Wastewater Treatment by Catalytic Wet Peroxidation Using Nano Gold-Based Catalysts: A Review

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Advanced Oxidation Processes
3. Catalytic Wet Peroxidation
4. Nano Gold-based Catalysts
4.1. Deposition/Precipitation
4.2. Co-Precipitation
4.3. Impregnation Method
4.4. Vapor-phase Deposition and Grafting Methods
4.5. Sol-Gel Method
4.6. Ion-Exchange Method
5. Application of CWPO using Gold Catalysts in Wastewater Treatment
5.1. Influence of the Catalyst Properties
5.2. Effect of the Operating Conditions
5.2.1. Catalyst Dose
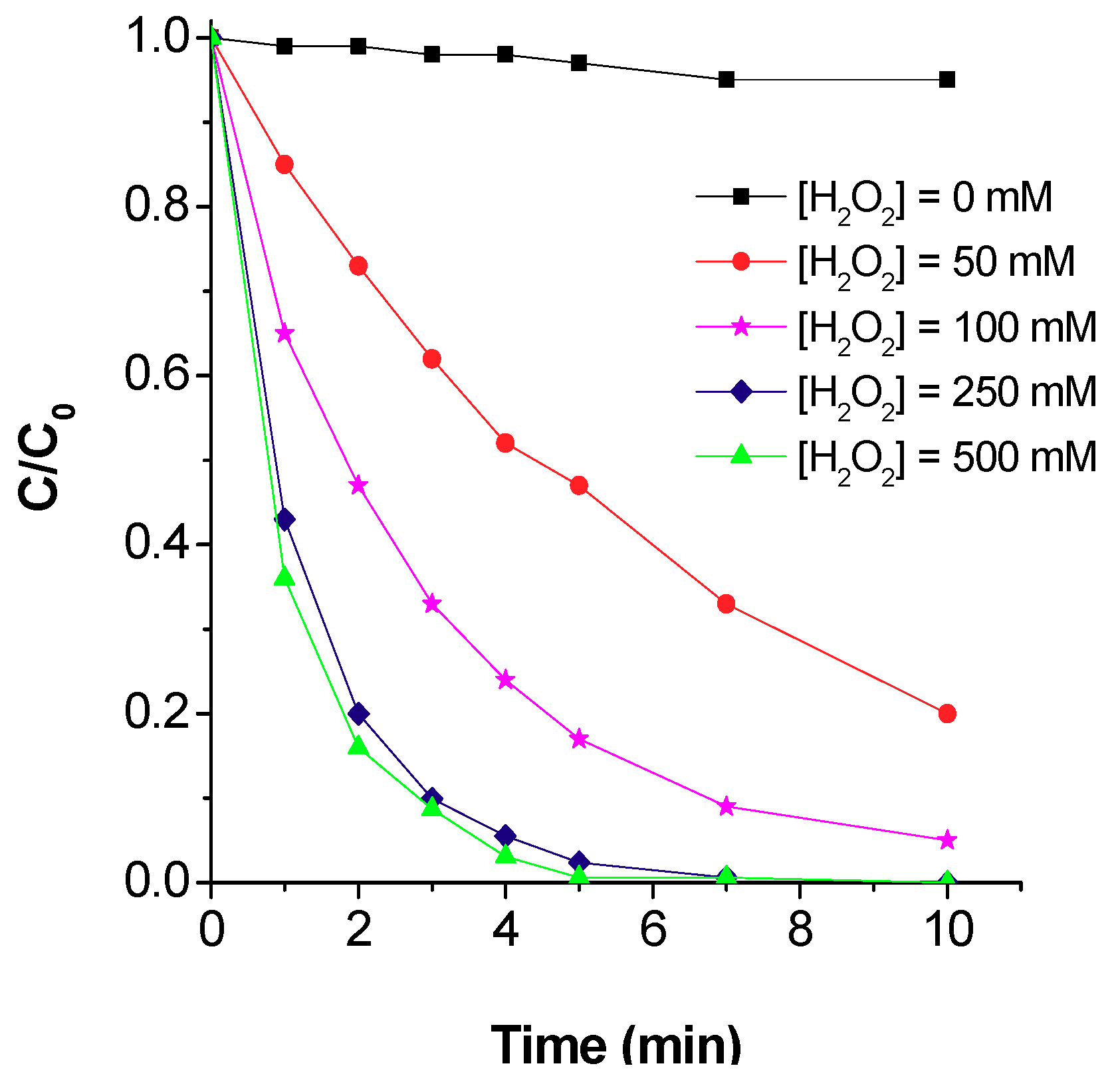
5.2.2. Hydrogen Peroxide Concentration
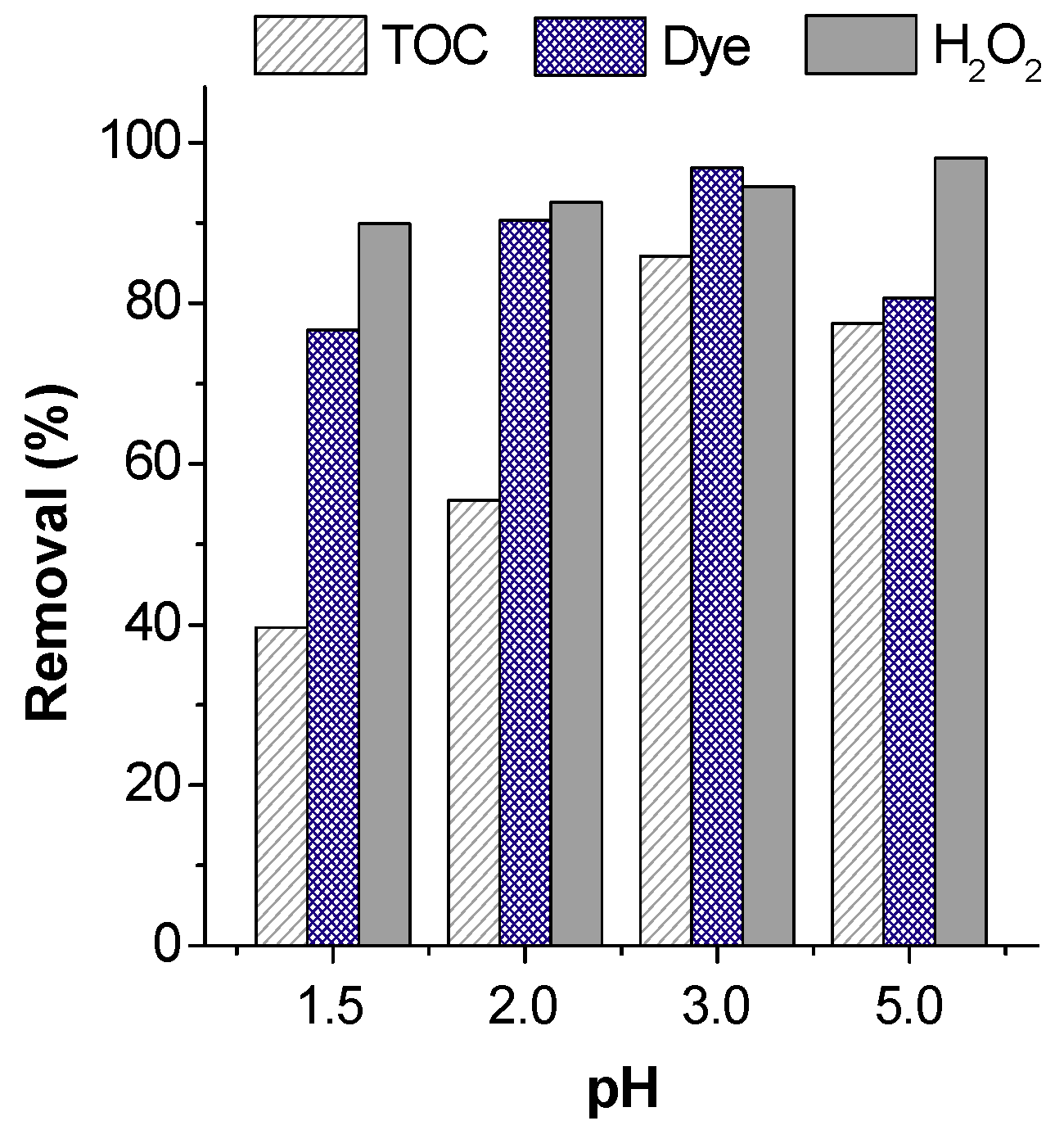
5.2.3. Initial pH
5.2.4. Temperature
5.2.5. Effect of radiation use
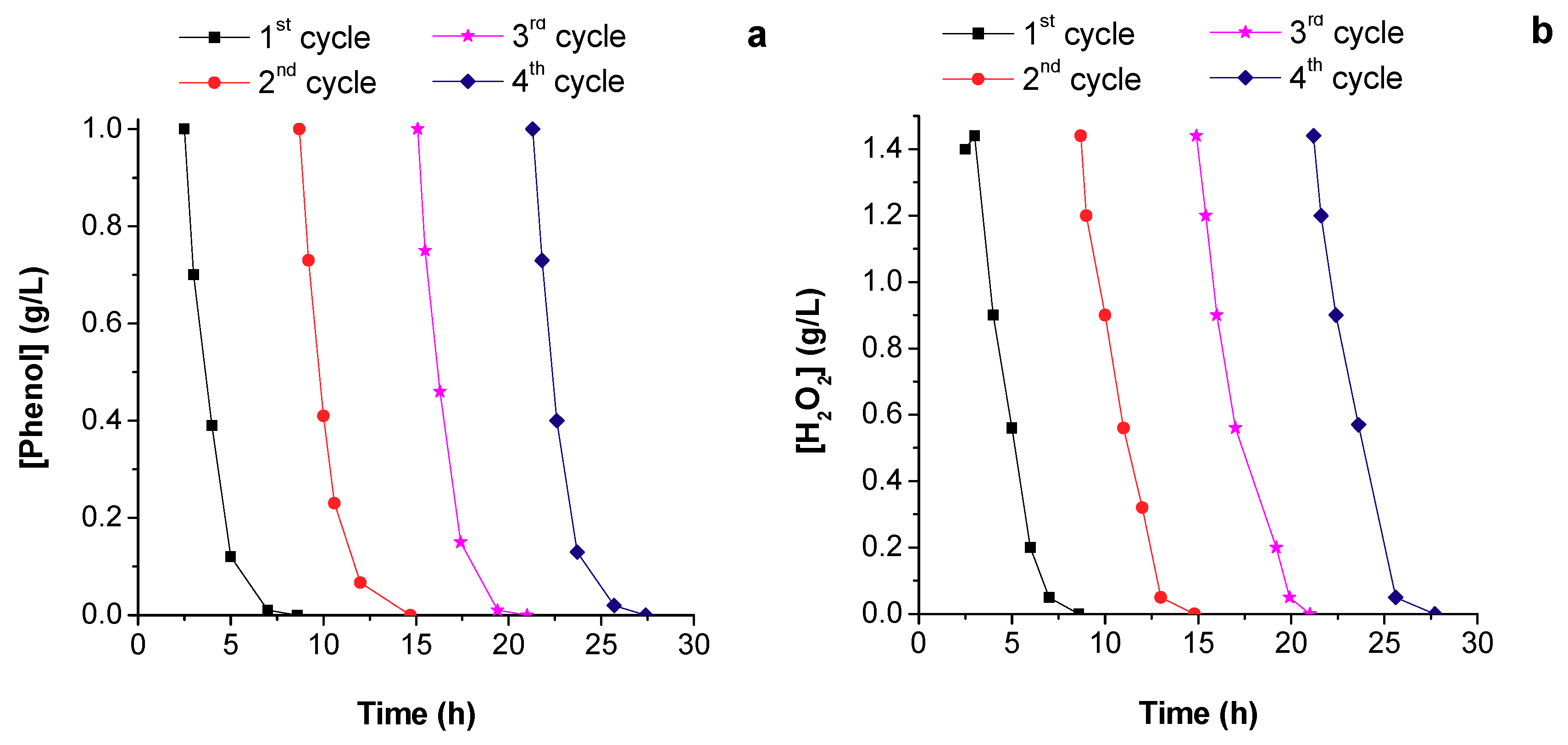
5.3. Catalyst Stability
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AO7 | Acid Orange dye |
| AOPs | Advanced Oxidation Processes |
| Au/AC | Gold on activated carbon |
| Au/C | Gold on carbon |
| Au/CNF | Gold on carbon nanofibers |
| Au/CNT | Gold on carbon nanotubes |
| Au/X40s | Gold on coconut shell carbon |
| Au/DNP | Gold on diamond nanoparticles |
| Au/F | Gold on diamond after thermal treatment at 420 °C in air atmosphere |
| Au/FH2 | Gold on diamond after thermal treatment at 420 °C in air atmosphere and at 500 °C in hydrogen atmosphere |
| Au/FN2 | Gold on diamond after thermal treatment at 420 °C in air atmosphere and at 500 °C in nitrogen atmosphere |
| Au/Hap | Gold on hydroxyapatite |
| Au/npD | Gold on nano power diamond |
| Au/HO-npD | Gold on nano power diamond previously treated with Fenton reagent |
| Au/FDU-15 | Gold on ordered mesoporous carbon |
| Au/PSAC | Gold on pitch-based spherical activated carbon |
| Au/SRAC | Gold on styrene-based activated carbon |
| Au/TN | Gold on titanium nanotubes functionalization with hydrogen peroxide |
| Au/TiO2-AD | Gold on titanium oxide prepared by adsorption method |
| BOD5 | Biological oxygen demand after 5 days |
| BPA | Bisphenol A |
| CWPO | Catalytic Wet Peroxidation |
| COD | Chemical oxygen demand |
| DM | Gold metal dispersion |
| DPPH | 1,1-diphenyl-2-picrylhydzazyl |
| EU-WFD | European Union Water Framework Directive |
| HR-TEM | High-resolution transmission electron microscopy |
| hν | Ultraviolet radiation |
| M | Transmission of metallic cations |
| MB | Methyl Blue dye |
| MO | Methyl Orange dye |
| NHE | Normal hydrogen electrode |
| OII | Orange II dye |
| RH | Organic matter |
| TOC | Total organic carbon |
| TOF | Turn off frequency |
| WGC | World Gold Council |
| X | Support |
References
- Zeng, G.-M.; Li, X.; Huang, J.-H.; Zhang, C.; Zhou, C.-F.; Niu, J.; Shi, L.-J.; He, S.-B.; Li, F. Micellar-enhanced ultrafiltration of cadmium and methylene blue in synthetic wastewater using sds. J. Hazard. Mater. 2011, 185, 1304–1310. [Google Scholar] [CrossRef]
- Cundy, A.B.; Hopkinson, L.; Whitby, R.L.D. Use of iron-based technologies in contaminated land and groundwater remediation: A review. Sci. Total Environ. 2008, 400, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.N.; Jin, B.; Chow, C.W.K.; Saint, C. Recent developments in photocatalytic water treatment technology: A review. Water Res. 2010, 44, 2997–3027. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, G.A. Organic compounds in sludge-amended soils and their potential for uptake by crop plants. Sci. Total Environ. 1996, 185, 71–81. [Google Scholar] [CrossRef]
- Li, X.; Zeng, G.-M.; Huang, J.-H.; Zhang, D.-M.; Shi, L.-J.; He, S.-B.; Ruan, M. Simultaneous removal of cadmium ions and phenol with meuf using sds and mixed surfactants. Desalination 2011, 276, 136–141. [Google Scholar] [CrossRef]
- Fatta-Kassinos, D.; Kalavrouziotis, I.K.; Koukoulakis, P.H.; Vasquez, M.I. The risks associated with wastewater reuse and xenobiotics in the agroecological environment. Sci. Total Environ. 2011, 409, 3555–3563. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zeng, G.M.; Huang, D.L.; Feng, C.L.; Hu, S.; Zhao, M.H.; Lai, C.; Wei, Z.; Huang, C.; Xie, G.X.; et al. Use of iron oxide nanomaterials in wastewater treatment: A review. Sci. Total Environ. 2012, 424, 1–10. [Google Scholar] [CrossRef]
- European Parliament & Council. Water Framework Directive 2000/60/ce; European Parliament & Counci: Brussels, Belgium, 2000; pp. 1–73. [Google Scholar]
- Oturan, M.A.; Aaron, J.-J. Advanced oxidation processes in water/wastewater treatment: Principles and applications. A review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes. J. Hazard. Mater. 2014, 275, 121–135. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. A review of imperative technologies for wastewater treatment I: Oxidation technologies at ambient conditions. Adv. Environ. Res. 2004, 8, 501–551. [Google Scholar] [CrossRef]
- Seow, T.W.; Lim, C.K.; Norb, M.H.M.; Mubarak, M.F.M.; Lam, C.Y.L.; Yahya, A.; Ibrahim, Z. Review on wastewater treatment technologies. Int. J. Appl. Environ. Sci. 2016, 11, 111–126. [Google Scholar]
- Ramalho, R.S. Introduction to Wastewater Treatment Processes; Academic Press: New York, NY, USA, 1977. [Google Scholar]
- Pant, D.; Adholeya, A. Biological approaches for treatment of distillery wastewater: A review. Bioresour. Technol. 2007, 98, 2321–2334. [Google Scholar] [CrossRef]
- Demirel, B.; Yenigun, O.; Onay, T.T. Anaerobic treatment of dairy wastewaters: A review. Process Biochem. 2005, 40, 2583–2595. [Google Scholar] [CrossRef]
- Wolfe, S.; Ingold, C.F. Oxidation of organic compounds by zinc permanganate. J. Am. Chem. Soc. 1983, 105, 7755–7757. [Google Scholar] [CrossRef]
- Xu, X.-R.; Li, H.-B.; Wang, W.-H.; Gu, J.-D. Decolorization of dyes and textile wastewater by potassium permanganate. Chemosphere 2005, 59, 893–898. [Google Scholar] [CrossRef]
- Calvosa, L.; Monteverdi, A.; Rindone, B.; Riva, G. Ozone oxidation of compounds resistant to biological degradation. Water Res. 1991, 25, 985–993. [Google Scholar] [CrossRef]
- Stasinakis, A.S. Use of selected advanced oxidation processes (aops) for wastewater treatment—A mini review. Glob. NEST J. 2008, 10, 376–385. [Google Scholar]
- Azbar, N.; Yonar, T.; Kestioglu, K. Comparison of various advanced oxidation processes and chemical treatment methods for COD and color removal from a polyester and acetate fiber dyeing effluent. Chemosphere 2004, 55, 35–43. [Google Scholar] [CrossRef]
- Lamarche, P.; Droste, R.L. Air-stripping mass transfer correlations for volatile organics. J. Am. Water Works Assoc. 1989, 81, 78–89. [Google Scholar] [CrossRef]
- Busca, G.; Berardinelli, S.; Resini, C.; Arrighi, L. Technologies for the removal of phenol from fluid streams: A short review of recent developments. J. Hazard. Mater. 2008, 160, 265–288. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, R.; Caprio, V.; Insola, A.; Marotta, R. Advanced oxidation processes (AOP) for water purification and recovery. Catal. Today 1999, 53, 51–59. [Google Scholar] [CrossRef]
- Poyatos, J.M.; Muñio, M.M.; Almecija, M.C.; Torres, J.C.; Hontoria, E.; Osorio, F. Advanced oxidation processes for wastewater treatment: State of the art. Water Air Soil Pollut. 2010, 205, 187–204. [Google Scholar] [CrossRef]
- Skoumal, M.; Cabot, P.-L.; Centellas, F.; Arias, C.; Rodríguez, R.M.; Garrido, J.A.; Brillas, E. Mineralization of paracetamol by ozonation catalyzed with Fe2+, Cu2+ and UVA light. Appl. Catal. B Environ. 2006, 66, 228–240. [Google Scholar] [CrossRef]
- Rosenfeldt, E.J.; Chen, P.J.; Kullman, S.; Linden, K.G. Destruction of estrogenic activity in water using UV advanced oxidation. Sci. Total Environ. 2007, 377, 105–113. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1934, 147, 332–351. [Google Scholar]
- Mahamuni, N.N.; Adewuyi, Y.G. Advanced oxidation processes (aops) involving ultrasound for waste water treatment: A review with emphasis on cost estimation. Ultrason. Sonochem. 2010, 17, 990–1003. [Google Scholar] [CrossRef]
- Herney-Ramirez, J.; Vicente, M.A.; Madeira, L.M. Heterogeneous photo-Fenton oxidation with pillared clay-based catalysts for wastewater treatment: A review. Appl. Catal. B Environ. 2010, 98, 10–26. [Google Scholar] [CrossRef]
- Esteves, B.M.; Rodrigues, C.S.D.; Madeira, L.M. Wastewater treatment by heterogeneous Fenton-like processes in continuous reactors. In Applications of Advanced Oxidation Processes (AOPs) in Drinking Water Treatment; Gil, A., Galeano, L.A., Vicente, M.Á., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 211–255. [Google Scholar]
- Pawłat, J.; Stryczewska Henryka, D.; Ebihara, K. Sterilization techniques for soil remediation and agriculture based on ozone and AOP. J. Adv. Oxid. Technol. 2010, 13, 138–145. [Google Scholar] [CrossRef]
- Flotron, V.; Delteil, C.; Padellec, Y.; Camel, V. Removal of sorbed polycyclic aromatic hydrocarbons from soil, sludge and sediment samples using the Fenton’s reagent process. Chemosphere 2005, 59, 1427–1437. [Google Scholar] [CrossRef]
- Tokumura, M.; Nakajima, R.; Znad, H.T.; Kawase, Y. Chemical absorption process for degradation of voc gas using heterogeneous gas–liquid photocatalytic oxidation: Toluene degradation by photo-Fenton reaction. Chemosphere 2008, 73, 768–775. [Google Scholar] [CrossRef]
- Liu, G.; Ji, J.; Huang, H.; Xie, R.; Feng, Q.; Shu, Y.; Zhan, Y.; Fang, R.; He, M.; Liu, S.; et al. UV/H2O2: An efficient aqueous advanced oxidation process for VOCs removal. Chem. Eng. J. 2017, 324, 44–50. [Google Scholar] [CrossRef]
- Domeño, C.; Rodríguez-Lafuente, Á.; Martos, J.; Bilbao, R.; Nerín, C. VOC removal and deodorization of effluent gases from an industrial plant by photo-oxidation, chemical oxidation, and ozonization. Environ. Sci. Technol. 2010, 44, 2585–2591. [Google Scholar] [CrossRef]
- Tokumura, M.; Shibusawa, M.; Kawase, Y. Dynamic simulation of degradation of toluene in waste gas by the photo-Fenton reaction in a bubble column. Chem. Eng. Sci. 2013, 100, 212–224. [Google Scholar] [CrossRef]
- Toor, R.; Mohseni, M. UV-H2O2 based AOP and its integration with biological activated carbon treatment for dbp reduction in drinking water. Chemosphere 2007, 66, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Shannon, M.A.; Bohn, P.W.; Elimelech, M.; Georgiadis, J.G.; Mariñas, B.J.; Mayes, A.M. Science and technology for water purification in the coming decades. Nature 2008, 452, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Comninellis, C.; Kapalka, A.; Malato, S.; Parsons, S.A.; Mantzavinos, I.P.D. Advanced oxidation processes for water treatment: Advances and trends for r&d. J. Chem. Technol. Biotechnol. 2008, 83, 769–776. [Google Scholar]
- Al Momani, F.A. Potential use of solar energy for waste activated sludge treatment. Int. J. Sustain. Eng. 2013, 6, 82–91. [Google Scholar] [CrossRef]
- Krzemieniewski, M.; Dębowski, M.; Janczukowicz, W.; Pesta, J. Effect of sludge conditioning by chemical methods with magnetic field application. Pol. J. Environ. Stud. 2003, 12, 595–605. [Google Scholar]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Brigda, R.J. Consider Fenton’s chemistry for wastewater treatment. Chem. Eng. Process. 1995, 91, 62–66. [Google Scholar]
- Ikehata, K.; Jodeiri Naghashkar, N.; Gamal El-Din, M. Degradation of aqueous pharmaceuticals by ozonation and advanced oxidation processes: A review. Ozone Sci. Eng. 2006, 28, 353–414. [Google Scholar] [CrossRef]
- Rice, R.G.; Netzer, A. Handbook of Ozone Technology and Applications; Ann Arbor Science Publishers: Butterworths, UK, 1982; Volume 1. [Google Scholar]
- Rodrigues, C.S.D.; Neto, A.R.; Duda, R.M.; de Oliveira, R.A.; Boaventura, R.A.R.; Madeira, L.M. Combination of chemical coagulation, photo-Fenton oxidation and biodegradation for the treatment of vinasse from sugar cane ethanol distillery. J. Clean. Prod. 2017, 142, 3634–3644. [Google Scholar] [CrossRef] [Green Version]
- Inchaurrondo, N.S.; Massa, P.; Fenoglio, R.; Font, J.; Haure, P. Efficient catalytic wet peroxide oxidation of phenol at moderate temperature using a high-load supported copper catalyst. Chem. Eng. J. 2012, 198, 426–434. [Google Scholar] [CrossRef]
- Maciel, R.; Sant’Anna, G.L.; Dezotti, M. Phenol removal from high salinity effluents using Fenton’s reagent and photo-Fenton reactions. Chemosphere 2004, 57, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Walling, C. Fenton’s reagent revisited. Acc. Chem. Res. 1975, 8, 125–131. [Google Scholar] [CrossRef]
- Gosu, V.; Dhakar, A.; Sikarwar, P.; Kumar, U.K.A.; Subbaramaiah, V.; Zhang, T.C. Wet peroxidation of resorcinol catalyzed by copper impregnated granular activated carbon. J. Environ. Manag. 2018, 223, 825–833. [Google Scholar] [CrossRef]
- Catrinescu, C.; Teodosiu, C.; Macoveanu, M.; Miehe-Brendlé, J.; Le Dred, R. Catalytic wet peroxide oxidation of phenol over fe-exchanged pillared beidellite. Water Res. 2003, 37, 1154–1160. [Google Scholar] [CrossRef]
- Neyens, E.; Baeyens, J. A review of classic fenton’s peroxidation as an advanced oxidation technique. J. Hazard. Mater. 2003, 98, 33–50. [Google Scholar] [CrossRef]
- Ribeiro, R.S.; Silva, A.M.T.; Figueiredo, J.L.; Faria, J.L.; Gomes, H.T. Catalytic wet peroxide oxidation: A route towards the application of hybrid magnetic carbon nanocomposites for the degradation of organic pollutants. A review. Appl. Catal. B Environ. 2016, 187, 428–460. [Google Scholar] [CrossRef]
- Perathoner, S.; Centi, G. Wet hydrogen peroxide catalytic oxidation (WHPCO) of organic waste in agro-food and industrial streams. Top. Catal. 2005, 33, 207–224. [Google Scholar] [CrossRef]
- Melero, J.A.; Martínez, F.; Botas, J.A.; Molina, R.; Pariente, M.I. Heterogeneous catalytic wet peroxide oxidation systems for the treatment of an industrial pharmaceutical wastewater. Water Res. 2009, 43, 4010–4018. [Google Scholar] [CrossRef] [PubMed]
- European Economic Community. List of Council Directives 76/4647; European Economic Community: Brussels, Belgium, 1982. [Google Scholar]
- Feng, J.; Hu, X.; Yue, P.L. Effect of initial solution ph on the degradation of orange II using clay-based Fe nanocomposites as heterogeneous photo-Fenton catalyst. Water Res. 2006, 40, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Kullmann, S.; Keller, H. Wastewater treatment with heterogeneous Fenton-type catalysts based on porous materials. J. Mater. Chem. 2010, 20, 9002–9017. [Google Scholar] [CrossRef]
- Dantas, T.L.P.; Mendonça, V.P.; José, H.J.; Rodrigues, A.E.; Moreira, R.F.P.M. Treatment of textile wastewater by heterogeneous Fenton process using a new composite Fe2O3/carbon. Chem. Eng. J. 2006, 118, 77–82. [Google Scholar] [CrossRef]
- Liou, R.-M.; Chen, S.-H.; Hung, M.-Y.; Hsu, C.-S.; Lai, J.-Y. Fe (III) supported on resin as effective catalyst for the heterogeneous oxidation of phenol in aqueous solution. Chemosphere 2005, 59, 117–125. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Zhao, G. Iron-copper bimetallic nanoparticles embedded within ordered mesoporous carbon as effective and stable heterogeneous Fenton catalyst for the degradation of organic contaminants. Appl. Catal. B Environ. 2015, 164, 396–406. [Google Scholar] [CrossRef]
- Subbaramaiah, V.; Srivastava, V.C.; Mall, I.D. Catalytic wet peroxidation of pyridine bearing wastewater by cerium supported SBA-15. J. Hazard. Mater. 2013, 248–249, 355–363. [Google Scholar] [CrossRef]
- Aravindhan, R.; Fathima, N.N.; Rao, J.R.; Nair, B.U. Wet oxidation of acid brown dye by hydrogen peroxide using heterogeneous catalyst Mn-Salen-Y zeolite: A potential catalyst. J. Hazard. Mater. 2006, 138, 152–159. [Google Scholar] [CrossRef]
- Hosseini, S.A.; Davodian, M.; Abbasian, A.R. Remediation of phenol and phenolic derivatives by catalytic wet peroxide oxidation over Co-Ni layered double nano hydroxides. J. Taiwan Inst. Chem. Eng. 2017, 75, 97–104. [Google Scholar] [CrossRef]
- Rodrigues, C.S.D.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Madeira, L.M. Wet peroxide oxidation of dye-containing wastewaters using nanosized Au supported on Al2O3. Catal. Today 2017, 280, 165–175. [Google Scholar] [CrossRef]
- Rodrigues, C.S.D.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Madeira, L.M. Orange II degradation by wet peroxide oxidation using Au nanosized catalysts: Effect of the support. Ind. Eng. Chem. Res. 2017, 56, 1988–1998. [Google Scholar] [CrossRef]
- Quintanilla, A.; García-Rodríguez, S.; Domínguez, C.M.; Blasco, S.; Casas, J.A.; Rodriguez, J.J. Supported gold nanoparticle catalysts for wet peroxide oxidation. Appl. Catal. B Environ. 2012, 111–112, 81–89. [Google Scholar] [CrossRef]
- Hassan, H.; Hameed, B.H. Fe–clay as effective heterogeneous Fenton catalyst for the decolorization of reactive blue 4. Chem. Eng. J. 2011, 171, 912–918. [Google Scholar] [CrossRef]
- Han, Y.-F.; Phonthammachai, N.; Ramesh, K.; Zhong, Z.; White, T. Removing organic compounds from aqueous medium via wet peroxidation by gold catalysts. Environ. Sci. Technol. 2008, 42, 908–912. [Google Scholar] [CrossRef]
- Ferentz, M.; Landau, M.V.; Vidruk, R.; Herskowitz, M. Fixed-bed catalytic wet peroxide oxidation of phenol with titania and Au/titania catalysts in dark. Catal. Today 2015, 241, 63–72. [Google Scholar] [CrossRef]
- Domínguez, C.M.; Quintanilla, A.; Casas, J.A.; Rodriguez, J.J. Kinetics of wet peroxide oxidation of phenol with a gold/activated carbon catalyst. Chem. Eng. J. 2014, 253, 486–492. [Google Scholar] [CrossRef]
- Martín, R.; Navalon, S.; Alvaro, M.; Garcia, H. Optimized water treatment by combining catalytic Fenton reaction using diamond supported gold and biological degradation. Appl. Catal. B Environ. 2011, 103, 246–252. [Google Scholar] [CrossRef]
- Navalon, S.; Martín, R.; Alvaro, M.; Garcia, H. Gold on diamond nanoparticles as a highly efficient Fenton catalyst. Angew. Chem. 2010, 122, 8581–8585. [Google Scholar] [CrossRef]
- Sempere, D.; Navalon, S.; Dančíková, M.; Alvaro, M.; Garcia, H. Influence of pretreatments on commercial diamond nanoparticles on the photocatalytic activity of supported gold nanoparticles under natural sunlight irradiation. Appl. Catal. B Environ. 2013, 142–143, 259–267. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid-liquid system. J. Chem. Soc. Chem. Commun. 1994, 7, 801–802. [Google Scholar] [CrossRef]
- Primo, A.; García, H. Chapter 18—Supported gold nanoparticles as heterogeneous catalysts. In New and Future Developments in Catalysis; Suib, S.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 425–449. [Google Scholar]
- Jiang, G.; Wang, L.; Chen, T.; Yu, H.; Chen, C. Preparation of gold nanoparticles in the presence of poly(benzyl ether) alcohol dendrons. Mater. Chem. Phys. 2006, 98, 76–82. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Thompson, D.T. Catalytic applications for gold nanotechnology. In Nanocatalysis; Heiz, U., Landman, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 377–489. [Google Scholar]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Haruta, M. Gold as a novel catalyst in the 21st century: Preparation, working mechanism and applications. Gold Bull. 2004, 37, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Bond, G.C.; Thompson, D.T. Catalysis by gold. Catal. Rev. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Hodge, N.A.; Kiely, C.J.; Whyman, R.; Siddiqui, M.R.H.; Hutchings, G.J.; Pankhurst, Q.A.; Wagner, F.E.; Rajaram, R.R.; Golunski, S.E. Microstructural comparison of calcined and uncalcined gold/iron-oxide catalysts for low-temperature CO oxidation. Catal. Today 2002, 72, 133–144. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Abad, A.; Almela, C.; Corma, A.; García, H. Efficient chemoselective alcohol oxidation using oxygen as oxidant. Superior performance of gold over palladium catalysts. Tetrahedron 2006, 62, 6666–6672. [Google Scholar] [CrossRef]
- Baatz, C.; Decker, N.; Prüße, U. New innovative gold catalysts prepared by an improved incipient wetness method. J. Catal. 2008, 258, 165–169. [Google Scholar] [CrossRef]
- Lorençon, E.; Ferreira, D.C.; Resende, R.R.; Krambrock, K. Amphiphilic gold nanoparticles supported on carbon nanotubes: Catalysts for the oxidation of lipophilic compounds by wet peroxide in biphasic systems. Appl. Catal. A Gen. 2015, 505, 566–574. [Google Scholar] [CrossRef]
- Lin, J.-N.; Wan, B.-Z. Effects of preparation conditions on gold/Y-type zeolite for CO oxidation. Appl. Catal. B Environ. 2003, 41, 83–95. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Herzing, A.A.; Kiely, C.J.; Carley, A.F.; Landon, P.; Hutchings, G.J. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. [Google Scholar] [CrossRef]
- Corma, A.; Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 2006, 313, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active nonmetallic au and pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Ma, S.; Liu, P.; Hrbek, J.; Evans, J.; Pérez, M. Activity of CeOx and TiOx nanoparticles grown on Au(111) in the water-gas shift reaction. Science 2007, 318, 1757–1760. [Google Scholar] [CrossRef]
- Pérez, P.; Soria, M.A.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Mendes, A.; Madeira, L.M. Application of Au/ TiO2 catalysts in the low-temperature water-gas shift reaction. Int. J. Hydrog. Energy 2016, 41, 4670–4681. [Google Scholar] [CrossRef]
- Scirè, S.; Minicò, S.; Crisafulli, C.; Satriano, C.; Pistone, A. Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts. Appl. Catal. B Environ. 2003, 40, 43–49. [Google Scholar] [CrossRef]
- Centeno, M.A.; Paulis, M.; Montes, M.; Odriozola, J.A. Catalytic combustion of volatile organic compounds on Au/CeO2/Al2O3 and Au/Al2O3 catalysts. Appl. Catal. A Gen. 2002, 234, 65–78. [Google Scholar] [CrossRef]
- Scirè, S.; Liotta, L.F. Supported gold catalysts for the total oxidation of volatile organic compounds. Appl. Catal. B Environ. 2012, 125, 222–246. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Chen, D.-H. Catalytic reduction of 4-nitrophenol by magnetically recoverable Au nanocatalyst. J. Hazard. Mater. 2009, 165, 664–669. [Google Scholar] [CrossRef]
- Aprile, C.; Corma, A.; Domine, M.E.; Garcia, H.; Mitchell, C. A cascade aerobic epoxidation of alkenes over Au/CeO2 and ti-mesoporous material by “in situ” formed peroxides. J. Catal. 2009, 264, 44–53. [Google Scholar] [CrossRef]
- Cojocaru, B.; Neaţu, Ş.; Sacaliuc-Pârvulescu, E.; Lévy, F.; Pârvulescu, V.I.; Garcia, H. Influence of gold particle size on the photocatalytic activity for acetone oxidation of Au/TiO2 catalysts prepared by dc-magnetron sputtering. Appl. Catal. B Environ. 2011, 107, 140–149. [Google Scholar] [CrossRef]
- Marino, T.; Molinari, R.; García, H. Selectivity of gold nanoparticles on the photocatalytic activity of TiO2 for the hydroxylation of benzene by water. Catal. Today 2013, 206, 40–45. [Google Scholar] [CrossRef]
- Martínez, F.; Calleja, G.; Melero, J.A.; Molina, R. Heterogeneous photo-Fenton degradation of phenolic aqueous solutions over iron-containing SBA-15 catalyst. Appl. Catal. B Environ. 2005, 60, 181–190. [Google Scholar] [CrossRef]
- Kuznetsova, E.V.; Savinov, E.N.; Vostrikova, L.A.; Parmon, V.N. Heterogeneous catalysis in the Fenton-type system FeZSM-5/H2O2. Appl. Catal. B Environ. 2004, 51, 165–170. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Machado, B.F.; Bacsa, R.R.; Serp, P.; Dražić, G.; Faria, J.L.; Figueiredo, J.L. Catalytic performance of Au/ZnO nanocatalysts for CO oxidation. J. Catal. 2010, 273, 191–198. [Google Scholar] [CrossRef]
- Ge, L.; Chen, T.; Liu, Z.; Chen, F. The effect of gold loading on the catalytic oxidation performance of CeO2/H2O2 system. Catal. Today 2014, 224, 209–215. [Google Scholar] [CrossRef]
- Drašinac, N.; Erjavec, B.; Dražić, G.; Pintar, A. Peroxo and gold modified titanium nanotubes for effective removal of methyl orange with CWPO under ambient conditions. Catal. Today 2017, 280, 155–164. [Google Scholar] [CrossRef]
- Yang, X.; Tian, P.-F.; Zhang, C.; Deng, Y.-Q.; Xu, J.; Gong, J.; Han, Y.-F. Au/carbon as Fenton-like catalysts for the oxidative degradation of bisphenol A. Appl. Catal. B Environ. 2013, 134–135, 145–152. [Google Scholar] [CrossRef]
- Alvaro, M.; Cojocaru, B.; Ismail, A.A.; Petrea, N.; Ferrer, B.; Harraz, F.A.; Parvulescu, V.I.; Garcia, H. Visible-light photocatalytic activity of gold nanoparticles supported on template-synthesized mesoporous titania for the decontamination of the chemical warfare agent soman. Appl. Catal. B Environ. 2010, 99, 191–197. [Google Scholar] [CrossRef]
- Rodrigues, C.S.D.; Silva, R.M.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Madeira, L.M. Dye-containing wastewater treatment by photo-assisted wet peroxidation using Au nanosized catalysts. J. Chem. Technol. Biotechnol. 2018, 93, 3223–3323. [Google Scholar] [CrossRef]
- Navalon, S.; Martin, R.; Alvaro, M.; Garcia, H. Sunlight-assisted Fenton reaction catalyzed by goldsupported on diamond nanoparticles as pretreatment forbiological degradation of aqueous phenol solutions. ChemSusChem 2011, 4, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Galindo, C.; Jacques, P.; Kalt, A. Photochemical and photocatalytic degradation of an indigoid dye: A case study of acid blue 74 (AB74). J. Photochem. Photobiol. A Chem. 2001, 141, 47–56. [Google Scholar] [CrossRef]
- Spinks, J.W.T.; Woods, R.J. An Introduction to Radiation Chemistry, 3rd ed.; John Wiley & Sons Inc.: New York, NY, USA, 1990. [Google Scholar]
- Fida, H.; Zhang, G.; Guo, S.; Naeem, A. Heterogeneous Fenton degradation of organic dyes in batch and fixed bed using la-fe montmorillonite as catalyst. J. Colloid Interface Sci. 2017, 490, 859–868. [Google Scholar] [CrossRef]
- Navalon, S.; de Miguel, M.; Martin, R.; Alvaro, M.; Garcia, H. Enhancement of the catalytic activity of supported gold nanoparticles for the fenton reaction by light. J. Am. Chem. Soc. 2011, 133, 2218–2226. [Google Scholar] [CrossRef] [PubMed]
- Zazo, J.A.; Pliego, G.; Blasco, S.; Casas, J.A.; Rodriguez, J.J. Intensification of the Fenton process by increasing the temperature. Ind. Eng. Chem. Res. 2011, 50, 866–870. [Google Scholar] [CrossRef]
- Huang, C.P.; Dong, C.; Tang, Z. Advanced chemical oxidation: Its present role and potential future in hazardous waste treatment. Waste Manag. 1993, 13, 361–377. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Species | Oxidation Potential (eV) |
|---|---|
| Fluorine | 3.03 |
| Hydroxyl radical | 2.80 |
| Atomic oxygen | 2.42 |
| Ozone | 2.07 |
| Hydrogen peroxide | 1.77 |
| Potassium permanganate | 1.67 |
| Hypobromous acid | 1.59 |
| Chlorine dioxide | 1.50 |
| Hypochlorous acid | 1.49 |
| Chlorine | 1.36 |
| Bromine | 1.09 |
| Iodine | 0.54 |
| Model Compound/Effluent | Catalyst | Operation Conditions | Efficiency of CWPO | Ref. |
|---|---|---|---|---|
| Orange II (OII) dye | Au/Al2O3 (0.7 wt.%) | pH = 3.0; T = 50 °C; [H2O2] = 6 mM; [catalyst] = 2.0 g/L; [OII] = 0.1 mM; t = 4 h | Dye removal = 98.9%; TOC removal = 49.8%; COD removal = 42.2%; H2O2 consumption = 95.0%; Specific Oxygen Uptake Rate = 27.8 mgO2/(gVSS.h); Inhibition of Vibrio Fischeri = 0.0%; Gold leaching < 0.04% | [66] |
| Acrylic Dyeing Wastewater | Au/Al2O3 (0.7 wt.%) | pH = 3.0; T = 50 °C; [H2O2] = 3.52 g/L; [catalyst] = 2.0 g/L; t = 4 h | Color removal = 34.4%; TOC removal = 42.9%; COD removal = 50.5%; H2O2 consumption = 98.8%; BOD5:COD = 0.23; Gold leaching < 0.04% | |
| OII dye | Au/Al2O3 (0.7 wt.%) | pH = 3.0; T = 30 °C; [H2O2] = 6 mM; [catalyst] = 2.0 g/L; [OII] = 0.1 mM; t = 16 h | Dye removal = 99.4%; TOC removal = 48.2%; H2O2 consumption = 96.1%; Gold leaching < 0.04% | [67] |
| Au/Fe2O3 (0.8 wt.%) | Dye removal = 51.4%; TOC removal = 36.9%; H2O2 consumption = 68.5%; Gold leaching < 0.04% | |||
| Au/Fe2O3 (4.0 wt.%) from WGC | Dye removal = 40.9%; TOC removal = 29.6%; Gold leaching < 0.04% | |||
| Au/TiO2 (1.6 wt.%) | Dye removal = 68.9%; TOC removal = 32.4%; H2O2 consumption = 91.7%; Gold leaching < 0.04% | |||
| Au/ZnO (1.2 wt.%) | Dye removal = 62.6%; TOC removal = 31.9%; H2O2 consumption = 96.1%; Gold leaching < 0.04% | |||
| Phenol | Au/TiO2 (0.8 wt.%) | [phenol] = 5.0 g/L; [catalyst] = 2.7 g/L; VH2O2 = 5 mL; t = 24 h; Vsolution = 45 mL | TOFphenol = 0.07*106 (h−1); TOFTOC = 0.07*106 (h−1); TOFH2O2 = 2.52*106 (h−1) | [68] |
| Au(3)/C (0.5 wt.%) | TOFphenol = 1.19*106 (h−1); TOFTOC = 1.08*106 (h−1); TOFH2O2 = 16.70*106 (h−1) | |||
| Au(5)/C (0.5 wt.%) | TOFphenol = 0.32*106 (h−1); TOFTOC = 0.25*106 (h−1); TOFH2O2 = 4.07*106 (h−1) | |||
| Au(7)/C (0.5 wt.%) | TOFphenol = 0.25*106 (h−1); TOFTOC = 0.25*106 (h−1); TOFH2O2 = 2.27*106 (h−1) | |||
| Au(10)/C (0.5 wt.%) | TOFphenol = 0.47*106 (h−1); TOFTOC = 0.43*106 (h−1); TOFH2O2 = 1.87*106 (h−1) | |||
| Phenol | Au/Hap (2.4 wt.% of Au) | pH = 2.0; T = 70 °C; VH2O2 with 30 wt.% = 1 mL; [catalyst] = 0.1 g/L; [phenol] = 100 mg/L; t = 2 h | Phenol removal = ~92.5% | [70] |
| Phenol | Au/TiO2 – AD (2.8wt.%) | [phenol] = 200 mg/L; [H2O2] = 1520 mg/L; pH = 2.5; T = 80 °C; P = 1 atm; LHSV = 3.8 h−1 | Phenol removal steady-state = 100.0%; TOC removal steady-state = ~65.0% | [71] |
| Au/TiO2 – AD (3.2 wt.%) | Phenol removal steady-state = 100.0%; TOC removal steady-state = ~80.0% | |||
| Phenol | Au/AC (0.8 wt.% of Au) | pH = 3.5; T = 80 °C; [H2O2] = 25 g/L [catalyst] = 2.5 g/L; [phenol] = 5 g/L; t = 22h | Phenol removal = 100%; TOC removal = 70% | [72] |
| Phenol | Au/DNP (1 wt.% of Au) | pH = 4.0; T = 50 °C; [H2O2] = 1.44 g/L; [catalyst] = 320 mg/L; [phenol] = 1 g/L; t = 7 h | Phenol removal = 100%; H2O2 consumption = 100%; BOD5:COD = 0.72 | [73] |
| Phenol | Au/CeO2 (1.0%) | pH = 4.0; Room temperature; [H2O2] = 200 mg/L; [Au] = 0.0025 mM; [phenol] = 100 mg/L; t = 24 h | Phenol removal = 7.0%; H2O2 consumption = 88.0%; Gold leaching = 0.8% | [74] |
| Au/Fe2O3 (1.5%) | Phenol removal = 3.0%; H2O2 consumption = 8.0%; Gold leaching = 0.7% | |||
| Au/TiO2 (1.5%) | Phenol removal = 3.0%; H2O2 consumption = 19.0%; Gold leaching = 0.5% | |||
| Au/C (0.8%) | Phenol removal = 7.0%; H2O2 consumption = 14.0%; Gold leaching = 5.8% | |||
| Au/npD (< 1.0%) | Phenol removal < 1.0%; H2O2 consumption = 6.0%; Gold leaching = 0.5% | |||
| Au/HO-npD (1.0%) | Phenol removal = 93.0%; H2O2 consumption = 48.0%; Gold leaching = 0.7% | |||
| Methyl Blue dye (MB) | Au/CNT (41.0 wt.%) | [MB dye] = 50 mg/L; [catalyst] = 0.5 g/L; [H2O2] = 500 mM; pH = 7.08; t = 120 min | MB removal = ~100% | [88] |
| 1,1-diphenyl-2-picrylhydrazyl (DPPH) | Au/CNT placed in water/cyclohexane mixture (1/10 v/v) (41.0 wt.%) | [DPPH] = 0.2 mM; [catalyst] = 1 g/L; [H2O2] = 250 mM; t = 10 min; Room temperature; W/O = 1:10 v/v | DPPH removal = 100% | |
| Acid Orange 7 (AO7) dye | Au/CeO2 (1 wt.% of Au) | [H2O2] = 20 mM; [catalyst] = 0.5 g/L; [dye] = 35 mg/L; t = 33 h | AO7 removal = 80% | [106] |
| Methyl Orange dye (MO) | Au/TN (1.0 wt.%) | [MO] = 50 mg/L; [catalyst] = 2 g/L; [H2O2] = 0.15 M; pH = 3.0; T = 80 °C; t = 240 min | MO removal = 85%; TOC removal = 83% | [107] |
| Bisphenol A (BPA) | Au/SRAC (3.0 wt.%) | [BPA] = 114 mg/L; [catalyst] = 125 mg/L; [H2O2] = 530 mg/L; pH = 3.0; T = 30 °C | BPA removal = 89.0%; H2O2 consumption = 44.1%; | [108] |
| Au/PSAC (3.0 wt.%) | BPA removal = 23.8%; H2O2 consumption = 8.3% | |||
| Au/CNF (3.0 wt.%) | BPA removal = 20.4%; H2O2 consumption = 14.5% | |||
| Au/FDU-15 (3.0 wt.%) | BPA removal = 32.4%; H2O2 consumption = 22.8% | |||
| Au/X40s (10.0 wt.%) | BPA removal = 14.5%; H2O2 consumption = 10.7% | |||
| Au/Fe2O3 (5.0 wt.%) | BPA removal = 10.1%; H2O2 consumption = 7.6% | |||
| Au/TiO2 (1.5 wt.%) | BPA removal = 5.3%; H2O2 consumption = 10.8% | |||
| Au-Fe2O3/Al2O3 (0.5 wt.%) | BPA removal = 6.6%; H2O2 consumption = 15.3% | |||
| Au/SRAC (1.5 wt.%) | [BPA] = 89 mg/L; [catalyst] = 125 mg/L; [H2O2] = 530 mg/L; pH = 3.0; T = 30 °C | BPA removal = ~80.0%; H2O2 consumption = ~40.0% |
| Catalyst | [Au]total (wt.%) | Au0 Fraction (%) | Auδ+ Fraction (%) | Au Size (nm) | TOF × 10−4 (h−1) | ||
|---|---|---|---|---|---|---|---|
| Phenol | TOC | H2O2 | |||||
| Au/TiO2 | 0.80 | 79 | 21 | 3.1 ± 1.8 | 0.07 | 0.07 | 2.52 |
| Au(3)/AC * | 0.13 | 69 | 31 | 5.1 ± 2.0 | 1.19 | 1.08 | 16.70 |
| Au(5)/AC * | 0.47 | 72 | 28 | 4.9 ± 1.0 | 0.32 | 0.25 | 4.07 |
| Au(7)/AC * | 0.48 | 71 | 29 | 6.8 ± 1.7 | 0.25 | 0.25 | 2.27 |
| Au(10)/AC * | 0.50 | 69 | 31 | 9.1 ± 1.1 | 0.47 | 0.43 | 1.87 |
| Model Compound/Effluent | Catalyst | Operation Conditions | Efficiency of CWPO assisted with Radiation | Ref. |
|---|---|---|---|---|
| Phenol | Au/FH2 (0.1%) | pH = 4.0; Room temperature; [phenol] = 100 mg/L; t = 3.5 h; Radiation: Sunlight | Phenol removal = 100%; H2O2 consumption = ~60% | [75] |
| Au/FN2 (0.5%) | pH = 4.0; Room temperature; [H2O2] = 200 mg/L; [phenol] = 100 mg/L; t = 8 h; Radiation: Sunlight | Phenol removal = ~20%; H2O2 consumption = ~20% | ||
| Au/F (0.5%) | Phenol removal = ~10%; H2O2 consumption = ~15% | |||
| Acid Orange 7 dye (AO7) | Au/CeO2 (1.0 at.%) | pH = 3.0; T = 30 °C; [H2O2] = 20 mM; [catalyst] = 0.5 g/L; [AO7] = 35 mg/L; t = 6 h; Radiation: Visible light | Dye removal = 100% | [106] |
| Orange II (OII) dye | Au/Al2O3 (0.7 wt.%) | pH = 3.0; T = 30 °C; [H2O2] = 6 mM; [catalyst] = 2.0 g/L; [OII] = 0.1 mM; t = 2 h; Radiation: UV/visible light (500 W/m2) | Dye removal = 96.8%; TOC removal = 80.5% | [110] |
| Au/Fe2O3 (0.8 wt.%) | Dye removal = 97.8%; TOC removal = 68.2% | |||
| Au/Fe2O3 (4.0 wt.%) from WGC | Dye removal = 96.9%; TOC removal = 58.4% | |||
| Au/TiO2 (1.6 wt.%) | Dye removal = 98.5%; TOC removal = 73.5% | |||
| Au/ZnO (1.2 wt.%) | Dye removal = 99.8%; TOC removal = 73.4% | |||
| Au/Al2O3 (0.7 wt.%) | pH = 3.0; T = 50 °C; [H2O2] = 3 mM; [catalyst] = 2.0 g/L; [OII] = 0.1 mM; t = 2 h; Radiation: UV/visible light (500 W/m2) | Dye removal = 99.3%; TOC removal = 90.9%; H2O2 consumption = 98.6%; Gold leaching < 0.5 mg/L | ||
| Acrylic dyeing wastewater | Au/Al2O3 (0.7 wt.%) | pH = 3.0; T = 50 °C; [H2O2] = 104 mM; [catalyst] = 2.0 g/L; t = 2 h; Radiation: UV/visible light (500 W/m2) | Color removal = 100%; TOC removal = 72.4%; COD removal = 70.0%; BOD5:COD = 0.5; Specific Oxygen Uptake Rate = 17.9 mgO2/(gVSS.h); Inhibition of Vibrio Fischeri = 0.0% | |
| Phenol | Au/HO-npD (1.0 wt%) | pH = 4.0; T = 30 °C; [H2O2] = 2.5 g/L; [catalyst] = 400 mg/L; [phenol] = 100 mg/L; t = 2 h; Radiation: Sunlight | Phenol removal = 100%; H2O2 consumption = 100%; COD removal = 69.7%; BOD5:COD = 0.4 | [111] |
| Phenol | Au/HO-npD (1.0 wt%) | pH = 4.0; [H2O2] = 200 mg/L; [catalyst] = 160 mg/L; [phenol] = 100 mg/L; t = 2 h; Radiation: Laser Flash (70 mJ/pulse) | Phenol removal = 100%; H2O2 consumption = ~90% | [115] |
| Au/CeO2 (1.0 wt%) | pH = 4.0; [H2O2] = 200 mg/L; [catalyst] = 160 mg/L; [phenol] = 100 mg/L; t = 3 h; Radiation: Laser Flash (70 mJ/pulse) | Phenol removal = ~15%; H2O2 consumption = ~100% | ||
| Au/TiO2 (1.0 wt%) | Phenol removal = ~10%; H2O2 consumption = ~80% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, C.S.D.; Silva, R.M.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Madeira, L.M. Wastewater Treatment by Catalytic Wet Peroxidation Using Nano Gold-Based Catalysts: A Review. Catalysts 2019, 9, 478. https://doi.org/10.3390/catal9050478
Rodrigues CSD, Silva RM, Carabineiro SAC, Maldonado-Hódar FJ, Madeira LM. Wastewater Treatment by Catalytic Wet Peroxidation Using Nano Gold-Based Catalysts: A Review. Catalysts. 2019; 9(5):478. https://doi.org/10.3390/catal9050478
Chicago/Turabian StyleRodrigues, Carmen S.D., Ricardo M. Silva, Sónia A.C. Carabineiro, F.J. Maldonado-Hódar, and Luís M. Madeira. 2019. "Wastewater Treatment by Catalytic Wet Peroxidation Using Nano Gold-Based Catalysts: A Review" Catalysts 9, no. 5: 478. https://doi.org/10.3390/catal9050478