Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water


J. Heyrovsky Institute of Physical Chemistry of the CAS, Dolejskova 3, 18223 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(9), 708; https://doi.org/10.3390/catal9090708
Submission received: 12 July 2019
/
Revised: 14 August 2019
/
Accepted: 20 August 2019
/
Published: 23 August 2019
(This article belongs to the Special Issue Photocatalytic Nanocomposite Materials)
Abstract
:The preparation of immobilized graphene-based photocatalyst layers is highly desired for environmental applications. In this study, the preparation of an immobilized reduced graphene oxide (rGO)/TiO2 composite by electrophoretic deposition (EPD) was optimized. It enabled quantitative deposition without sintering and without the use of any dispersive additive. The presence of rGO had beneficial effects on the photocatalytic degradation of 4-chlorophenol in an aqueous solution. A marked increase in the photocatalytic degradation rate was observed, even at very low concentrations of rGO. Compared with the TiO2 and GO/TiO2 reference layers, use of the rGO/TiO2 composite (0.5 wt% of rGO) increased the first-order reaction rate constant by about 70%. This enhanced performance was due to the increased formation of hydroxyl radicals that attacked the 4-chlorophenol molecules. The direct charge transfer mechanism had only limited effect on the degradation. Thus, EPD-prepared rGO/TiO2 layers appear to be suitable for environmental application.
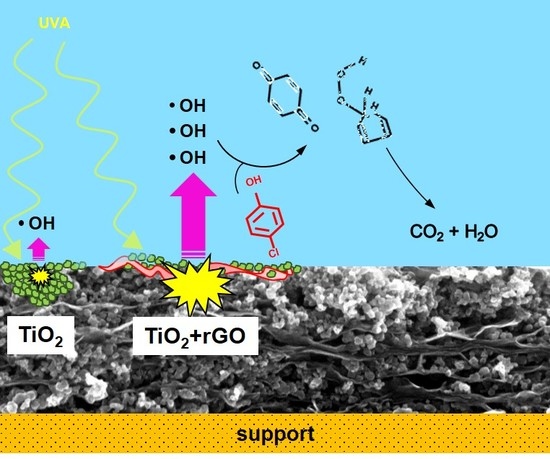
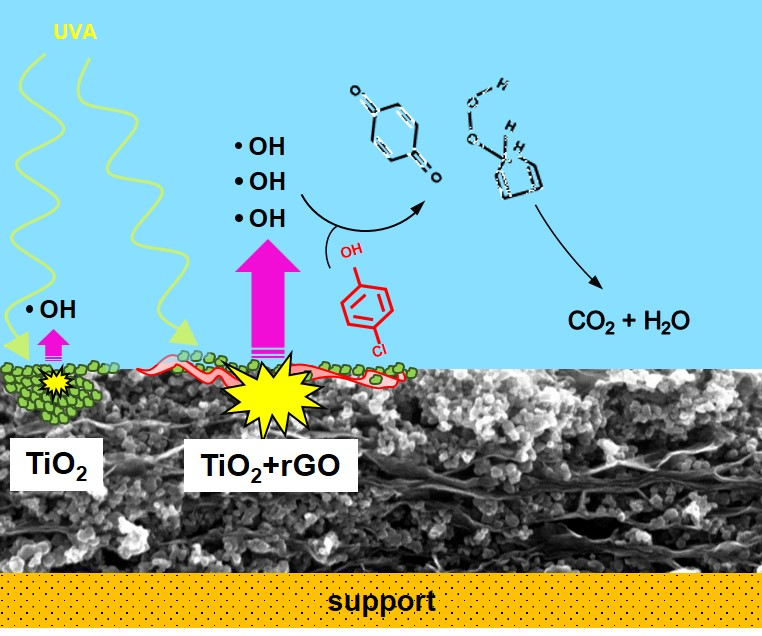
1. Introduction
Graphene-based nanomaterials possess unique properties that can be applied to environmental remediation [1,2,3,4,5]. The interest in these nanomaterials originates from graphene’s unique physicochemical properties [6], notably its exceptionally high surface area, electron and thermal mobility [7,8,9], and mechanical strength [10]. One of the most popular approaches for preparing graphene-based nanomaterials involves the use of graphene oxide (GO) because of its lower production costs. GO is an oxidized form of graphene, showing a high density of oxygen functional groups (carboxyl, hydroxyl, carbonyl, and epoxy) in the carbon lattice. However, the oxidation of graphite to GO introduces defects into the carbon structure that result in significantly altered physicochemical properties.
By the reduction of GO, a large fraction of its oxygen content can be removed, which leads to a substantial change in its physicochemical properties [11,12]. The formed reduced graphene oxide (rGO) possesses enhanced electronic (higher mobility of electrons), optical, and surface properties (decreased wettability) [4]. Moreover, reduction also results in an altered chemical structure, with carbon vacancies, residual oxygen content, and clustered pentagon and heptagon carbon structures [13]. Thus, the complete reduction of GO is challenging. Despite these defects, the physicochemical properties of rGO represent a partial restoration that is closer to pristine graphene than GO.
Restoring the physicochemical properties of graphene is vital for the production of graphene-based composite materials [14,15,16,17]. However, the improved performance of graphene-based composites often relies on synergistic interactions between rGO and the materials attached to its surface. Therefore, the quality of the graphene-based materials used will have an important impact on the performance of the final nanocomposite.
In the environmental field, graphene-based nanocomposites have been used to develop novel adsorbents [8] and photocatalysts [2,18,19]. Adsorption is a fast, inexpensive, and effective method for the removal of contaminants from aquatic environments [20]. It is based on physicochemical interactions between the adsorbent and contaminant. Although rGO-based adsorbents can remove contaminants from water, this technique does not degrade the compounds, which require further disposal. Thus, for the complete mineralization of organic pollutants, photocatalytic treatment is often more suitable [21].
Composite graphene-based photocatalysts have been mostly tested in the form of suspensions, [1,22] which is favorable from the point of view of the mass transport of reactants and degradation products. However, for environmental remediation and clean-up applications, immobilized photocatalysts are often preferable for several reasons: (i) they do not require complicated separation or filtration steps, (ii) a thin layer deposited on a suitable substrate can be easily incorporated into continuous flow systems, and, last but not least, (iii) the aggregation, or even agglomeration, of particles can be prevented. Despite this, to the best of our knowledge, no study has reported the preparation of an immobilized rGO-based photocatalyst for the removal of environmental pollutants.
As a relevant environmental pollutant, we selected 4-chlorophenol, which represents serious health hazards due to its bioaccumulation and high toxicity. The US Environmental Protection Agency (EPA) has issued a list of 11 phenolic compounds considered as highly polluting materials; among them, chlorophenols are the most toxic and carcinogenic. European Union (EU) legislation has also addressed this issue, setting a maximum allowed phenol concentration of 0.5 µg L−1 in tap water. Our previous studies showed that this toxic compound can be efficiently degraded by photocatalysis [21,23,24,25,26,27,28,29].
In this study, we optimized the suspension and process parameters of the electrophoretic deposition (EPD) process to prepare an rGO-based photocatalyst immobilized on stainless-steel substrates for the photocatalytic degradation of 4-chlorophenol. Our criteria were rGO/TiO2 layer homogeneity, good mechanical stability in aqueous media, strong adhesion to the substrate, and, in particular, high photocatalytic activity. In the degradation of 4-chlorophenol, our nanocomposite was much more effective than pristine TiO2.
2. Results
2.1. Preparation of Layers by EPD
In order to quantitatively prepare rGO/TiO2 photocatalytic layers of the desired mass without using additives (e.g., binders, surfactants), EPD should be carried out under highly specific conditions. Using novel mixtures of organic solvents and a strong electric field, we found that quantitative deposition was feasible. Furthermore, the layers prepared were ready for photocatalytic testing immediately after EPD and fast volatile solvent evaporation. Neither heating for drying nor calcination for layer consolidation was necessary because the deposited composite did not contain any additional impurity and was mechanically stable when immersed in aqueous media.
Using our method, all of the particles dispersed in an organic solvent mixture were completely deposited on the substrate used. We identified tetrahydrofuran (THF) as the most suitable solvent for rGO/TiO2 deposition because of its low toxicity, sufficient volatility, and suitable electric properties (especially low relative permittivity and low conductivity). Particles of TiO2, dispersed in ethanol and injected into THF, and of rGO, dispersed in 2-propanol and injected into THF, exhibited positive and negative charges, respectively. However, in the presence of positively charged TiO2 particles, rGO did not migrate to the anode because the positive charge of TiO2 prevailed, and, consequently, the composite was deposited on the cathode. Particle settling was negligible due to their low concentration (< 0.5 g L−1) and small particle size. The low conductivity of THF enabled the application of high electric field strength (300 V cm−1). The initial electric current did not exceed 30 µA, and during EPD it decreased approximately two-fold due to the resistance of the deposited layer.
To investigate the stability of the layers in the stirred aqueous media under UV irradiation, the leakage of particles from the layer was determined and the layers with negligible leakage were selected for the photocatalytic testing. The reason for this mechanical stability was the combination of the high electric field strength and electrostatic interaction between the rGO-TiO2 aggregates and the electrode surface, which enabled to achieve the required cohesion of the layer and its adhesion to the electrode surface. Concerning the good homogeneity of the rGO-TiO2 deposit, the cathodic electrophoretic deposition was better than the anodic one. The reason was that the rGO-TiO2 aggregates were already formed in the liquid phase and not when they reached the relevant electrode, to which they adhered.
2.2. Properties of Graphene Materials
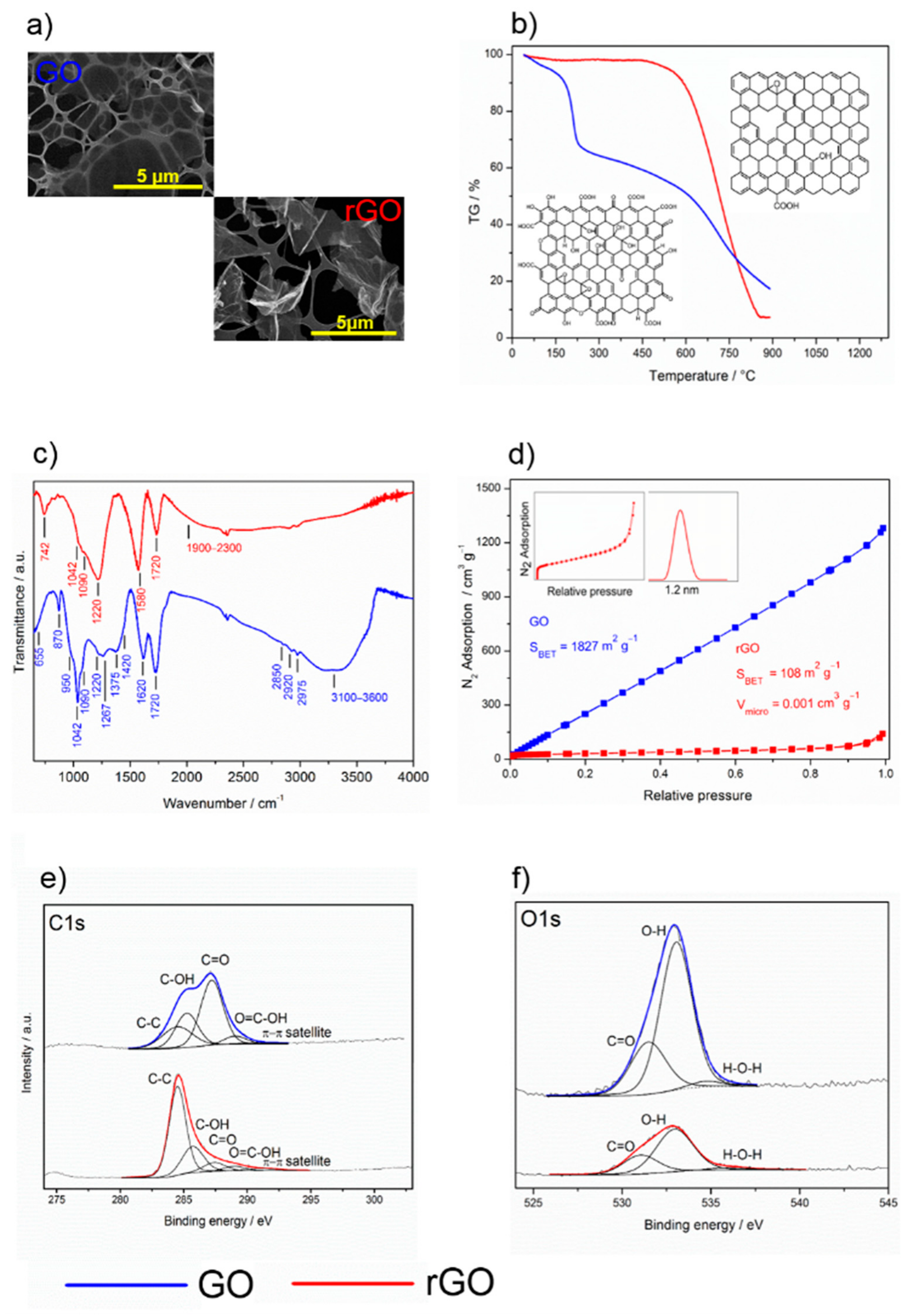
The morphological properties of the graphene materials, GO and rGO, were quite different. The two-dimensional character of the starting graphene oxide powder, GO, was confirmed by field emission scanning electron microscopy (FE-SEM) (Figure 1a) and high resolution transmission electron microscopy (HR-TEM). It was characterized by a very large surface area of approximately 1800 m2 g−1 (Figure 1c), which is in reasonable agreement with the theoretical surface area of graphene sheets [30]. The isotherms did not exhibit any stepwise character, which was clearly due to the heterogeneous structure of the surface (due to the presence of defects and various oxygen functional groups). The reduction of GO in a stream of hydrogen at 400 °C brought about drastic morphological changes. The BET surface area of the rGO sample decreased to 108 m2 g−1, the range of the validity of the BET equation being very narrow, only 0.05–0.15 P/P0. That this material is to some extent microporous was revealed by the Broekhoff–de Boer t-plot. The micropore volume determined by the back-extrapolation of the linear multilayer section was about 0.012 cm3 g−1. Assuming that this linear section represents unrestricted multilayer adsorption on the external surface, its approximate area was around 83 m2 g−1. Thus, it is reasonable to hypothesize that the change in the morphology may have been due to the sintering of carbon particles or the formation of holes because of the high-temperature reduction process.
Thermogravimetric analysis (TGA) showed that rGO had much higher thermal stability than GO due to its lower content of oxygenated functional groups. In GO (Figure 1b), a slight decrease in mass (8%) occurred between 100 and 200 °C, which can be associated with the removal of hydroxyls and adsorbed water. After this slight reduction, significant mass loss (25%) followed, indicating the exothermal release of CO and CO2 from the most labile functional groups. At temperatures below 800 °C, the total mass loss was about 80%. However, in contrast to GO, rGO showed very high thermal stability with a total mass loss of only 10% at temperatures below 800 °C, corresponding to the low content of oxygen functional groups in its structure (Figure 1b).
Fourier transformed infrared spectroscopy (FT-IR) spectroscopy showed that the spectra of the GO and rGO layers differed considerably. The GO spectrum indicated the presence of oxygen-containing functional groups together with sorbed water (Figure 1d), in accordance with the published data [31,32]. The bands at 1375 and 1090 cm−1 correspond to deformation vibrations of sorbed water while the broad bands at 1900–2300 and 3100–3600 cm−1 are associated with bridging hydroxyls (O–H), obviously overlapped by the sorbed water molecules [33]. The absorption bands at 1720 and 1620 cm−1 show C=O stretching of carbonyl groups, which are adjacent to the hexagonal graphene lattice. The overlapped bands at 1420 and 950 cm−1 are attributed to deformations of carboxyl O–H groups, while the peak centered at 1220 cm−1 is assigned to a C–O carboxylic stretch. The absorption band with a strong intensity at 1042 cm−1 can be associated with alkoxy functional groups (C–O). The band at 1267 cm−1 can be attributed to the epoxy ring stretch. The band at 870 cm−1 can be assigned either to ether symmetric C–O–C or epoxide symmetric C–O–C ring deformation vibrations. Moreover, the GO structure was represented by out-of-plane vibrations of the aromatic ring at 655 cm−1, as well as vibrations of alkyl groups (C–H) at higher frequencies of 2850, 2920, and 2975 cm−1. After GO reduction, the FT-IR spectra changed significantly, reflecting the elimination of the oxygen functional groups. Absorption at 3100–3600 cm−1 (bridging hydroxyls) decreased noticeably with the diminishing intensity of the 1620 cm−1 and 1375 cm−1 absorption bands, which was due to the substantial removal of sorbed water. Furthermore, carboxyl (950 and 1420 cm−1), alkoxy (1042 cm−1), and ether/epoxide (870 cm−1) bands were substantially reduced. Contrary, the new absorption bands at 1580 cm−1, corresponding to C–C vibrations, and at 742 cm−1, corresponding to vibration of the sp2 C–H aromatic bond, appeared. This suggests the restoration of the conjugated aromatic graphene structure. However, the distinguishable absorption band associated with C–O carboxylic groups (1220 cm−1) at the edges of the planes remained as well as the carbonyl groups (1720 cm−1). Overall, the data obtained by IR spectroscopy shows that reduction of GO under hydrogen atmosphere at a high temperature resulted in a substantial decrease of the concentration of a majority of oxygenated groups, while that of carboxylic groups appeared to be considerably less prominent.
X-ray photoelectron spectroscopy (XPS) verified the FT-IR data by confirming the elemental composition before and after reduction. Both C1s (Figure 1e, Table 1) and O1s (Figure 1f, Table 2) spectra confirmed the substantial reduction of oxygen functional groups of the GO sample. The O/C ratio before and after reduction was 0.53 and 0.19, respectively, which means that 60% of the oxygen groups were reduced. Deconvolution of O1s spectra resulted in three peaks centered at 531.3, 533.0, and 535.3 eV assigned to carbonyl, hydroxyls, and sorbed water molecules [34]. The high-resolution C1s spectra clearly show the difference between GO and its reduced form. In the GO lattice structure, the carbon atoms were bonded either to the carbon-forming C–C conjugation (284.5 eV) or to the oxygen-forming carboxyl (288.6 eV), carbonyl (287.4 eV), and hydroxyl (285.6 eV) functional groups. These assignments are in qualitative agreement with those in the literature, even though there is a certain vagueness in the exact binding energy value determination for individual components [34,35,36,37,38,39]. For both GO and rGO, the asymmetry of the C–C peak was observed, which was probably related to the screening of the core holes left by photoelectrons. Increase of aromatic sp2 (284.5 eV) in the C1s spectrum after reduction indicates that the delocalized π conjugations in rGO were significantly restored.
2.3. Properties of rGO/TiO2 Composite Layers
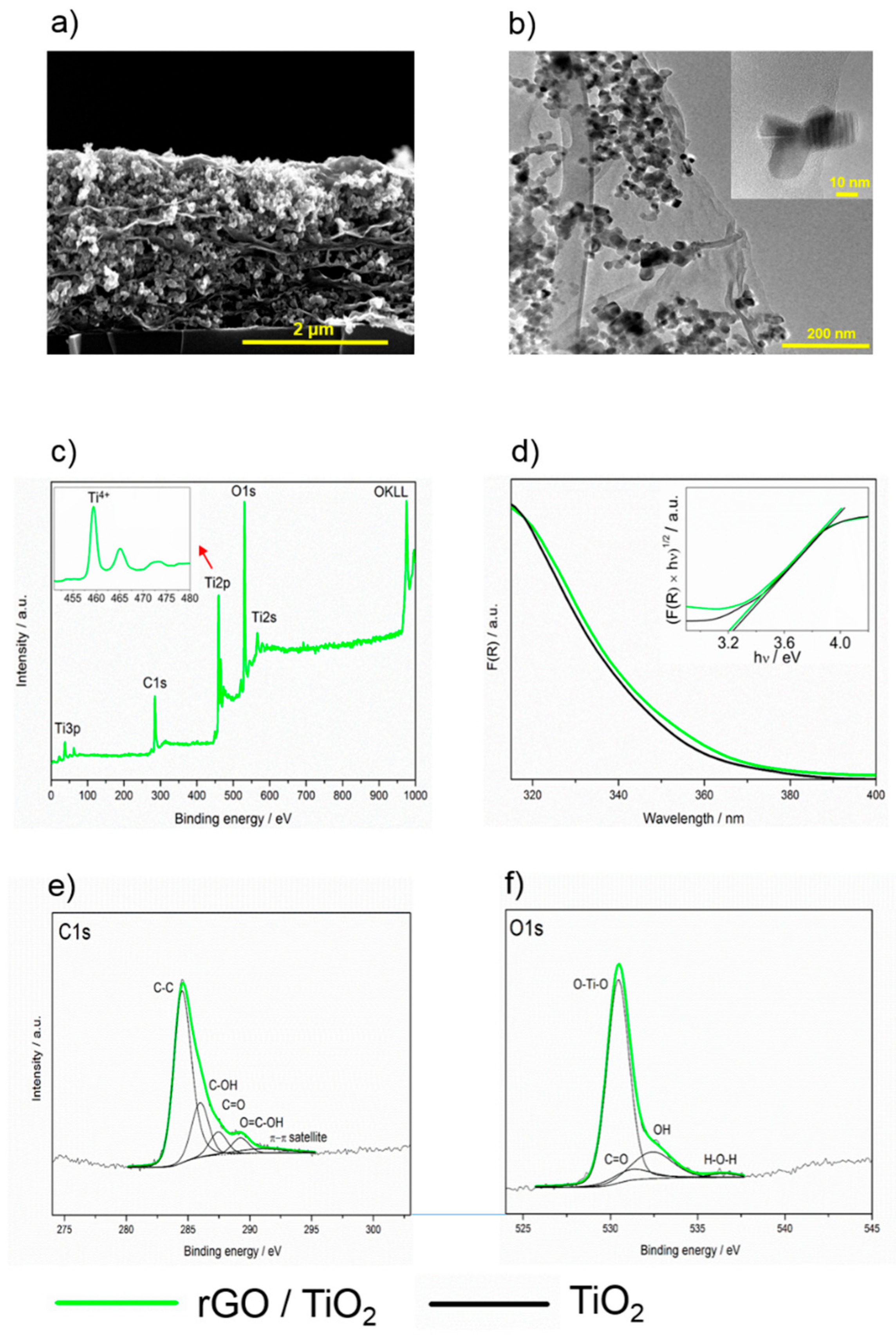
SEM and HR-TEM images of composite layers are shown in Figure 2a,b, respectively. The SEM image shows that the rGO sheets are homogeneously distributed within the whole volume of the layer, preferentially parallel with the steel substrate. The HR-TEM image proves the heterojunction between TiO2 particles and rGO sheets. In the inset to the HR-TEM image, the contact of an individual TiO2 crystal with a single rGO is shown.
The survey XPS spectra of the rGO/TiO2 composite layer show the presence of carbon, titanium, and oxygen atoms, while no other elements were identified on the surface at the detection limit (Figure 2c). The high-resolution C1s spectra showed a similar level of reduction to bare rGO (compare Figure 1e and Figure 2e), with the exception of the peak assigned to carboxyl groups which seem to be more pronounced. This confirmed the data obtained by infrared spectroscopy. The high-resolution O1s spectra showed the presence of differently bonded oxygen, dominantly bonded with Ti (Figure 2f). The Ti2p XPS spectrum (inset in Figure 2c) with the peak for Ti 2p3/2 centered at 459.4 eV, and spin-orbit-splitting of 5.6 eV documents the presence of fully oxygen-coordinated Ti4+ species.
The diffuse reflectance spectra of the rGO/TiO2 showed absorption edges below 400 nm. The spectra were plotted using the Kubelka–Munk F(R) function expressed by Equation (1).
where R, α, and s are the diffuse reflectance, the absorption, and scattering coefficients, respectively [40]. The optical band gap energy was determined using Equation (2).
where F(R), h, ν, Eg, and n are the Kubelka–Munk function, the Planck constant, the oscillation frequency, the band gap energy, and the constant relating to the mode of transition, respectively. The minimal-energy state in the conduction band and the maximal-energy state in the valence band are each characterized by a k-vector in the Brillouin zone. Two types of band-to-band transitions are suggested: first, direct transitions, where the k-vectors are the same and the participation of a phonon is not required to conserve momentum; second, indirect transitions, where at least one phonon participates in the absorption or emission of a photon to conserve momentum. The constant n equals 1/2 for the allowed direct, 3/2 for the forbidden direct, or 2 for the allowed indirect transitions. We checked the linearity of the plots of against using n = 1/2 (allowed direct) to assess the mode of the transition of a given crystal. More details are provided in the review by Ohtani [41]. Using the Tauc plot, the optical band gap energies Eg for the TiO2 and rGO/TiO2 layers were calculated [42]. The absorption data were fitted according to Equation (2). For the direct band gap (n = 1/2),satisfactory fit was obtained, giving Eg values of 3.20 and 3.25 for rGO/TiO2 and TiO2, respectively (Figure 2d).
2.4. Photocatalytic Performance of rGO/TiO2 Layers
As a test reaction, we used the photocatalytic degradation of 4-chlorophenol because this compound is a common water pollutant that exhibits good chemical stability, undergoes negligible photolysis, and possesses relatively low adsorption on the photocatalyst surface.
First, the effect of the stability of the deposited layers on their photocatalytic performance was determined. Experiments showed that the layers deposited on the stainless-steel surface exhibited very good adhesion, with no leaking of titania or carbonaceous particles during the photocatalytic tests. No particles were detected in the solution after the experiments.
Having determined the adhesion properties of the composite, we investigated the photocatalytic performance of the GO/TiO2 and TiO2 layers. As they exhibited similar activities, the doping with GO did not produce any substantial improvement in photocatalytic activity. This performance is due to the physicochemical properties of GO, which is an insulant with abundant oxygenated surface groups. This drawback was removed by the reduction of GO, which transformed the insulant GO into more or less highly conductive rGO.
The improved photocatalytic performance is shown in Figure 3a, in which the decrease in 4-chlorophenol concentration using rGO/TiO2 was statistically significant much faster. The first-order reaction rate constant for rGO/TiO2 was 0.0053 min−1, which was 70% higher than that for GO/TiO2 or TiO2. When reused, the layers showed no deterioration in photocatalytic performance, which is very important from the application point of view (Figure 3b).
Generally, a photocatalytic process involves two different mechanisms: a direct transfer of charge carriers and an attack by radicals, especially by hydroxyl ones. Depending on the reaction conditions, these mechanisms may act separately or jointly.
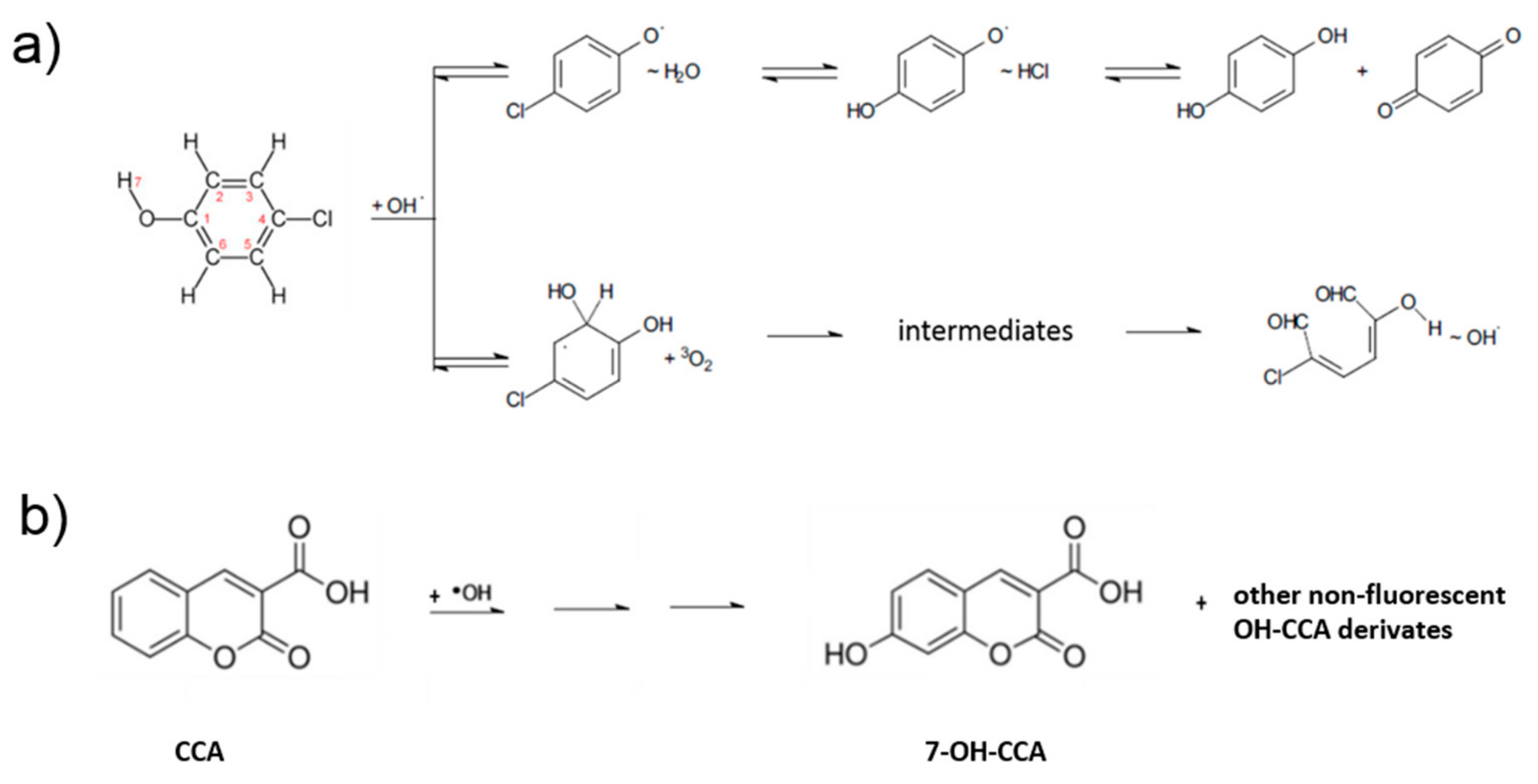
To explain the mechanism of 4-chlorophenol photocatalytic degradation, in our previous studies, we employed experimental and theoretical, using density function theory (DFT), approaches. Our results therein showed that the hydroxylation and opening of the aromatic ring can occur in parallel with the release of the hydroperoxyl and hydroxyl radicals [23,27]. The restored OH radical can either further oxidize the primary ring opening product or attack another molecule of 4-chlorophenol. These DFT results were in good agreement with the photocatalytic degradation experiments conducted using both ZnO and TiO2 photocatalysts [23,27]. According to the parallel mechanism, the products of 4-chlorophenol degradation are the experimentally identified aromatics hydroquinone and benzoquinone, as well as various ring-opening compounds formed under the restoration of OH radicals (Figure 4a).
In this study, the enhanced performance of the rGO/TiO2 photocatalyst can be explained by an increased formation of radicals owing to a better separation of charge carriers. This phenomenon can be caused by the downward bending of the TiO2 conduction band due to the presence of rGO, which can suppress the charge carrier recombination. The recombination can be further suppressed by the high mobility of electrons over the graphene surface. The freed oxidative holes can react either directly with the pollutant molecules or indirectly through the OH radicals, which are strong oxidants.
Due to the high reactivity and short lifetime of OH radicals in water, their direct detection is very difficult. Thus, for their indirect detection, fluorescence spectroscopy was employed using coumarin-3-carboxylic acid. After the UV irradiation of TiO2, this acid reacts with OH radicals to produce 7-OH-coumarin-3-carboxy acid (Figure 4b), emitting strong fluorescence at 442 nm. Compared with the TiO2 sample, rGO/TiO2 exhibited higher fluorescence intensity (Figure 3c).
Additional experiments performed to confirm the predominant mechanism of 4-CP degradation on the doped and undoped TiO2 layers showed that indirect action through radicals played the major role. However, its predominance only became clear when 2-propanol was used as a radical scavenger. Due to the similar rate constants of the reaction of the hydroxyl radical with 2-propanol [43] and with 4-chlorophenol [44], a 1000-fold excess of 2-propanol would practically stop the degradation of 4-chlorophenol if only an OH-attack is responsible for it. However, as can be seen in Figure 3d, the photocatalytic degradation of 4-chlorophenol only slowed down, 6.7 times for rGO/TiO2, 4.2 times for GO/TiO2, and 3.6 times for pristine TiO2 layer (pseudo first-order reaction rate constants of 0.00079 ± 0.00004, 0.00073 ± 0.00004, and 0.00085 ± 0.00005 min−1, respectively), but continued. These similar rate constants suggest that direct charge transfer participated similarly in the photocatalytic degradation of 4-chlorophenol in each case and that the presence of the rGO dopant did not influence this mechanism. However, the presence of rGO positively influenced the photocatalytic degradation of 4-chlorophenol in the absence of 2-propanol (pseudo first-order rate constants of 0.0031 and 0.0053 min−1 for TiO2 layers without and with rGO, respectively), which therefore is probably due to enhanced radical production.
3. Materials and Methods
Materials. The commercial 2–4 layered graphene oxide and Aeroxide TiO2 P 25 were purchased from Cheap Tubes Inc. (Grafton, USA) and Evonik Industries (Essen, Germany), respectively. Organic solvents (p.a. purity) acetone, tetrahydrofuran, heptane, 2-propanol, and ethanol were supplied by Penta (Prague, Czech Republic). Sheets of glossy stainless steel AISI 304 (25 × 50 × 1 mm3) served as electrodes.
Preparation of rGO. The commercial GO sample was reduced in a stream of hydrogen (10 mL/min) at 400 °C for 8 h to obtain reduced graphene oxide rGO used for the deposition of composite layers.
Preparation of rGO/TiO2 composite layers. Prior to EPD, electrodes were washed ultrasonically in deionized water and acetone. During EPD, the electric field was adjusted to 300 V cm−1 using DC power supply EV245 (Consort, Turnhout, Belgium). Polished stainless-steel plates served as the working (cathode) and counter electrode (anode). The distance between the vertically placed electrodes was 11 mm. The deposition mixture was prepared by a two-step process. First, titanium dioxide (2 or 4 g/L) and rGO (up to 0.4 g/L) were dispersed in ethanol or 2-propanol. Then a small volume (0.25 or 0.5 mL) of these dispersions was injected into tetrahydrofuran (10 mL). The titania loading on the support was between 0.03 and 2.5 mg cm−2. Ten minutes of the EPD process was found sufficient for the deposition of all dispersed particles.
Characterization. The elements present in titania, graphene oxide, and reduced graphene oxide, as well as their chemical state, were identified by X-ray photoelectron spectrometer (East Grinstead, Great Britain) (VG ESCA3 MkII) with a base pressure better than 10−9 mbar. For the excitation of the electrons, Al Kα radiation was used. The energy of electrons was analyzed using a hemispherical analyzer, which operated at a constant pass energy of 20 eV. The XPS spectra were calibrated by setting the C1s peak belonging to the sp2 component at a binding energy of 284.5 eV. The estimated error in binding energy determination was ±0.1 eV. The surface atomic content was accomplished assuming a homogenous distribution of atoms and Scofield photoionization cross-section. The crystallinity of titania powders was measured using a D5000 high-resolution X-ray diffractometer (Siemens, Munich, Germany) operated at 40 kV and 45 mA with Cu Kα radiation (λ = 1.5406 Å). The texture properties of powders and layers were determined by the analysis of adsorption isotherms of nitrogen or krypton at approximately 77 K performed with a Micrometrics 3FLEX volumetric adsorption unit. The surface profile of layers was studied with a FE-SEM instrument Hitachi S-4800 (Hitachi High-Technologies Corporation, Tokyo, Japan) and HR-TEM instrument JEM-2100Plus (JEOL Ltd., Tokyo, Japan). The optical properties of the layers were measured with a Lambda 950 UV-Vis-NIR spectrometer (Perkin Elmer, Waltham, USA) equipped with Spectralon and gold integration spheres for diffuse reflectance measurements in the UV-Vis-NIR region and with a Fourier transform infrared spectrophotometer Nicolet 6700. The thermogravimetric measurements were carried out in a flow of Ar at a temperature ramp of 10 K/min using a TGA-MS Netzsch STA449 apparatus (Selb, Germany).
Photocatalytic experiment. Before the photocatalytic experiments, the samples to be tested were cleaned overnight by UV light of a dominant wavelength of 365 nm with an irradiation intensity of 2.0 mW cm−2 to decompose any residual organic matter on them. The 4-chlorophenol in aqueous solution (0.1 mmol L−1) was photocatalytically degraded at 25 °C on TiO2 and doped TiO2 layers (TiO2 mass of 0.5 mg, rGO mass of 0.025 mg, substrate area of 6 cm2) in a 25 mL quartz cell. As the top of the liquid in the cell was open to air and the solution was intensively stirred, the concentration of dissolved oxygen was constant during the experiment. A Sylvania Lynx-S 11 W BLB lamp irradiated the layer surface with UV light (365 nm) at a low power density of 1.0 mW cm−2. Prior to the photocatalytic experiments, the dissolved 4-chlorophenol was equilibrated with the photocatalyst surface for three hours. For each experiment, six aliquots each of 100 µL were taken from the solution in the reaction cell at regular time intervals and analyzed by high-performance liquid chromatography. The first-order reaction rate constants for the 4-chlorophenol degradation were calculated using nonlinear regression fitting of the kinetic curves. In the mechanistic study, the scavenger (2-propanol) was added at the very beginning of the experiment in a concentration of 10−1 M. For the detection of OH radicals formed during the irradiation of titanium dioxide and its composite containing rGO with 365 nm light, we employed a fluorescence spectroscopy (Fluorolog 3, Horiba, Kyoto, Japan). In this experiment, an aqueous 10−4 M solution of coumarin-3-carboxylic acid reacted with OH radicals to produce 7-OH-coumarin-3-carboxylic acid, which was excited at 365 nm and emitted a strong fluorescence at 442 nm. The photoluminescence (PL) spectra of the layers were measured using continuous PL waves of the 4th harmonic (266 nm) line of the YAG: Nd laser and dispersed with a HR250 monochromator (Jobin-Yvon, Horiba, Kyoto, Japan) coupled with a UV-enhanced intensified charge coupled device (ICCD) (Roper Scientific, Planegg, Germany). Under pulsed laser excitation, PL spectra were recorded in a pseudo CW mode with a continuous integration of the intensity in 300 ms corresponding to three full illumination pulses.
Data analysis and statistics. The photocatalytic experiments were done in three replicates. Significant differences between compared samples were determined by using Student’s t test where p values were used as the threshold for statistical significance. Normality of the data and homogeneity of variance were tested using the Pearson’s chi-squared test of goodness of fit and the Levene’s test (StatSoft CR, Zlicin, Czech Republic), respectively. The tests showed that the data fulfilled the assumptions for the parametric statistics applied (i.e., normality of the data and homogeneity of variance).
4. Conclusions
We have shown that our optimized EPD technique enables the fast and simple preparation of an immobilized rGO/TiO2 composite photocatalyst and that the addition of rGO, even at low concentrations (0.5%), can substantially increase the rate of pollutant degradation compared with GO/TiO2 and pristine TiO2. The reduction of GO with hydrogen at higher temperatures led to marked changes in morphological and chemical properties. In terms of morphology, rGO exhibited a much smaller surface area accompanied by the formation of micropores, which is very different from graphene itself. Concerning the chemical properties, such as thermal stability and the content of surface oxygenated groups, they approached those of graphene. The presence of rGO in the composite photocatalyst led to the increased formation of hydroxyl radicals, which explains the enhancement in photocatalytic performance. This mechanism dominated the performance of the composite while the degradation due to the direct charge transfer mechanism was smaller. The present study, together with our previous ones [21,23,24,25,26,27,28,29], form the basis for extensive and complex research into the photocatalytic performance of composites containing different allotropes of TiO2 and carbonaceous dopants, including fullerene, carbon nanotubes, reduced graphene oxide, and graphene itself.
Author Contributions
Conceptualization, J.R.; methodology, L.B., J.P.; validation, J.R., R.Z., M.R., L.B., J.P.; investigation, M.R., L.B., J.P.; resources, J.R.; data curation, J.R., R.Z., M.R., L.B., J.P.; writing—original draft preparation, J.R., R.Z.; writing—review and editing, J.R., R.Z.; supervision, J.R.; project administration, J.R.; funding acquisition, J.R.
Funding
This research was funded by the Czech Science Foundation (GACR, Grant No. 19-21801S).
Acknowledgments
This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic and The European Union—European Structural and Investments Funds in the frame of Operational Programme Research, Development and Education—project Pro-NanoEnviCz (Project No. CZ.02.1.01/0.0/0.0/16_013/0001821), which provided an access to the Micromeritics 3Flex apparatus. Finally, the authors thank Craig Alfred Riddell, B.A.M.Sc. (UCT, Prague) for an improvement of the readability of the text.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Niu, P.; Zhang, L.; Liu, G.; Cheng, H.M. Graphene-like carbon nitride nanosheets for improved photocatalytic activities. Adv. Funct. Mater. 2012, 22, 4763–4770. [Google Scholar] [CrossRef]
- Tu, W.; Zhou, Y.; Zou, Z. Versatile graphene-promoting photocatalytic performance of semiconductors: Basic principles, synthesis, solar energy conversion, and environmental applications. Adv. Funct. Mater. 2013, 23, 4996–5008. [Google Scholar] [CrossRef]
- Jeon, S.J.; Kang, T.W.; Ju, J.M.; Kim, M.J.; Park, J.H.; Raza, F.; Han, J.; Lee, H.R.; Kim, J.H. Modulating the Photocatalytic Activity of Graphene Quantum Dots via Atomic Tailoring for Highly Enhanced Photocatalysis under Visible Light. Adv. Funct. Mater. 2016, 26, 8211–8219. [Google Scholar] [CrossRef]
- Chabot, V.; Higgins, D.; Yu, A.; Xiao, X.; Chen, Z.; Zhang, J. A review of graphene and graphene oxide sponge: Material synthesis and applications to energy and the environment. Energy Environ. Sci. 2014, 7, 1564–1596. [Google Scholar] [CrossRef]
- Li, F.; Jiang, X.; Zhao, J.; Zhang, S. Graphene oxide: A promising nanomaterial for energy and environmental applications. Nano Energy 2015, 16, 488–515. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Avouris, P. Graphene: Electronic and photonic properties and devices. Nano Lett. 2010, 10, 4285–4294. [Google Scholar] [CrossRef]
- Bolotin, K.I.; Sikes, K.J.; Jiang, Z.; Klima, M.; Fudenberg, G.; Hone, J.; Kim, P.; Stormer, H.L. Ultrahigh electron mobility in suspended graphene. Solid State Commun. 2008, 146, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Morozov, S.V.; Novoselov, K.S.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Gao, W. The chemistry of graphene oxide. In Graphene Oxide Reduction Recipes, Spectroscope and Application; Springer: Cham, Switzerland, 2015; pp. 61–95. [Google Scholar]
- Pei, S.; Cheng, H.M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Liu, L.; Qing, M.; Wang, Y.; Chen, S. Defects in Graphene: Generation, Healing, and Their Effects on the Properties of Graphene: A Review. J. Mater. Sci. Technol. 2015, 31, 599–606. [Google Scholar] [CrossRef]
- Huang, X.; Qi, X.; Boey, F.; Zhang, H. Graphene-based composites. Chem. Soc. Rev. 2012, 41, 666–686. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Yu, J.; Jaroniec, M. Graphene-based semiconductor photocatalysts. Chem. Soc. Rev. 2012, 41, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Eda, G.; Chhowalla, M. Graphene-based composite thin films for electronics. Nano Lett. 2009, 9, 814–818. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, J.C. Graphene-based photocatalytic composites. RSC Adv. 2011, 1, 1426–1434. [Google Scholar] [CrossRef]
- Li, N.; Liu, G.; Zhen, C.; Li, F.; Zhang, L.; Cheng, H.M. Battery performance and photocatalytic activity of mesoporous anatase TiO2 nanospheres/graphene composites by template-free self-assembly. Adv. Funct. Mater. 2011, 21, 1717–1722. [Google Scholar] [CrossRef]
- Oishi, I.; Kim, S.; Yoshii, K.; Esteban, C.R.; Izpisua Belmonte, J.C. Cre-LoxP-regulated expression of monoclonal antibodies driven by an ovalbumin promoter in primary oviduct cells. BMC Biotechnol. 2011, 11, 19. [Google Scholar] [CrossRef]
- Dąbrowski, A. Adsorption-from theory to practice. Adv. Colloid Interface Sci. 2001, 93, 135–224. [Google Scholar] [CrossRef]
- Zouzelka, R.; Remzova, M.; Brabec, L.; Rathousky, J. Photocatalytic performance of porous TiO2 layers prepared by quantitative electrophoretic deposition from organic solvents. Appl. Catal. B Environ. 2018, 227. [Google Scholar] [CrossRef]
- Liu, J.; Bai, H.; Wang, Y.; Liu, Z.; Zhang, X.; Sun, D.D. Self-assembling TiO2 nanorods on large graphene oxide sheets at a two-phase interface and their anti-recombination in photocatalytic applications. Adv. Funct. Mater. 2010, 20, 4175–4181. [Google Scholar] [CrossRef]
- Guérin, V.M.; Zouzelka, R.; Bibova-Lipsova, H.; Jirkovsky, J.; Rathousky, J.; Pauporté, T. Experimental and DFT study of the degradation of 4-chlorophenol on hierarchical micro-/nanostructured oxide films. Appl. Catal. B Environ. 2015, 168–169, 132–140. [Google Scholar] [CrossRef]
- Zouzelka, R.; Kusumawati, Y.; Remzova, M.; Rathousky, J.; Pauporté, T. Photocatalytic activity of porous multiwalled carbon nanotube-TiO2 composite layers for pollutant degradation. J. Hazard. Mater. 2016, 317, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.; Fattakhova-Rohlfing, D.; Kayaalp, B.E.; Rathouský, J.; Bein, T. Tailoring the morphology of mesoporous titania thin films through biotemplating with nanocrystalline cellulose. J. Am. Chem. Soc. 2014, 136, 5930–5937. [Google Scholar] [CrossRef] [PubMed]
- Zita, J.; Krýsa, J.; Černigoj, U.; Lavrenčič-Štangar, U.; Jirkovský, J.; Rathouský, J. Photocatalytic properties of different TiO2 thin films of various porosity and titania loading. Catal. Today 2011, 161, 29–34. [Google Scholar] [CrossRef]
- Hynek, J.; Kalousek, V.; Žouželka, R.; Bezdička, P.; Dzik, P.; Rathouský, J.; Demel, J.; Lang, K. High photocatalytic activity of transparent films composed of zno nanosheets. Langmuir 2014, 30, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.; Fravventura, M.C.; Fattakhova-Rohlfing, D.; Rathouský, J.; Movsesyan, L.; Ganter, P.; Savenije, T.J.; Bein, T. Nanocellulose-Templated Porous Titania Scaffolds Incorporating Presynthesized Titania Nanocrystals. Chem. Mater. 2015, 27, 6205–6212. [Google Scholar] [CrossRef]
- Kusumawati, Y.; Pauporté, T.; Viana, B.; Zouzelka, R.; Remzova, M.; Rathousky, J. Mesoporous TiO2/graphene composite films for the photocatalytic degradation of eco-persistent pollutants. In Proceedings of the Oxide-based Materials and Devices VIII, San Francisco, CA, USA, 29 January–1 February 2017; Volume 10105. [Google Scholar]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 183, 153–160. [Google Scholar] [CrossRef]
- Shahhriary, L.; Athawale, A.A. Graphene Oxide Synthesized by using Modified Hummers Approach. Int. J. Renew. Energy Environ. Eng. 2014, 2, 58–63. [Google Scholar]
- Rattana, T.; Chaiyakun, S.; Witit-Anun, N.; Nuntawong, N.; Chindaudom, P.; Oaew, S.; Kedkeaw, C.; Limsuwan, P. Preparation and characterization of graphene oxide nanosheets. Procedia Eng. 2012, 32, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yan, J.; Wang, T.; Zhao, Z.-J.; Zhang, J.; Gong, J.; Guan, N. Sub-10 nm rutile titanium dioxide nanoparticles for efficient visible-light-driven photocatalytic hydrogen production. Nat. Commun. 2015, 6, 5881. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the thermal deoxygenation of graphene oxide using high-resolution in situ X-ray-based spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar] [CrossRef]
- Mattevi, C.; Eda, G.; Agnoli, S.; Miller, S.; Mkhoyan, K.A.; Celik, O.; Mastrogiovanni, D.; Granozzi, G.; Carfunkel, E.; Chhowalla, M. Evolution of electrical, chemical, and structural properties of transparent and conducting chemically derived graphene thin films. Adv. Funct. Mater. 2009, 19, 2577–2583. [Google Scholar] [CrossRef]
- Akhavan, O. The effect of heat treatment on formation of graphene thin films from graphene oxide nanosheets. Carbon 2010, 48, 509–519. [Google Scholar] [CrossRef]
- Jeong, H.K.; Yun, P.L.; Lahaye, R.J.W.E.; Park, M.H.; Kay, H.A.; Ick, J.K.; Yang, C.W.; Chong, Y.P.; Ruoff, R.S.; Young, H.L. Evidence of graphitic AB stacking order of graphite oxides. J. Am. Chem. Soc. 2008, 130, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Velamakanni, A.; Bozoklu, G.; Park, S.; Stoller, M.; Piner, R.D.; Stankovich, S.; Jung, I.; Field, D.A.; Ventrice, C.A.; et al. Chemical analysis of graphene oxide films after heat and chemical treatments by X-ray photoelectron and Micro-Raman spectroscopy. Carbon 2009, 47, 145–152. [Google Scholar] [CrossRef]
- Vanquickenborne, L.G.; Vranckx, J.; Görller-walrand, C. On the Kinetic Trans Effect in Square Planar Transition Metal Complexes. J. Am. Chem. Soc. 1974, 96, 4121–4125. [Google Scholar] [CrossRef]
- Kubelka, P. New Contributions to the Optics of Intensely Light-Scattering Materials. Part I. J. Opt. Soc. Am. 1948, 38, 448–457. [Google Scholar] [CrossRef]
- Ohtani, B. Photocatalysis A to Z-What we know and what we do not know in a scientific sense. J. Photochem. Photobiol. C Photochem. Rev. 2010, 11, 157–178. [Google Scholar] [CrossRef]
- Tauc, J. Optical Properties and electronic structure of amorphous. Mater. Res. Bull. 1968, 3, 37–46. [Google Scholar] [CrossRef]
- Motohashi, N.; Saito, Y. Competetive measurement of rate constants for hydroxyl radical reactions using radiolytic hydroxylation of benzoate. Chem. Pharm. Bull. (Tokyo) 1993, 41, 1842–1845. [Google Scholar] [CrossRef]
- Stafford, U.; Gray, K.A.; Kamat, P.V. Radiolytic and TiO2-assisted photocatalytic degradation of 4-chlorophenol. A comparative study. J. Phys. Chem. 1994, 98, 6343–6351. [Google Scholar] [CrossRef]
Figure 1.
Physicochemical properties of graphene oxide (GO, blue) and reduced graphene oxide (rGO, red). (a) SEM images, (b) TGA curves, (c) FT-IR spectra, (d) physical sorption of nitrogen, (e) C1s, and (f) O1s XPS spectra.
Figure 1.
Physicochemical properties of graphene oxide (GO, blue) and reduced graphene oxide (rGO, red). (a) SEM images, (b) TGA curves, (c) FT-IR spectra, (d) physical sorption of nitrogen, (e) C1s, and (f) O1s XPS spectra.

Figure 2.
(a) SEM cross-section image of a composite layer containing TiO2 and rGO, (b) HR-TEM image of the composite, (c) survey XPS spectrum of the rGO/TiO2 composite layer, (d) UV/vis spectra of the rGO/TiO2 and TiO2 layers, (e) C1s and (f) O1s spectra rGO/TiO2 composite layer.
Figure 2.
(a) SEM cross-section image of a composite layer containing TiO2 and rGO, (b) HR-TEM image of the composite, (c) survey XPS spectrum of the rGO/TiO2 composite layer, (d) UV/vis spectra of the rGO/TiO2 and TiO2 layers, (e) C1s and (f) O1s spectra rGO/TiO2 composite layer.

Figure 3.
(a) Course of the photocatalytic degradation of 4-chlorophenol on TiO2 and rGO/TiO2 photocatalysts, (b) reusability of TiO2 and rGO/TiO2 photocatalysts, (c) fluorescence spectra of 7-OH-coumarin-3-carboxylic acid after irradiation of TiO2 and rGO/TiO2 with 365 nm light for 30 s, (d) effect of OH radical scavenger (2-propanol) on the course of the 4-chlorophenol photocatalytic degradation on TiO2, GO/TiO2, and rGO/TiO2.
Figure 3.
(a) Course of the photocatalytic degradation of 4-chlorophenol on TiO2 and rGO/TiO2 photocatalysts, (b) reusability of TiO2 and rGO/TiO2 photocatalysts, (c) fluorescence spectra of 7-OH-coumarin-3-carboxylic acid after irradiation of TiO2 and rGO/TiO2 with 365 nm light for 30 s, (d) effect of OH radical scavenger (2-propanol) on the course of the 4-chlorophenol photocatalytic degradation on TiO2, GO/TiO2, and rGO/TiO2.

Figure 4.
(a) Simplified scheme of the photocatalytic 4-chlorophenol degradation. (b) Reaction of OH radical with coumarin 3-carboxy acid to form fluorescent molecule 7-hydroxy coumarin 3-carboxy acid.
Figure 4.
(a) Simplified scheme of the photocatalytic 4-chlorophenol degradation. (b) Reaction of OH radical with coumarin 3-carboxy acid to form fluorescent molecule 7-hydroxy coumarin 3-carboxy acid.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Amount of various oxygenated groups before and after reduction of GO, calculated from the C1s XPS spectra.
Table 1.
Amount of various oxygenated groups before and after reduction of GO, calculated from the C1s XPS spectra.
| Sample | C–C/% | C–OH/% | C=O/% | O=C–OH/% |
|---|---|---|---|---|
| GO | 22.7 | 24.6 | 46.8 | 5.8 |
| rGO | 63.5 | 23.9 | 8.2 | 4.3 |
| rGO/TiO2 | 66.7 | 19.4 | 8.2 | 5.6 |
Table 2.
Amount of various oxygenated groups before and after reduction of GO, calculated from the O1s XPS spectra.
Table 2.
Amount of various oxygenated groups before and after reduction of GO, calculated from the O1s XPS spectra.
| Sample | C=O/% | C–OH/% | H–O–H/% |
|---|---|---|---|
| GO | 27.6 | 69.8 | 2.6 |
| rGO | 28.5 | 67.7 | 3.8 |
| rGO/TiO2 | 26.6 | 66.5 | 6.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zouzelka, R.; Remzova, M.; Plsek, J.; Brabec, L.; Rathousky, J. Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water. Catalysts 2019, 9, 708. https://doi.org/10.3390/catal9090708
AMA Style
Zouzelka R, Remzova M, Plsek J, Brabec L, Rathousky J. Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water. Catalysts. 2019; 9(9):708. https://doi.org/10.3390/catal9090708
Chicago/Turabian StyleZouzelka, Radek, Monika Remzova, Jan Plsek, Libor Brabec, and Jiri Rathousky. 2019. "Immobilized rGO/TiO2 Photocatalyst for Decontamination of Water" Catalysts 9, no. 9: 708. https://doi.org/10.3390/catal9090708
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.