Theoretical Determination of Size Effects in Zeolite-Catalyzed Alcohol Dehydration


Abstract
:1. Introduction
2. Results and Discussion
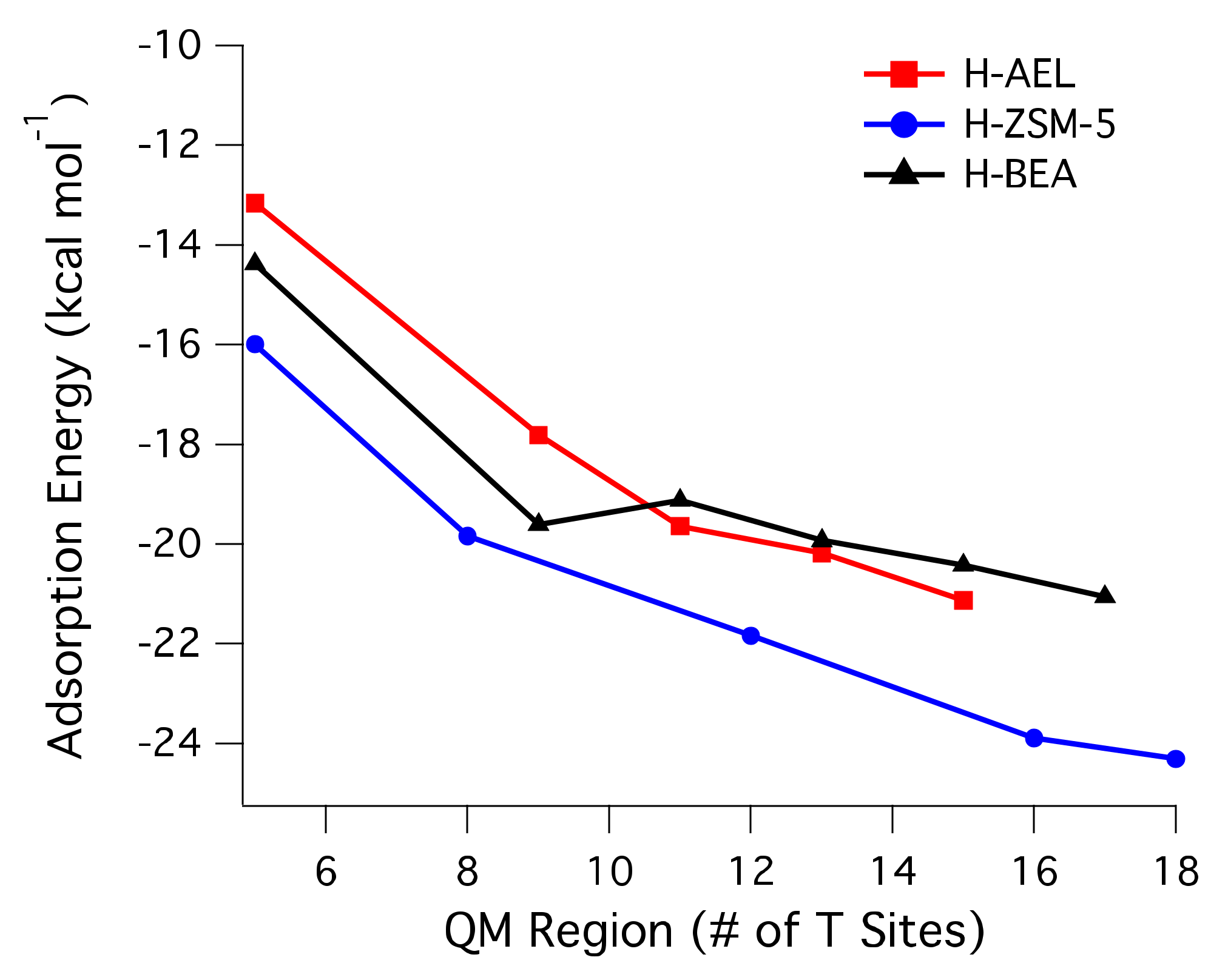
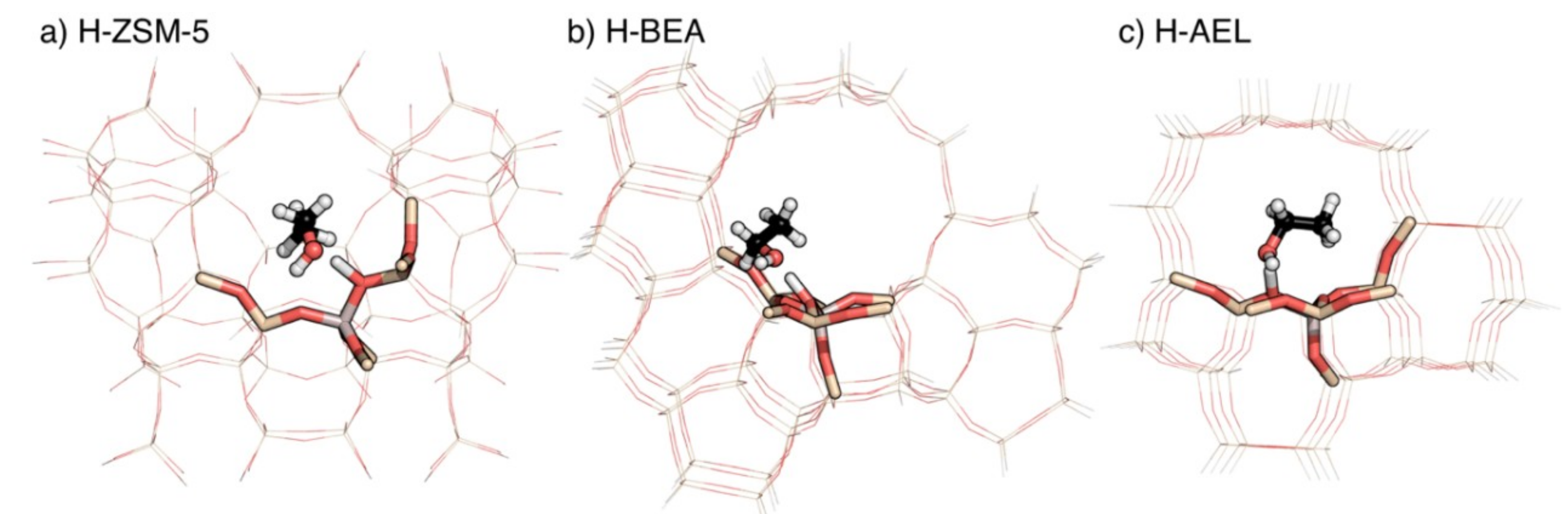
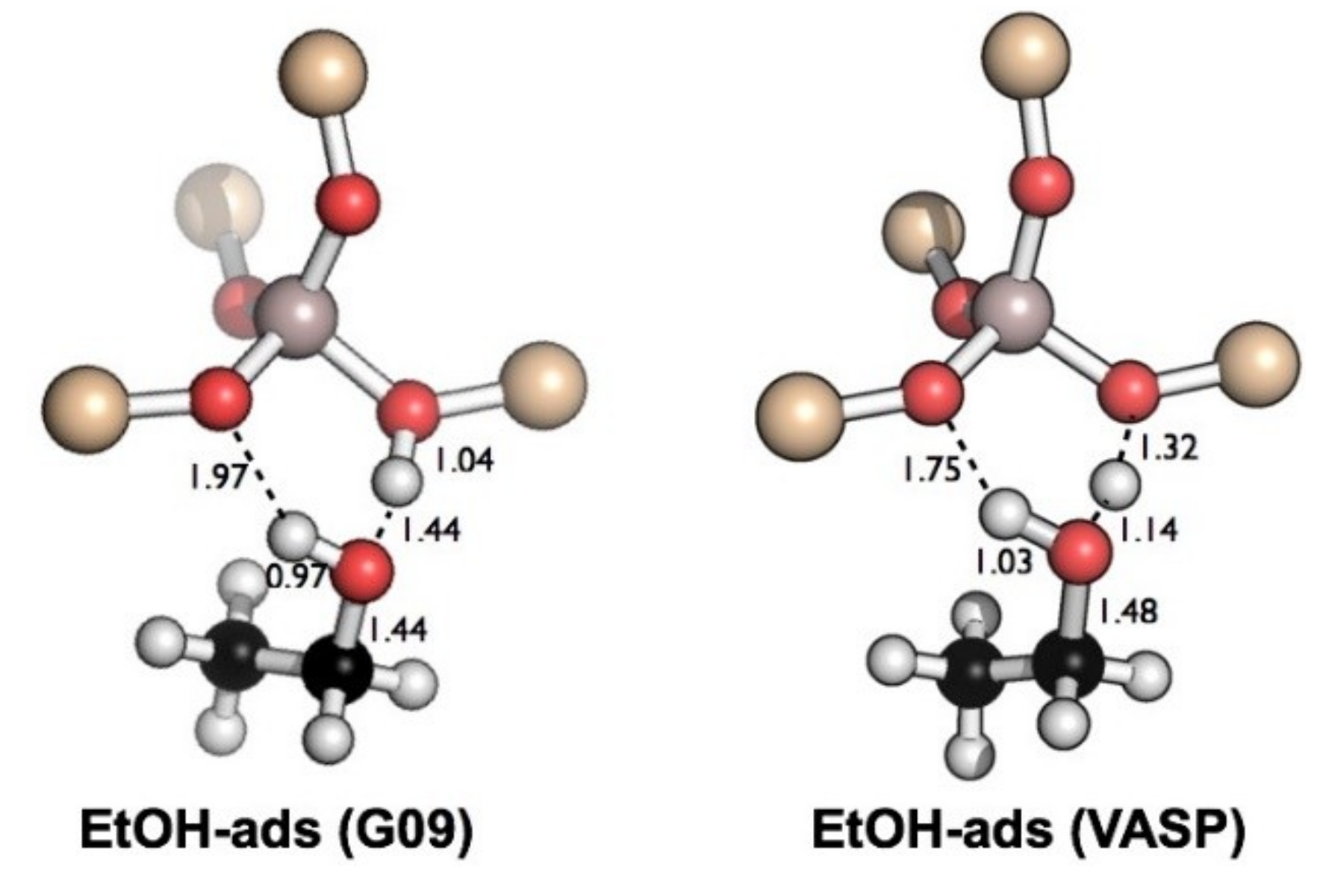
2.1. Adsorption Energies
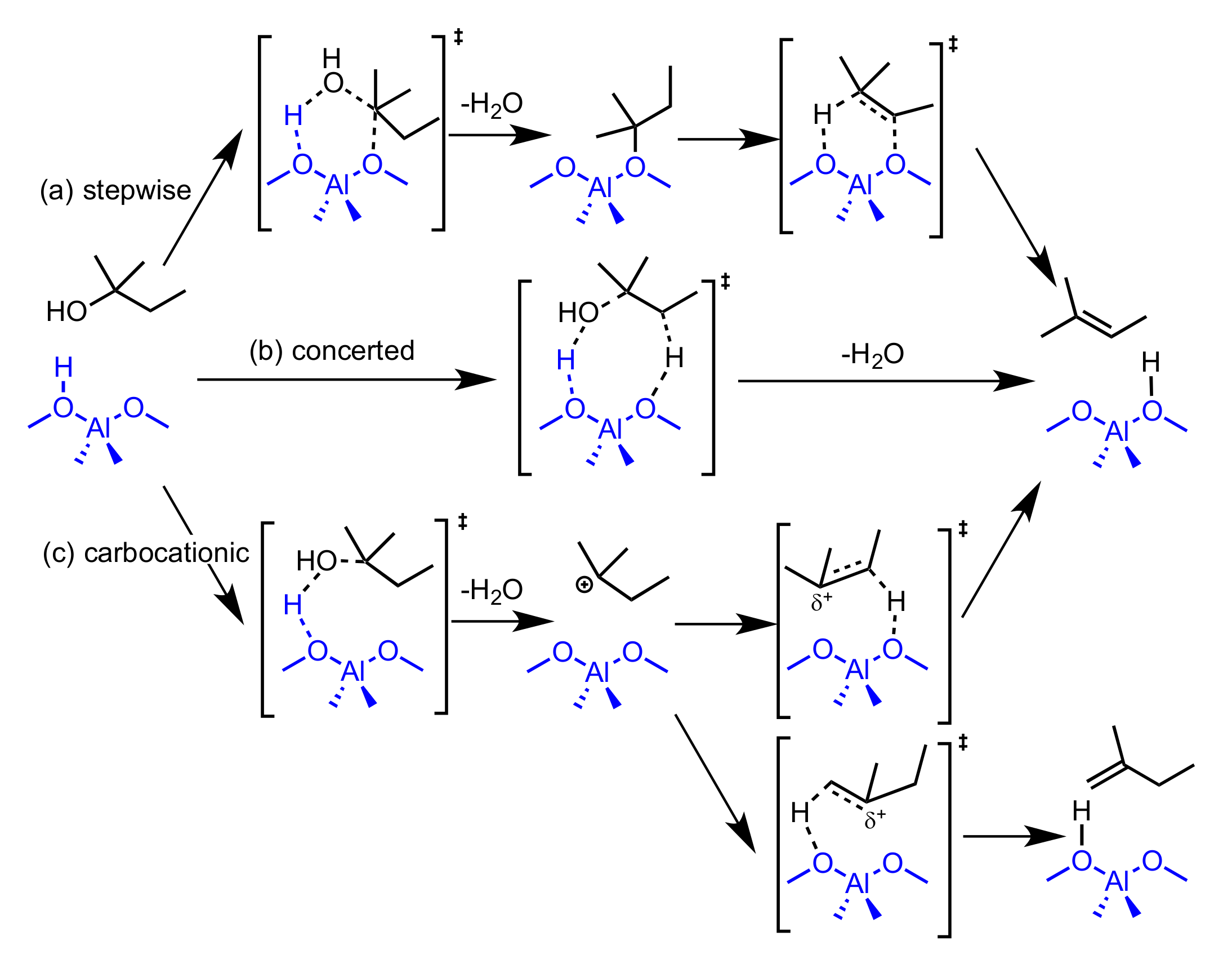
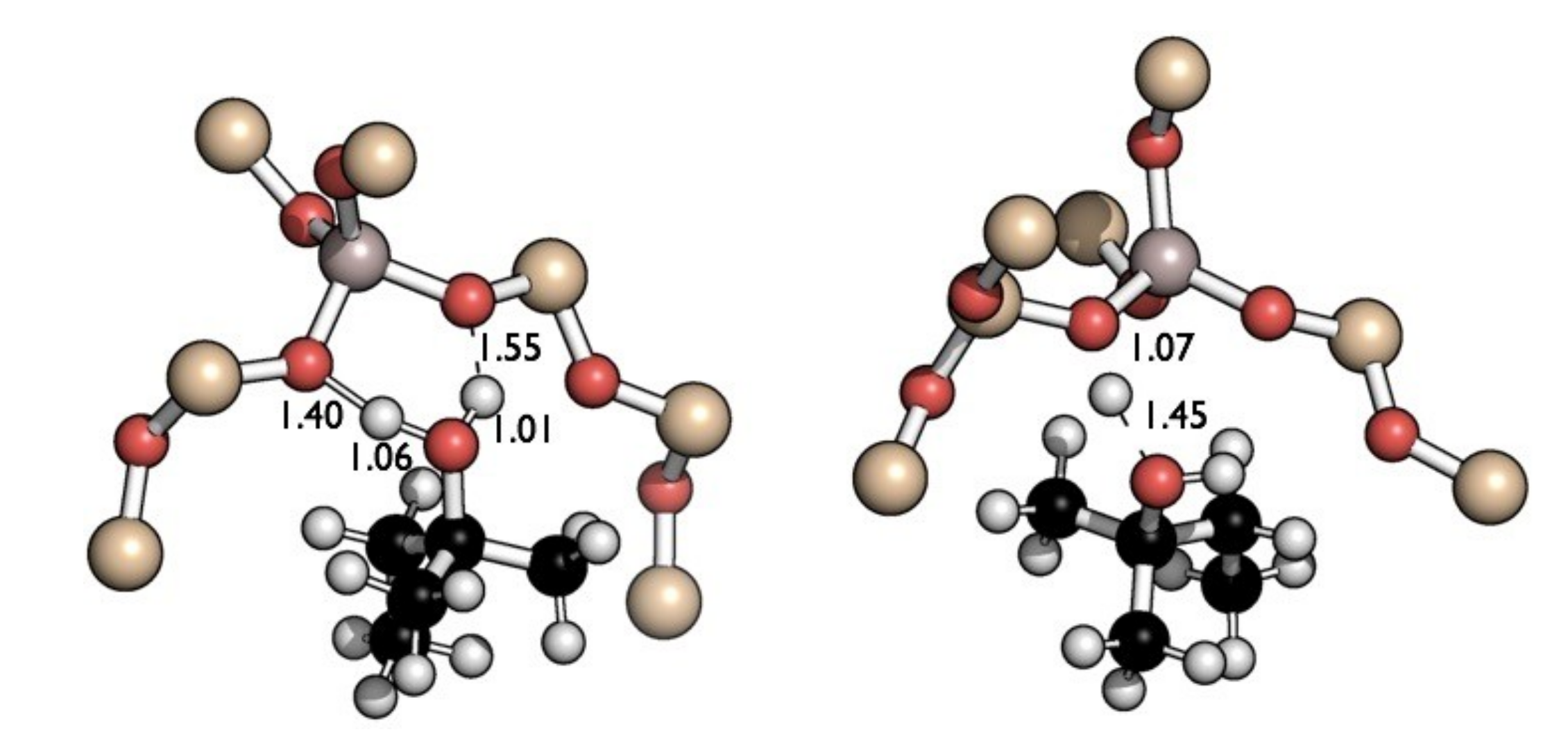
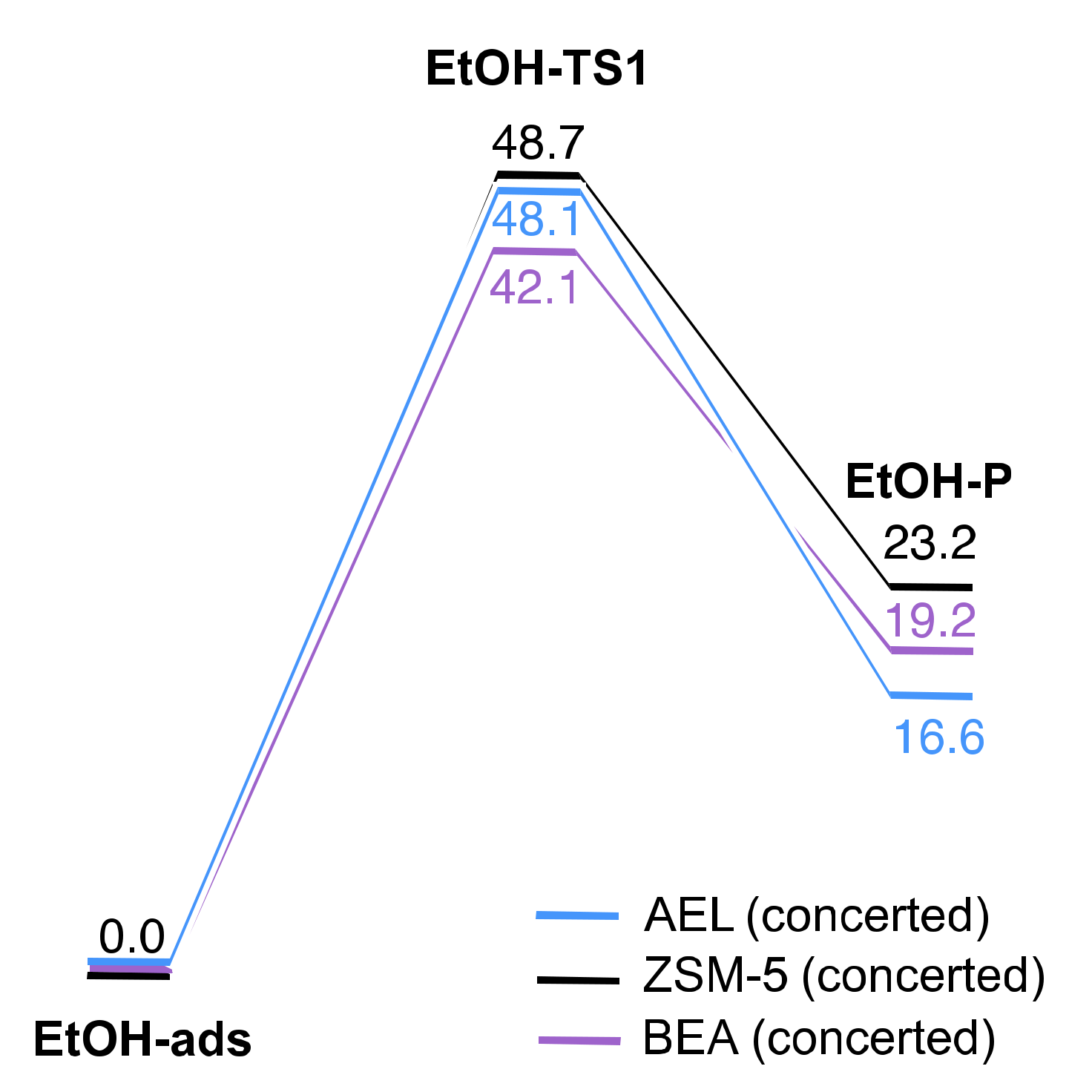
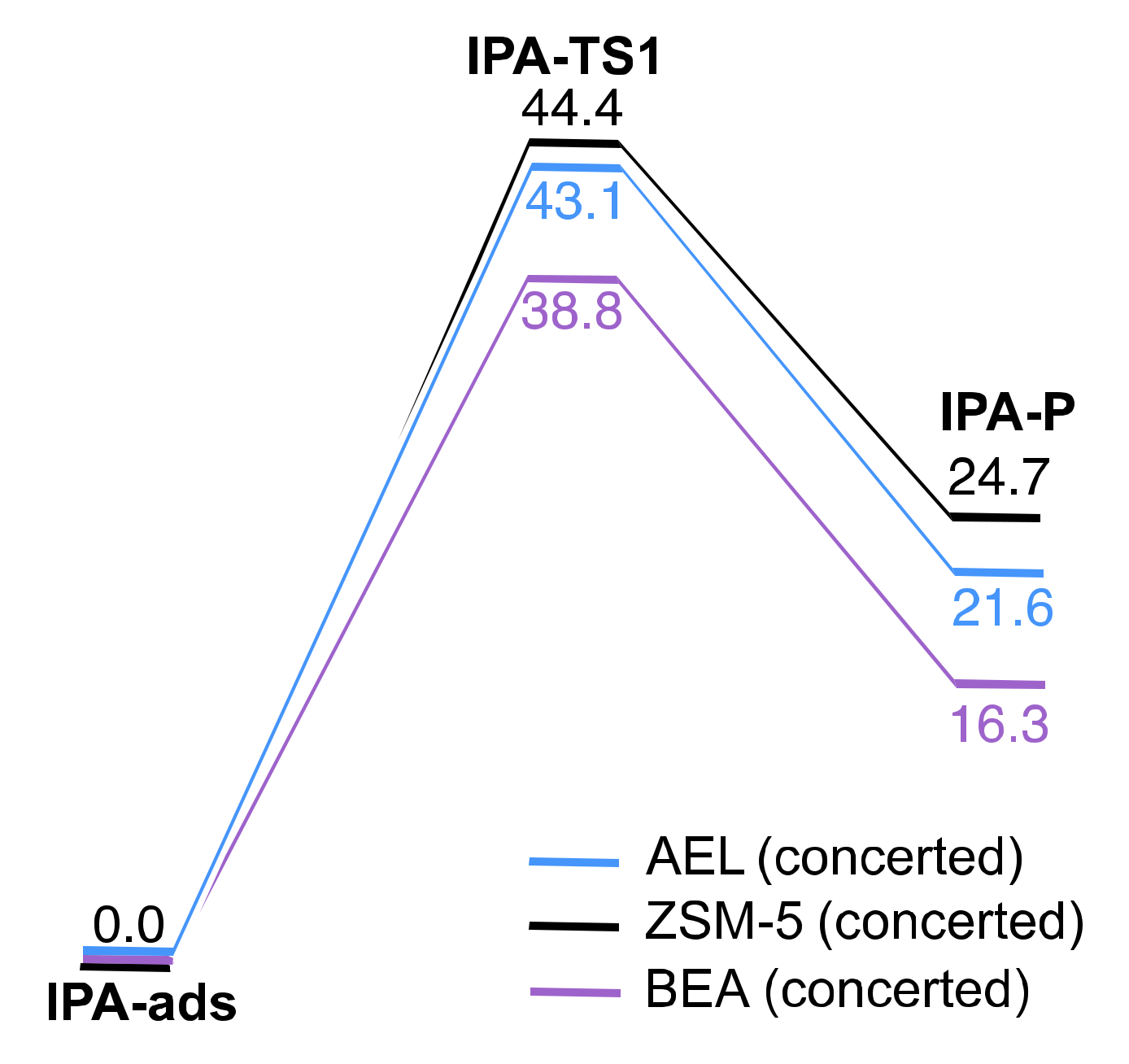
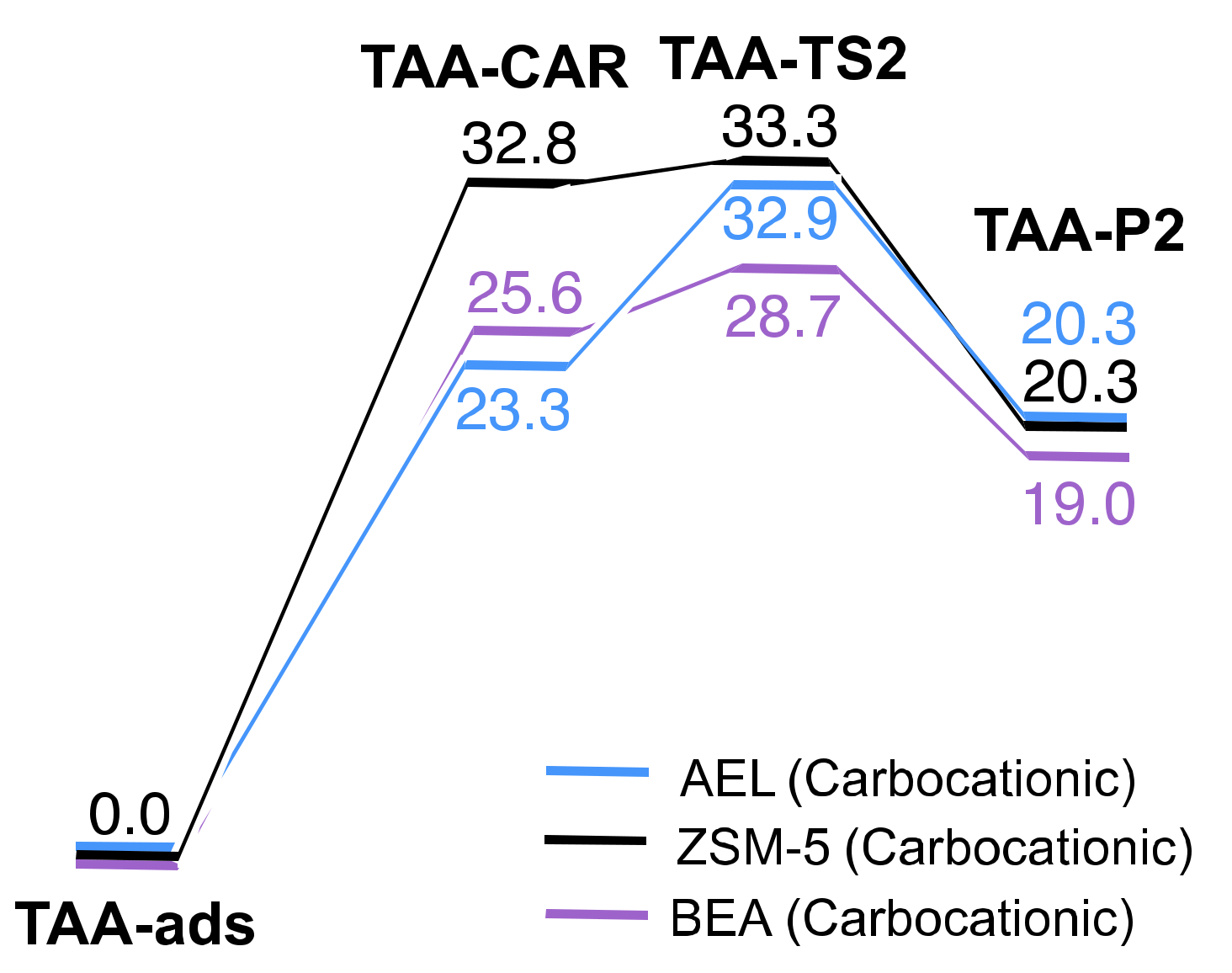
2.2. Reaction Coordinate
2.3. Molecular Diffusion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McKendry, P. Energy production from biomass (part 1): Overview of biomass. Bioresour. Technol. 2002, 83, 37–46. [Google Scholar] [CrossRef]
- Oasmaa, A.; Koponen, P. Physical Characterisation of Biomass-Based Pyrolysis Liquids; Espoo: Espoo, Finland, 1997. [Google Scholar]
- Oasmaa, A.; Peacocke, C. A Guide to physical Property Characterisation of Biomass-Derived Fast Pyrolysis Liquids; Technical Research Centre of Finland Espoo: Espoo, Finland, 2001. [Google Scholar]
- Oasmaa, A.; Peacocke, C. Properties and Fuel Use of Biomass-Derived Fast Pyrolysis Liquids; VTT Publications: Espoo, Finland, 2010; Volume 731, p. 79. [Google Scholar]
- Ruddy, D.A.; Schaidle, J.A.; Ferrell Iii, J.R.; Wang, J.; Moens, L.; Hensley, J.E. Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: Catalyst development through the study of model compounds. Green Chem. 2014, 16, 454–490. [Google Scholar] [CrossRef]
- Triantafillidis, C.S.; Evmiridis, N.P. Dealuminated H-Y Zeolites: Influence of the Number and Type of Acid Sites on the Catalytic Activity for Isopropanol DehydrationInd. Ind. Eng. Chem. Res. 2000, 39, 3233–3240. [Google Scholar] [CrossRef]
- Turek, W.; Haber, J.; Krowiak, A. Dehydration of isopropyl alcohol used as an indicator of the type and strength of catalyst acid centres. Appl. Surf. Sci. 2005, 252, 823–827. [Google Scholar] [CrossRef]
- Dutta, A.; Sahir, A.; Tan, E.; Humbird, D.; Snowden-Swan, L.J.; Meyer, P.; Ross, J.; Sexton, D.; Yap, R.; Lukas, J.L. Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels, Thermochemical Research Pathways with In Situ and Ex Situ Upgrading of Fast Pyrolysis Vapors; NREL (National Renewable Energy Laboratory (NREL): Golden, CO, USA, 2015. [Google Scholar]
- Liu, C.; Evans, T.J.; Cheng, L.; Nimlos, M.R.; Mukarakate, C.; Robichaud, D.J.; Assary, R.S.; Curtiss, L.A. Catalytic upgrading of biomass-derived compounds via C-C coupling reactions: Computational and experimental studies of furan and acetaldehyde reactions in HZSM-5. J. Phys. Chem. C 2015, 119, 24025–24035. [Google Scholar] [CrossRef]
- Kim, S.; Evans, T.J.; Mukarakate, C.; Bu, L.; Beckham, G.T.; Nimlos, M.R.; Paton, R.S.; Robichaud, D.J. Furan Production from Glycoaldehyde over HZSM-5. ACS Sustain. Chem. Eng. 2016, 4, 2615–2623. [Google Scholar] [CrossRef]
- Jae, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbach, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- Greenhalf, C.E.; Nowakowski, D.J.; Harms, A.B.; Titiloye, J.O.; Bridgwater, A.V. A comparative study of straw, perennial grasses and hardwoods in terms of fast pyrolysis products. Fuel 2013, 108, 216–230. [Google Scholar] [CrossRef]
- Bokade, V.V.; Yadav, G.D. Heteropolyacid supported on montmorillonite catalyst for dehydration of dilute bio-ethanol. Appl. Clay Sci. 2011, 53, 263–271. [Google Scholar] [CrossRef]
- Fan, D.; Dai, D.J.; Wu, H.S. Ethylene Formation by Catalytic Dehydration of Ethanol with Industrial Considerations. Materials 2013, 6, 101–115. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Aguado, R.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols. Ind. Eng. Chem. Res. 2004, 43, 2610–2618. [Google Scholar] [CrossRef]
- Johansson, R.; Hruby, S.L.; Rass-Hansen, J.; Christensen, C.H. The Hydrocarbon Pool in Ethanol-to-Gasoline over HZSM-5 Catalysts. Catal. Lett. 2009, 127, 1–6. [Google Scholar] [CrossRef]
- Bun, S.; Nishiyama, S.; Tsuruya, S.; Masai, M. ETHANOL CONVERSION OVER ION-EXCHANGED ZSM-5 ZEOLITES. Appl. Catal. 1990, 59, 13–29. [Google Scholar] [CrossRef]
- Calsavara, V.; Baesso, M.L.; Camargo Fernandes-Machado, N.R. Transformation of ethanol into hydrocarbons on ZSM-5 zeolites modified with iron in different ways. Fuel 2008, 87, 1628–1636. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.; Tao, L.; Dai, B.; Yang, M.; Chen, Z.; Zhu, X. Dehydration reaction of bio-ethanol to ethylene over modified SAPO catalysts. J. Ind. Eng. Chem. 2010, 16, 717–722. [Google Scholar] [CrossRef]
- Dumrongsakda, P.; Ruangpornvisuti, V. Theoretical Investigation of Ethanol Conversion to Ethylene over H-ZSM-5 and Transition Metals-Exchanged ZSM-5. Catal. Lett. 2012, 142, 143–149. [Google Scholar] [CrossRef]
- Inaba, M.; Murata, K.; Takahara, I. Effect of Fe-loading and reaction temperature on the production of olefins from ethanol by Fe/H-ZSM-5 zeolite catalysts. React. Kinet. Catal. Lett. 2009, 97, 19–26. [Google Scholar] [CrossRef]
- Kim, S.; Robichaud, D.J.; Beckham, G.T.; Paton, R.S.; Nimlos, M.R. Ethanol Dehydration in HZSM-5 Studied by Density Functional Theory: Evidence for a Concerted Process. J. Phys. Chem. A 2015, 119, 3604–3614. [Google Scholar] [CrossRef]
- Madeira, F.F.; Gnep, N.S.; Magnoux, P.; Maury, S.; Cadran, N. Ethanol transformation over HFAU, HBEA and HMFI zeolites presenting similar Bronsted acidity. Appl. Catal. A Gen. 2009, 367, 39–46. [Google Scholar] [CrossRef]
- Maihom, T.; Khongpracha, P.; Sirijaraensre, J.; Limtrakul, J. Mechanistic Studies on the Transformation of Ethanol into Ethene over Fe-ZSM-5 Zeolite. Chem. Phys. Chem. 2013, 14, 101–107. [Google Scholar] [CrossRef]
- Mao, R.L.; Levesque, P.; McLaughlin, G.; Dao, L.H. Ethylene from ethanol over zeolite catalysts. Appl. Catal. 1987, 34, 163–179. [Google Scholar]
- Pan, Q.; Ramanathan, A.; Snavely, W.K.; Chaudhari, R.V.; Subramaniam, B. Intrinsic Kinetics of Ethanol Dehydration Over Lewis Acidic Ordered Mesoporous Silicate, Zr-KIT-6. Top. Catal. 2014, 57, 1407–1411. [Google Scholar] [CrossRef]
- Phillips, C.B.; Datta, R. Production of ethylene from hydrous ethanol on H-ZSM-5 under mild conditions. Ind. Eng. Chem. Res. 1997, 36, 4466–4475. [Google Scholar] [CrossRef]
- Phung, T.K.; Hernandez, L.P.; Lagazzo, A.; Busca, G. Dehydration of ethanol over zeolites, silica alumina and alumina: Lewis acidity, Bronsted acidity and confinement effects. Appl. Catal. A Gen. 2015, 493, 77–89. [Google Scholar] [CrossRef]
- Schulz, J.; Bandermann, F. Conversion of Ethanol over Zeolite H-ZSM-5. Chem. Eng. Technol. 1994, 17, 179–186. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y. Recent Advances in Catalytic Conversion of Ethanol to Chemicals. ACS Catal. 2014, 4, 1078–1090. [Google Scholar] [CrossRef]
- Takahara, I.; Saito, M.; Inaba, M.; Murata, K. Dehydration of ethanol into ethylene over solid acid catalysts. Catal. Lett. 2005, 105, 249–252. [Google Scholar] [CrossRef]
- Takahara, I.; Saito, M.; Matsuhashi, H.; Inaba, M.; Murata, K. Increase in the number of acid sites of a H-ZSM5 zeolite during the dehydration of ethanol. Catal. Lett. 2007, 113, 82–85. [Google Scholar] [CrossRef]
- Tavan, Y.; Hosseini, S.H.; Ghavipour, M.; Nikou, M.R.K.; Shariati, A. From laboratory experiments to simulation studies of methanol dehydration to produce dimethyl ether-Part I: Reaction kinetic study. Chem. Eng. Process. 2013, 73, 144–150. [Google Scholar] [CrossRef]
- Tret’yakov, V.F.; Nhu, C.T.Q.; Tret’yakov, K.V.; Sil’chenkova, O.N.; Matyshak, V.A. Conversion of ethanol on HZSM-5 modified zeolite, according to data from in situ spectrokinetic studies. Russ. J. Phys. Chem. A 2013, 87, 941–944. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, Y. Dehydration of Ethanol to Ethylene. Ind. Eng. Chem. Res. 2013, 52, 9505–9514. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Yang, X.; Zhang, F. Comparison of four catalysts in the catalytic dehydration of ethanol to ethylene. Micropor. Mesopor. Mater. 2008, 116, 210–215. [Google Scholar] [CrossRef]
- Bond, G.C.; Frodsham, S.J.; Jubb, P.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Compensation effect in isopropanol dehydration over heteropoly acid catalysts at a gas-solid interface. J. Catal. 2012, 293, 158–164. [Google Scholar] [CrossRef]
- Lopes, J.F.; Silva, J.C.M.; Cruz, M.T.M.; Carneiro, J.W.d.M.; De Almeida, W.B. DFT study of ethanol dehydration catalysed by hematite. RSC Adv. 2016, 6, 40408–40417. [Google Scholar] [CrossRef]
- Roy, S.; Mpourmpakis, G.; Hong, D.-Y.; Vlachos, D.G.; Bhan, A.; Gorte, R.J. Mechanistic Study of Alcohol Dehydration on γ-Al2O3. ACS Catal. 2012, 2, 1846–1853. [Google Scholar] [CrossRef]
- Zavelev, D.E.; Chistyakov, A.V.; Zhidomirov, G.M.; Gubanov, M.A.; Tsodikov, M.V.; Moiseev, I.I. Mechanism of the reductive dehydration of ethanol into C3+ alkanes over the commercial alumina—platinum catalyst AP-64. Kinet. Catal. 2016, 57, 95–103. [Google Scholar] [CrossRef]
- Alexopoulos, K.; John, M.; Van der Borght, K.; Galvita, V.; Reyniers, M.-F.; Marin, G.B. DFT-based microkinetic modeling of ethanol dehydration in H-ZSM-5. J. Catal. 2016, 339, 173–185. [Google Scholar] [CrossRef]
- Stepanov, A.G.; Zamaraev, K.I.; Thomas, J.M. 13C CP/MAS and2H NMR study of tert-butyl alcohol dehydration on H-ZSM-5 zeolite. Evidence for the formation of tert-butyl cation and tert-butyl silyl ether intermediates. Catal. Lett. 1992, 13, 407–422. [Google Scholar] [CrossRef]
- Stepanov, A.G.; Romannikov, V.N.; Zamaraev, K.I. 13C CP/MAS NMR study of isobutyl alcohol dehydration on H-ZSM-5 zeolite. Evidence for the formation of stable isobutyl silyl ether intermediate. Catal. Lett. 1992, 13, 395–405. [Google Scholar] [CrossRef]
- Stepanov, A.G.; Zamaraev, K.I. 13C solid state NMR evidence for the existence of isobutyl carbenium ion in the reaction of isobutyl alcohol dehydration in H-ZSM-5 zeolite. Catal. Lett. 1993, 19, 153–158. [Google Scholar] [CrossRef]
- Yuan, S.P.; Wang, J.G.; Li, Y.W.; Jiao, H. Brønsted Acidity of Isomorphously Substituted ZSM-5 by B., Al, Ga, and Fe. Density Functional Investigations. J. Phys. Chem. A 2002, 106, 8167–8172. [Google Scholar] [CrossRef]
- Trombetta, M.; Busca, G.; Storaro, L.; Lenarda, M.; Casagrande, M.; Zambon, A. Surface acidity modifications induced by thermal treatments and acid leaching on microcrystalline H-BEA zeolite. A FTIR, XRD and MAS-NMR study. Phys. Chem. Chem. Phys. 2000, 2, 3529–3537. [Google Scholar] [CrossRef]
- Xu, B.; Sievers, C.; Hong, S.B.; Prins, R.; van Bokhoven, J.A. Catalytic activity of Brønsted acid sites in zeolites: Intrinsic activity, rate-limiting step, and influence of the local structure of the acid sites. J. Catal. 2006, 244, 163–168. [Google Scholar] [CrossRef]
- Richardson, J.W., Jnr; Pluth, J.J.; Smith, J.V. Rietveld profile analysis of calcined AlPO4–11 using pulsed neutron powder diffraction. Acta Crystallogr. B 1988, 44, 367–373. [Google Scholar] [CrossRef]
- Alexopoulos, K.; Lee, M.-S.; Liu, Y.; Zhi, Y.; Liu, Y.; Reyniers, M.-F.; Marin, G.B.; Glezakou, V.-A.; Rousseau, R.; Lercher, J.A. Anharmonicity and Confinement in Zeolites: Structure, Spectroscopy, and Adsorption Free Energy of Ethanol in H-ZSM-5. J. Phys. Chem. C 2016, 120, 7172–7182. [Google Scholar] [CrossRef]
- Van der Mynsbrugge, J.; Hemelsoet, K.; Vandichel, M.; Waroquier, M.; Van Speybroeck, V. Efficient Approach for the Computational Study of Alcohol and Nitrile Adsorption in H-ZSM-5. J. Phys. Chem. C 2012, 116, 5499–5508. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-C.; Gorte, R.J.; Farneth, W.E. Calorimetric Study of Alcohol and Nitrile Adsorption Complexes in H-ZSM-5. J. Phys. Chem. B 1997, 101, 3811–3817. [Google Scholar] [CrossRef]
- Nguyen, C.M.; Reyniers, M.-F.; Marin, G.B. Theoretical study of the adsorption of C1-C4 primary alcohols in H-ZSM-5. Phys. Chem. Chem. Phys. 2010, 12, 9481–9493. [Google Scholar] [CrossRef]
- Li, Y.-P.; Gomes, J.; Mallikarjun Sharada, S.; Bell, A.T.; Head-Gordon, M. Improved Force-Field Parameters for QM/MM Simulations of the Energies of Adsorption for Molecules in Zeolites and a Free Rotor Correction to the Rigid Rotor Harmonic Oscillator Model for Adsorption Enthalpies. J. Phys. Chem. C 2015, 119, 1840–1850. [Google Scholar] [CrossRef]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 31 July 2019).
- Nguyen, C.M.; Reyniers, M.-F.; Marin, G.B. Theoretical Study of the Adsorption of the Butanol Isomers in H-ZSM-5. J. Phys. Chem. C 2011, 115, 8658–8669. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.S. A correlation of reaction rates. J. Am. Chem. Soc. 1955, 77, 334–338. [Google Scholar] [CrossRef]
- Leffler, J.E. The enthalpy-entropy relationship and its implications for organic chemistry. J. Org. Chem. 1955, 20, 1202–1231. [Google Scholar] [CrossRef]
- Ferguson, G.A.; Cheng, L.; Bu, L.; Kim, S.; Robichaud, D.J.; Nimlos, M.R.; Curtiss, L.A.; Beckham, G.T. Carbocation Stability in H-ZSM5 at High Temperature. J. Phys. Chem. A 2015, 119, 11397–11405. [Google Scholar] [CrossRef]
- Jones, A.J.; Zones, S.I.; Iglesia, E. Implications of transition state confinement within small voids for acid catalysis. J. Phys. Chem. C 2014, 118, 17787–17800. [Google Scholar] [CrossRef]
- Xiao, J.; Wei, J. Diffusion mechanism of hydrocarbons in zeolites-I. Theory. Chem. Eng. Sci. 1992, 47, 1123–1141. [Google Scholar] [CrossRef]
- Bu, L.; Nimlos, M.R.; Robichaud, D.J.; Kim, S. Diffusion of aromatic hydrocarbons in hierarchical mesoporous H-ZSM-5 zeolite. Catal. Today 2018, 312, 73–81. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Bu, L.; Nimlos, M.R.; Robichaud, D.J.; Kim, S. Diffusion of Biomass Pyrolysis Products in H-ZSM-5 by Molecular Dynamics Simulations. J. Phys. Chem. C 2017, 121, 500–510. [Google Scholar] [CrossRef]
- Farberow, C.A.; Cheah, S.; Kim, S.; Miller, J.T.; Gallagher, J.R.; Hensley, J.E.; Schaidle, J.A.; Ruddy, D.A. Exploring low-temperature dehydrogenation at ionic Cu sites in beta zeolite to enable alkane recycle in dimethyl ether homologation. ACS Catal. 2017, 7, 3662–3667. [Google Scholar] [CrossRef]
- Knott, B.C.; Nimlos, C.T.; Robichaud, D.J.; Nimlos, M.R.; Kim, S.; Gounder, R. Consideration of the aluminum distribution in zeolites in theoretical and experimental catalysis research. ACS Catal. 2017, 8, 770–784. [Google Scholar] [CrossRef]
- Cook, S.J.; Chakraborty, A.K.; Bell, A.T.; Theodorou, D.N. Structural and electronic features of a Broensted acid site in H-ZSM-5. J. Phys. Chem. 1993, 97, 6679–6685. [Google Scholar] [CrossRef]
- Lonsinger, S.R.; Chakraborty, A.K.; Theodorou, D.N.; Bell, A.T. The effects of local structural relaxation on aluminum siting within H-ZSM-5. Catal. Lett. 1991, 11, 209–217. [Google Scholar] [CrossRef]
- Vjunov, A.; Fulton, J.L.; Huthwelker, T.; Pin, S.; Mei, D.; Schenter, G.K.; Govind, N.; Camaioni, D.M.; Hu, J.Z.; Lercher, J.A. Quantitatively Probing the Al Distribution in Zeolites. J. Am. Chem. Soc. 2014, 136, 8296–8306. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Boekfa, B.; Choomwattana, S.; Khongpracha, P.; Limtrakul, J. Effects of the Zeolite Framework on the Adsorptions and Hydrogen-Exchange Reactions of Unsaturated Aliphatic, Aromatic, and Heterocyclic Compounds in ZSM-5 Zeolite: A Combination of Perturbation Theory (MP2) and a Newly Developed Density Functional Theory (M06-2X) in ONIOM Scheme. Langmuir 2009, 25, 12990–12999. [Google Scholar] [CrossRef] [PubMed]
- Rayne, S.; Forest, K. Performance of the M062X density functional against the ISOL set of benchmark isomerization energies for large organic molecules. Nat. Preced. 2010. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harl, J.; Schimka, L.; Kresse, G. Assessing the quality of the random phase approximation for lattice constants and atomization energies of solids. Phys. Rev. B 2010, 81, 115126. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for \textit{ab initio} total-energy calculations using a plane-wave basis set. Phy. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- GoodVibes, v2.0.3. Available online: http://doi.org/10.5281/zenodo.595246 (accessed on 26 September 2018).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expt. 400 K [51] | Expt. 300 K [49] | PBE-D pbc, QHA [49] | PBE-D pbc, HA [49] | PBE-D pbc [52] | M06-2X Cluster [50] | This Work, ONIOM |
|---|---|---|---|---|---|---|
| −31.1 | −21.3 | −25.6 | −30.1 | −31.5 | −25.8 | −23.9 |
| Molecule Size (Å) | Cavity Size (Å) [54] | ||
|---|---|---|---|
| Ethanol | 4.1 × 4.5 × 6.5 | H-AEL | 5.64 |
| IPA | 5.0 × 5.7 × 6.1 | H-ZSM-5 | 6.36 |
| TAA | 5.8 × 6.7 × 7.0 | H-BEA | 6.68 |
| Substrate | ONIOM | VASP | ||||
|---|---|---|---|---|---|---|
| H-AEL | H-ZSM-5 | H-BEA | H-AEL | H-ZSM-5 | H-BEA | |
| Ethanol | −17.8 | −19.8 | −19.6 | −18.3 | −21.0 | −22.7 |
| IPA | −16.3 | −16.6 | −18.1 | −18.8 | −14.2 | −23.5 |
| TAA | −6.1 | −9.8 | −16.0 | −15.4 | −17.5 | −20.3 |
| TAA TS Size (Å) | Cavity Size (Å) | |
|---|---|---|
| H-AEL | 5.2 × 6.4 × 10.8 | 5.64 |
| H-ZSM-5 | 5.7 × 6.7 × 8.9 | 6.36 |
| H-BEA | 5.5 × 7.2 × 8.2 | 6.68 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunz, L.Y.; Bu, L.; Knott, B.C.; Liu, C.; Nimlos, M.R.; Assary, R.S.; Curtiss, L.A.; Robichaud, D.J.; Kim, S. Theoretical Determination of Size Effects in Zeolite-Catalyzed Alcohol Dehydration. Catalysts 2019, 9, 700. https://doi.org/10.3390/catal9090700
Kunz LY, Bu L, Knott BC, Liu C, Nimlos MR, Assary RS, Curtiss LA, Robichaud DJ, Kim S. Theoretical Determination of Size Effects in Zeolite-Catalyzed Alcohol Dehydration. Catalysts. 2019; 9(9):700. https://doi.org/10.3390/catal9090700
Chicago/Turabian StyleKunz, Larissa Y., Lintao Bu, Brandon C. Knott, Cong Liu, Mark R. Nimlos, Rajeev S. Assary, Larry A. Curtiss, David J. Robichaud, and Seonah Kim. 2019. "Theoretical Determination of Size Effects in Zeolite-Catalyzed Alcohol Dehydration" Catalysts 9, no. 9: 700. https://doi.org/10.3390/catal9090700