, 10, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Uytdenhouwen, Y.; Van Alphen, S.; Michielsen, I.; Meynen, V.; Cool, P.; Bogaerts, A. A packed-bed DBD micro plasma reactor for CO2 dissociation: Does size matter? Chem. Eng. J. 2018, 348, 557–568. [Google Scholar] [CrossRef]
- Whitehead, J.C. Plasma–catalysis: The known knowns, the known unknowns and the unknown unknowns. J. Phys. D Appl. Phys. 2016, 49, 243001. [Google Scholar] [CrossRef]
- Pinhão, N.; Moura, A.; Branco, J.B.; Neves, J. Influence of gas expansion on process parameters in non-thermal plasma plug-flow reactors: A study applied to dry reforming of methane. Int. J. Hydrog. Energy 2016, 41, 9245–9255. [Google Scholar] [CrossRef]
- Snoeckx, R.; Heijkers, S.; Van Wesenbeeck, K.; Lenaerts, S.; Bogaerts, A. CO2 conversion in a dielectric barrier discharge plasma: N2 in the mix as a helping hand or problematic impurity? Energy Environ. Sci. 2016, 9, 30–39. [Google Scholar] [CrossRef]
- Dullien, F.A. Porous Media-Fluid Transport and Pore Structure; Academic Press: Cambridge, MA, USA, 1991; ISBN 9780122236518. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Reactor | Operating Conditions | Implementing Packing and/or Catalysts | Conclusion | Highest Conversion | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gap (mm) | Power (Watt) | Flow (mL/min) | Reactor Volume (cm3) | Frequency | SEI (kJ/L) | Packing/Catalyst | Shape | Packing Size | ||||
| CO2 splitting/non-packed | 1.5 | 70 | 150 | 21.9 | 5–65 kHz | 28 | No influence of frequency | 10% | [42] | |||
| 2 | 100–200 | 50–500 | 13.56 | 10–90 kHz | 12–240 | Conversion  when flow rate when flow rate  , Tgas , P . Best frequency depends on power , Tgas , P . Best frequency depends on power | 30% | [15] | ||||
| 2 | 10–97 | 50–2000 | 15.1 | 16.2–28.6 kHz | 0.3–116 | Conversion when flow rate , barrier thickness , frequency and power | 35% | [16] | ||||
| 4 | 21.6–35.3 | 40 | 30.17 | 13 kHz | 32.4–53 | Conversion when power and discharge length Cokes: small on inner electrode | 13% | [43] | ||||
| 4.5, 3.5, 2.5 or 2 | 60 | 50 | 17.67 | 26.5 kHz | 72 | Conversion when flow rate | 12% | [32] | ||||
| CO2 splitting/packed | 4 | 21.6–35.3 | 40 | 30.17 | 13 kHz | 32.4–53 | Silica gel | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 14% | [43] |
| Quartz | Pellets with rigid edges | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 16% | ||||||||
| γ-Al2O3 | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 16% | ||||||||
| α-Al2O3 | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 15% | ||||||||
| CaTiO3 | Beads | 20–40 mesh | silica gel < α-Al2O3 < quartz ≈ γ-Al2O3 < CaTiO3 Cokes: limited on inner electrode | 20.5% | ||||||||
| 3 | 20–50 | 50 | 10.1 | 9 kHz | 24–60 | Glass | Beads | 1 mm | Glass < BaTiO3 | 22% (16% without packing) | [44] | |
| BaTiO3 | Beads | 1 mm | Glass < BaTiO3 | 28% (16% without packing) | ||||||||
| 4.5, 3.5, 2.5 or 2 | 60 | 50 | 17.67 | 26.5 kHz | 72 | Glass wool | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 10% | [32] | |
| Quartz wool | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 10% | ||||||||
| SiO2 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 16% | ||||||||
| ZrO2 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 19% | ||||||||
| α-Al2O3 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 17% | ||||||||
| BaTiO3 | Beads | 1.25–2.24 mm | Conversion when # contact points , void space volumes and bead/gap size ratio . Impact of the packing material (chemistry and physical), also influenced by setup | 26% | ||||||||
| Study | Reactor | Operating Conditions | Implementing Packing and/or Catalysts | Selectivity | Conclusion | Highest Conversion | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gap (mm) | Power (Watt) | Flow (mL/min) | Reactor volume (cm3) | Frequency | Ratio CO2/CH4 | T | SEI (kJ/L) | Packing/Catalyst | Shape | Packing size | Highest achieved selectivity per component | ||||
| DRM/ non-packed | 3 | 30–60 | 25–100 | 11.4 | 30–40 kHz | 1 | / | 18–144 | 45% CO, 29% H2, 5% C2H2/C2H4, 22% C2H6, 2% C3H6, 12% C3H8 (estimation) | Conversion when flow rate and P | 50.4% CH4, 30.5% CO2 | [30] | |||
| 1 | 25–75 | 30–75 | 4.4 | 30 kHz | 0.66–3 | / | 20–150 | 76% CO, 57% H2 | Conversion when flow rate , P . CH4 conversion , CO2 conversion ≈ when ratio | 59.7% CH4, 36.9% CO2 | [17] | ||||
| 1 | 80–130 | 10–40 | 4.7 | 20 kHz | 0.25–1 | / | 120–780 | 73.8% CO, 65.9% H2, 18.0% C2, 10.2% C3, 6.2% C4 | Conversion when flow rate , P . CH4 conversion , CO2 conversion ≈ when ratio | 64% CH4, 34% CO2 | [29] | ||||
| 3 | 10 | 40 | 2.12 | 12 kHz | 4 | / | 15 | 20% CO, 34% H2, <1% C2H2, <1% C2H4, 12% C2H6, 1% C3H8, <1% C4H10, 11.9% methanol, 11.9% ethanol, 33.7% acetic acid, 1.6% acetone, 0% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 18% CH4, 15% CO2 | [12] | ||||
| DRM/ packed | 3 | 30–60 | 25–100 | 11.4 | 30–40 kHz | 1 | / | 18–144 | 10 wt% Ni@γ-Al2O3 | Pellets | 0.5–1.7 mm | 55% CO, 33% H2, 10% C2H2/C2H4, 47% C2H6, 2% C3H6, 25% C3H8 (estimation) | Conversion when pellet size , quartz wool is best, impact packing on selectivity | 40.2% CH4, 30.5% CO2 | [30] |
| 1 | 25–75 | 30–75 | 4.4 | 30 kHz | 1 | 450 °C | 20–150 | 12% Ni/γ-Al2O3 | ? | ? | 43% CO, 53% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 30% CH4, 24% CO2 | [17] | |
| 12% Cu/γ-Al2O3 | ? | ? | 50% CO, 31% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 7% CH4, 5% CO2 | ||||||||||
| 1%Cu-12% Ni/γ-Al2O3 | ? | ? | 45% CO, 51% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 33% CH4, 25% CO2 | ||||||||||
| DRM/ packed | 5%Cu-12% Ni/γ-Al2O3 | ? | ? | 47% CO, 54% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 37% CH4, 24% CO2 | |||||||||
| 12%Cu-12% Ni/γ-Al2O3 | ? | ? | 75% CO, 56% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 69% CH4, 75% CO2 | ||||||||||
| 16%Cu-12% Ni/γ-Al2O3 | ? | ? | 64% CO, 57% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 43% CH4, 47% CO2 | ||||||||||
| 5% Ni-12%Cu/γ-Al2O3 | ? | ? | 75% CO, 56% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 43% CH4, 45% CO2 | ||||||||||
| 16% Ni-12%Cu/γ-Al2O3 | ? | ? | 71% CO, 58% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 57% CH4, 57% CO2 | ||||||||||
| 20% Ni-12%Cu/γ-Al2O3 | ? | ? | 62% CO, 58% H2 | 12% Cu–12% Ni/γ-Al2O3 performs best, Ni content influences CO selectivity | 35% CH4, 32% CO2 | ||||||||||
| 1 | 130 | 30 | 4.7 | 20 kHz | 1 | / | 260 | γ-Al2O3 | Crushed flakes | 10–20 mesh | 49.2% CO, 51% H2, 9.7% C2, 5.5% C3, 3% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 57.6% CH4, 30.9% CO2 | [29] | |
| 2 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 60.6% CO, 52.3% H2, 9.8% C2, 5.9% C3, 3.2% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.4% CH4, 32.7% CO2 | ||||||||||
| 5 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 60.9% CO, 51.9% H2, 10.1% C2, 5.9% C3, 3.2% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.7% CH4, 33.5% CO2 | ||||||||||
| 7 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 63.9%CO, 53.5% H2, 10.6% C2, 6.1% C3, 3.6% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.5% CH4, 32.6% CO2 | ||||||||||
| 10 wt% Ni @ γ-Al2O3 | Crushed flakes | 10–20 mesh | 61.4% CO, 53% H2, 10.6% C2, 6.2% C3, 3.4% C4 | Packing: CO2 conversion , CH4 conversion . After activation: conversion , selectivity for H2 and C2 | 55.2% CH4, 32.7% CO2 | ||||||||||
| 3 | 10 | 40 | 2.12 | 9 kHz | 1 | / | 15 | γ-Al2O3 | ? | ? | 23% CO, 55% H2, <1% C2H2, <1% C2H4, 20% C2H6, 2% C3H8, <1% C4H10, 13% methanol, 9% ethanol, 20% acetic acid, 2% acetone, 0% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 15% CH4, 12.5% CO2 | [12] | |
| DRM/packed | Cu/γ-Al2O4 | ? | ? | 14% CO, 35% H2, <1% C2H2, <1% C2H4, 15% C2H6, 2% C3H8, <1% C4H10, 11% methanol, 11% ethanol, 42% acetic acid, 2% acetone, 0% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 16% CH4, 7.5% CO2 | |||||||||
| Au/γ-Al2O5 | ? | ? | 20% CO, 42% H2, <1% C2H2, <1% C2H4, 16% C2H6, 2% C3H8, <1% C4H10, 10% methanol, 10% ethanol, 30% acetic acid, 2% acetone, 5% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 16% CH4, 15% CO2 | ||||||||||
| Pt/γ-Al2O6 | ? | ? | 20% CO, 40% H2, <1% C2H2, <1% C2H4, 17% C2H6, 2% C3H8, <1% C4H10, 10% methanol, 9% ethanol, 25% acetic acid, 2% acetone, 11% HCHO | Impact depends on catalyst, both and conversion and differs from pure packing | 17.5% CH4, 13% CO2 | ||||||||||
| 5.9 | 40 | 80 | ? | 300 Hz | 0.07–1 | RT-600 °C | 30 | Glass | Beads | 2 mm | 70% CO, 19.5% H2, 42.9% C2, 15% C3, 8.7% C4 | CH4 concentration = C2 . Influence catalyst only > 200 ◦C, for CO2. Effect glass = Al2O3 | 25% CH4, 56.1% CO2 | [27] | |
| γ-Al2O3 | Beads | 2 mm | 70% CO, 19.5% H2, 42.9% C2, 15% C3, 8.7% C4 | CH4 concentration = C2 . Influence catalyst only > 200 ◦C, for CO2. Effect glass = Al2O3 | 25% CH4, 56.1% CO2 | ||||||||||
| La2O3/γ-Al2O3 | Beads | 2 mm | 70% CO, 19.5% H2, 42.9% C2, 15% C3, 8.7% C4 | CH4 concentration = C2 . Influence catalyst only > 200 ◦C, for CO2. | 25% CH4, 56.1% CO2 | ||||||||||
| 2 | 40–240 | 40 | ? | 5–20 kHz | 1 | / | 60–360 | Ni/γ-Al2O3 | Nano-particles | 100 nm | 86% CO, 73% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 64.6% CH4, 58% CO2 | [18] | |
| Ni-Fe/γ-Al2O3 | Nano-particles | 100 nm | 87% CO, 74% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 68.7% CH4, 60.5% CO2 | ||||||||||
| Ni-Fe/SiO2 | Nano-particles | 100 nm | 88% CO, 75% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 73.5% CH4, 62.7% CO2 | ||||||||||
| NiFe2O4 | Nano-particles | 100 nm | 89% CO, 77% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 77.4% CH4, 67.1% CO2 | ||||||||||
| NiFe2O4#SiO2 | Nano-particles | 100 nm | 90% CO, 81% H2 | NiFe2O4#SiO2 conversion and selectivity , carbon deposit | 80% CH4, 70.3% CO2 | ||||||||||
| 2 | 150 | 40 | ? | 5–100 kHz | 1 | / | 225 | Ni/SiO2 | ? | ? | 87% CO, 73% H2 | Packing: conversion , selectivity | 65% CH4, 52% CO2 | [19] | |
| LaNiO3/SiO2 | ? | ? | 89% CO, 79% H2 | Packing: conversion , selectivity | 82% CH4, 69% CO2 | ||||||||||
| DRM/packed | LaNiO3 | ? | ? | 90% CO, 81% H2 | Packing: conversion , selectivity | 84% CH4, 72% CO2 | |||||||||
| LaNiO3@SiO2 | ? | ? | 92% CO, 84% H2 | Packing: conversion , selectivity | 88% CH4, 78% CO2 | ||||||||||
| 4.5 | 50 | 50 | ? | 30–40 kHz | 1 | / | 60 | Ni/Al2O3 | Pellets | 0.85–5 mm | 25% CO, 45% H2, 10% C2, 5% C3 | non-packed: filamentary discharge, packed: combination of surface discharges microdischarges, breakdown voltage and conversion | 18% CH4, 13% CO2 | [25] | |
| 3.5 | 1.4–4.8 | 40 | 27.2 | 50 Hz | 0.5–2 | / | 2–7.2 | Ni/Al2O3 | Pellets | 1 mm | 35% CO, 56% H2 | Conversion with packing. Conversion when ratio | 52% CH4, 43% CO2 (38% CH4, 23% CO2 non-packed) | [20] | |
| 3 | 19 | 16.7–33.3 | ? | 6 kHz | 1 | 130–340 °C | 34–68 | Al2O3 | ? | 1–2 mm | 19% CO, 24% H2, 0.6% C2H2/C2H4, 10% C2H6, 0.3% C3H6, 6% C3H8, 1.3% CH3OH | Conversion with packing | 52% CH4, 31% CO2 | [21] | |
| Fe/Al2O3 | ? | 1–2 mm | 14% CO, 21% H2, 1.3% C2H2/C2H4, 9% C2H6, 0.3% C3H6, 5% C3H8, 1% CH3OH | No effect of T or flow rate, Conversion with packing | 46% CH4, 20% CO2 | ||||||||||
| zeolite NaY | ? | ? | 10% CO, 21% H2, 1% C2H2/C2H4, 6% C2H6, 0.2% C3H6, 3% C3H8, 0% CH3OH | No effect of T or flow rate, Conversion with packing | 49% CH4, 19% CO2 | ||||||||||
| zeolite Na ZSM-5 | ? | ? | 5% CO, 21% H2, 0.1% C2H2/C2H4, 9% C2H6, 0% C3H6, 5% C3H8, 0% CH3OH | Conversion with packing | 65% CH4, 40% CO2 | ||||||||||
| 4 | 15–60 | 5–50 | ? | 1–100 kHz | 1 | 325–525 °C | 18–720 | Ni/γ-Al2O3 | Grains | 70–100 mesh | ? | Conversion with packing (fluidized bed) | 48% CH4, 40% CO2 | [22] | |
| 3 | 19 | 16.7–33.3 | ? | 5.7–6 kHz | 1–2 | 120–290 °C | 34–68 | Al2O3 | ? | 1–2 mm | 38% CO, 28% H2, 11% C2, 6% C3, 4% C4, 2% CH3OH | Conversion with packing | 55% CH4, 31% CO2 | [26] | |
| Pd/Al2O3 | ? | 1–2 mm | 40% CO, 29% H2, 15% C2, 5% C3, 3% C4, 1% CH3OH | Conversion with packing | 51% CH4, 28% CO2 | ||||||||||
| Ag/Al2O3 | ? | 1–2 mm | 38% CO, 29% H2, 10% C2, 5% C3, 4% C4, 2% CH3OH | Conversion with packing | 52% CH4, 30% CO2 | ||||||||||
| 4.5 | 10–40 | 50 | 16.5 | 30–40 kHz | 1 | / | 12–48 | Quartz wool | ? | ? | 28% CO, 22% H2, 1% C2H2/C2H4, 7% C2H6, 0.5% C3H6, 4% C3H8 (estimation) | CH4 conversion: quartz wool> no packing > Al2O3 > zeolite 3A | 30% CH4, 12% CO2 | [28] | |
| γ-Al2O3 | pellets | 500–850 μm | 32% CO, 18% H2, 2% C2H2/C2H4, 8% C2H6, 0.5% C3H6, 4% C3H8 (estimation) | CH4 conversion: quartz wool> no packing > Al2O3 > zeolite 3A | 23% CH4, 8% CO2 | ||||||||||
| DRM/ packed | zeolite 3A | beads | 2 mm | 22% CO, 30% H2, 19% C2H2/C2H4, 8% C2H6, 1% C3H6, 6% C3H8 (estimation) | CH4 conversion: quartz wool> no packing > Al2O3 > zeolite 3A | 7% CH4, 3% CO2 | |||||||||
| 2.5 | 7.5–15 | 25–200 | 11.6 | 50 Hz | 0.11–9 | / | 2–36 | Ni/γ-Al2O3 | ? | ? | 37% CO, 33% H2, 22% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 . | 19% CH4, 9% CO2 | [23] | |
| Co/γ-Al2O4 | ? | ? | 42% CO, 43% H2, 30% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 | 15% CH4, 8% CO2 | ||||||||||
| Cu/γ-Al2O5 | ? | ? | 43% CO, 44% H2, 30% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 | 14% CH4, 8% CO2 | ||||||||||
| Mn/γ-Al2O6 | ? | ? | 35% CO, 34% H2, 24% C2H6 | Ni/γ-Al2O3 and Mn/γ-Al2O3: CH4 conversion , yields CO and H2 | 18% CH4, 10% CO2 | ||||||||||
| 7.5 | 46–106 | 25–100 | 100 | 25 kHz | 1 | 110 °C | 28–254 | BaTiO3 | Beads | 3 mm | 50% CO, 56% H2 | BaTiO3 size = conversions | 33% CH4, 20% CO2 | [24] | |
| Ni/SiO2 | Pellets | 2–3 mm | 56% CO, 54% H2 | Packing = conversions | 20% CH4, 12% CO2 | ||||||||||
| NiFe/SiO2 | Pellets | 2–3 mm | 54% CO, 56% H2 | Packing = conversions | 28% CH4, 15% CO2 | ||||||||||
| CH4 Conversion/CO2 Conversion | CO/H2 | ||
|---|---|---|---|
| 1.25–1.4 mm | ZrO2 | 1.7 | 5.5 |
| SiO2 | 1.7 | 4.8 | |
| α-Al2O3 | 1.5 | 9.5 | |
| BaTiO3 | 1.6 | 6.0 | |
| 1.6–1.8 mm | ZrO2 | 1.8 | 5.9 |
| SiO2 | 1.9 | 4.7 | |
| α-Al2O3 | 1.8 | 8.8 | |
| BaTiO3 | 1.9 | 6.3 | |
| 2.0–2.24 mm | ZrO2 | 2.2 | 6.4 |
| SiO2 | 1.2 | 5.3 | |
| α-Al2O3 | 1.5 | 9.0 | |
| BaTiO3 | 2.0 | 6.9 | |
| γ-Al2O3 | 2.3 | 8.3 | |
| Non-packed reactor | 50 mL/min | 1.9 | 7.9 |
| 192 mL/min | 2.0 | 7.2 |
| CO | H2 | C2H6 | C2H4 | C2H2 | C3H8 | C2H6O | C2H5OH | CH2O | CH3OH | CO/H2 Ratio | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethane | Ethene | Ethyne | Propane | DME | Ethanol | Formaldehyde | Methanol | |||||
| Non-packed (50 mL/min) | 64.2 | 8.1 | 15.9 | 2.7 | 4.1 | 4.3 | 0.2 | 0.22 | 0.14 | 0.15 | 7.9 | |
| Non-packed (192 mL/min) | 53.5 | 7.4 | 23.8 | 2.8 | 5.6 | 4.0 | 0.7 | 0.27 | 1.63 | 0.28 | 7.2 | |
| ZrO2 | 1.25–1.4 | 58.8 | 10.8 | 11.6 | 1.6 | 11.9 | 2.9 | 0.9 | 0.33 | 0.87 | 0.36 | 5.5 |
| 1.6–1.8 | 60.4 | 10.2 | 12.2 | 1.5 | 10.9 | 3.1 | 0.7 | 0.33 | 0.42 | 0.31 | 5.9 | |
| 2.0–2.24 | 63.4 | 9.8 | 12.0 | 1.2 | 8.6 | 3.3 | 0.5 | 0.34 | 0.32 | 0.32 | 6.4 | |
| SiO2 | 1.25–1.4 | 49.3 | 10.3 | 11.7 | 2.7 | 20.3 | 2.9 | 0.9 | 0.24 | 1.44 | 0.26 | 4.8 |
| 1.6–1.8 | 51.9 | 11.0 | 11.8 | 2.0 | 18.0 | 3.3 | 0.8 | 0.28 | 0.51 | 0.26 | 4.7 | |
| 2.0–2.24 | 56.3 | 10.6 | 13.0 | 13.1 | 3.7 | 0.6 | 0.31 | 0.41 | 0.33 | 5.3 | ||
| α-Al2O3 | 1.25–1.4 | 71.3 | 7.5 | 14.0 | 0.5 | 2.2 | 2.9 | 0.5 | 0.28 | 0.29 | 0.43 | 9.5 |
| 1.6–1.8 | 70.9 | 8.0 | 13.6 | 0.6 | 2.7 | 2.7 | 0.4 | 0.25 | 0.36 | 0.41 | 8.8 | |
| 2.0–2.24 | 72.2 | 8.0 | 12.9 | 0.5 | 2.2 | 2.9 | 0.4 | 0.29 | 0.29 | 0.46 | 9 | |
| BaTiO3 | 1.25–1.4 | 53.1 | 8.9 | 11.1 | 2.5 | 13.8 | 2.4 | 1.6 | 0.40 | 5.28 | 0.86 | 6 |
| 1.6–1.8 | 59.0 | 9.3 | 12.4 | 2.1 | 11.4 | 2.5 | 1.3 | 0.37 | 0.96 | 0.69 | 6.3 | |
| 2.0–2.24 | 61.4 | 8.9 | 13.7 | 1.8 | 9.0 | 2.8 | 0.8 | 0.33 | 0.59 | 0.57 | 6.9 | |
| γ-Al2O3 | 2.0–2.24 | 70.1 | 8.5 | 15.4 | 0.6 | 2.0 | 0.4 | 0.0 | 3.0 | 0.0 | 0.0 | 8.3 |
| Parameter | Specification |
|---|---|
| Gap (mm) | 4.5 |
| Frequency (kHz) | 23.5 |
| Power (Watt) | 100 |
| Gas flow rate (mL/min) | 50 (or 192, for non-packed, to have the same residence time as in the packed reactor) |
| Type of material | Non-packed reactor versus SiO2, α-Al2O3, γ-Al2O3, ZrO2 and BaTiO3 |
| Diameter spheres (mm) a | 1.25–1.4; 1.6–1.8; 2.0–2.24 |
| CO2/CH4 ratio | 1/1 |
| Temperature | Ambient (no external heating) |
| Pressure | Atmospheric (±1.2 atm) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michielsen, I.; Uytdenhouwen, Y.; Bogaerts, A.; Meynen, V. Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material. Catalysts 2019, 9, 51. https://doi.org/10.3390/catal9010051
Michielsen I, Uytdenhouwen Y, Bogaerts A, Meynen V. Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material. Catalysts. 2019; 9(1):51. https://doi.org/10.3390/catal9010051
Chicago/Turabian StyleMichielsen, Inne, Yannick Uytdenhouwen, Annemie Bogaerts, and Vera Meynen. 2019. "Altering Conversion and Product Selectivity of Dry Reforming of Methane in a Dielectric Barrier Discharge by Changing the Dielectric Packing Material" Catalysts 9, no. 1: 51. https://doi.org/10.3390/catal9010051