
Completion of Crystallographic Data for the Series of 4-Halogenated-1H-pyrazoles: Crystal Structure Determination of 4-Iodo-1H-pyrazole and Spectroscopic Comparison


Abstract
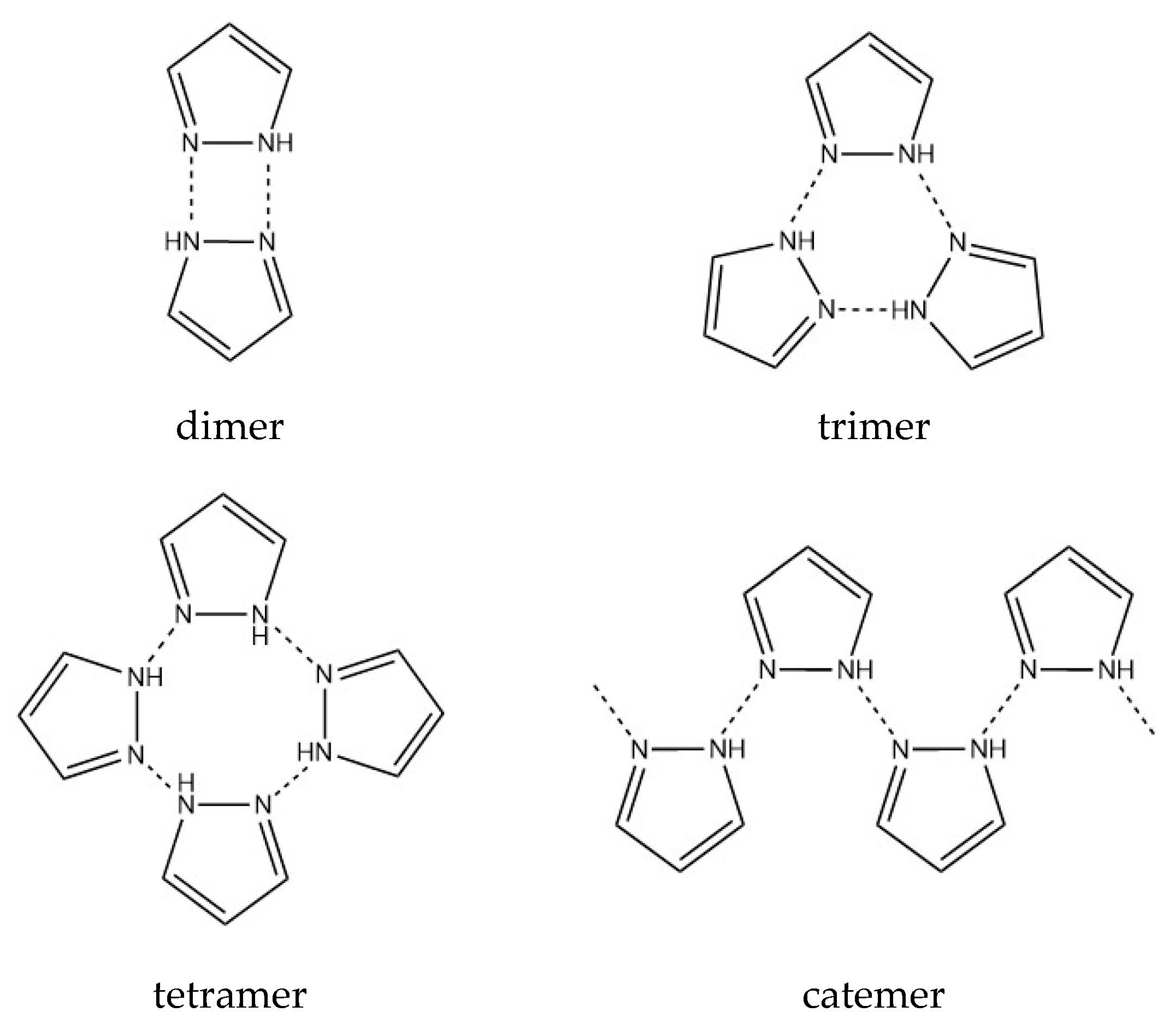
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DFT Calculations
2.3. X-ray Crystallography and Data Collection
3. Results and Discussion
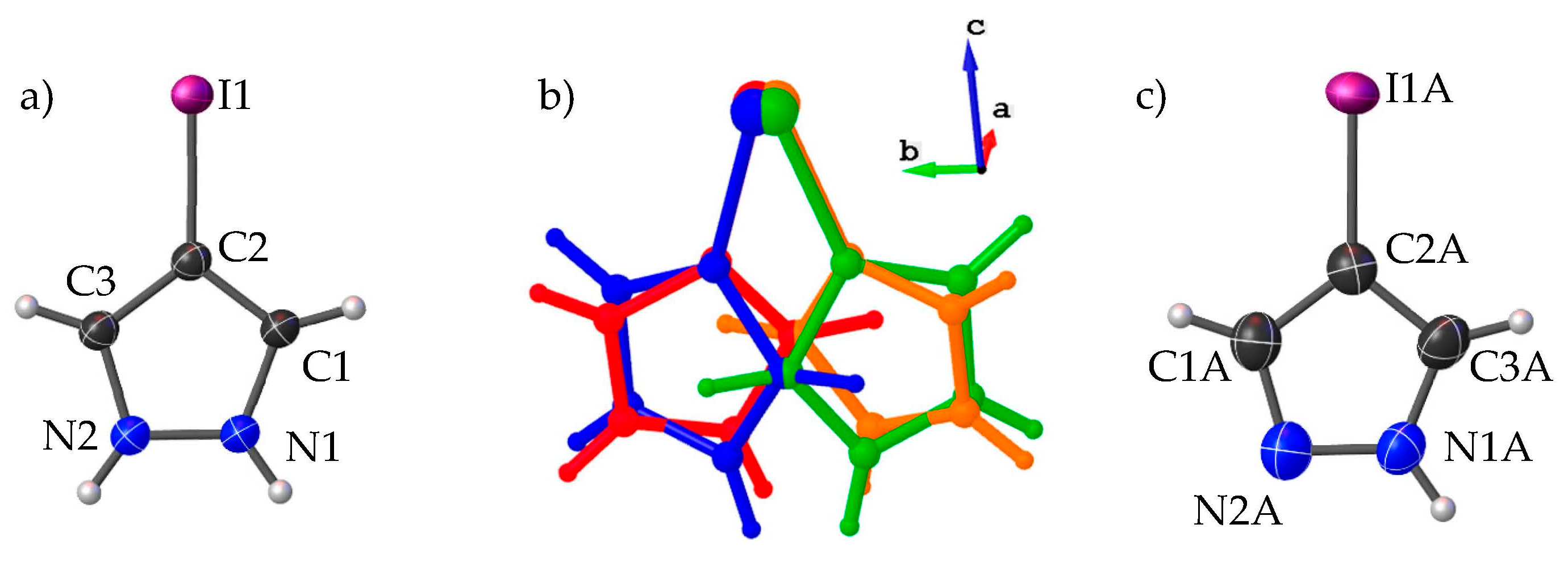
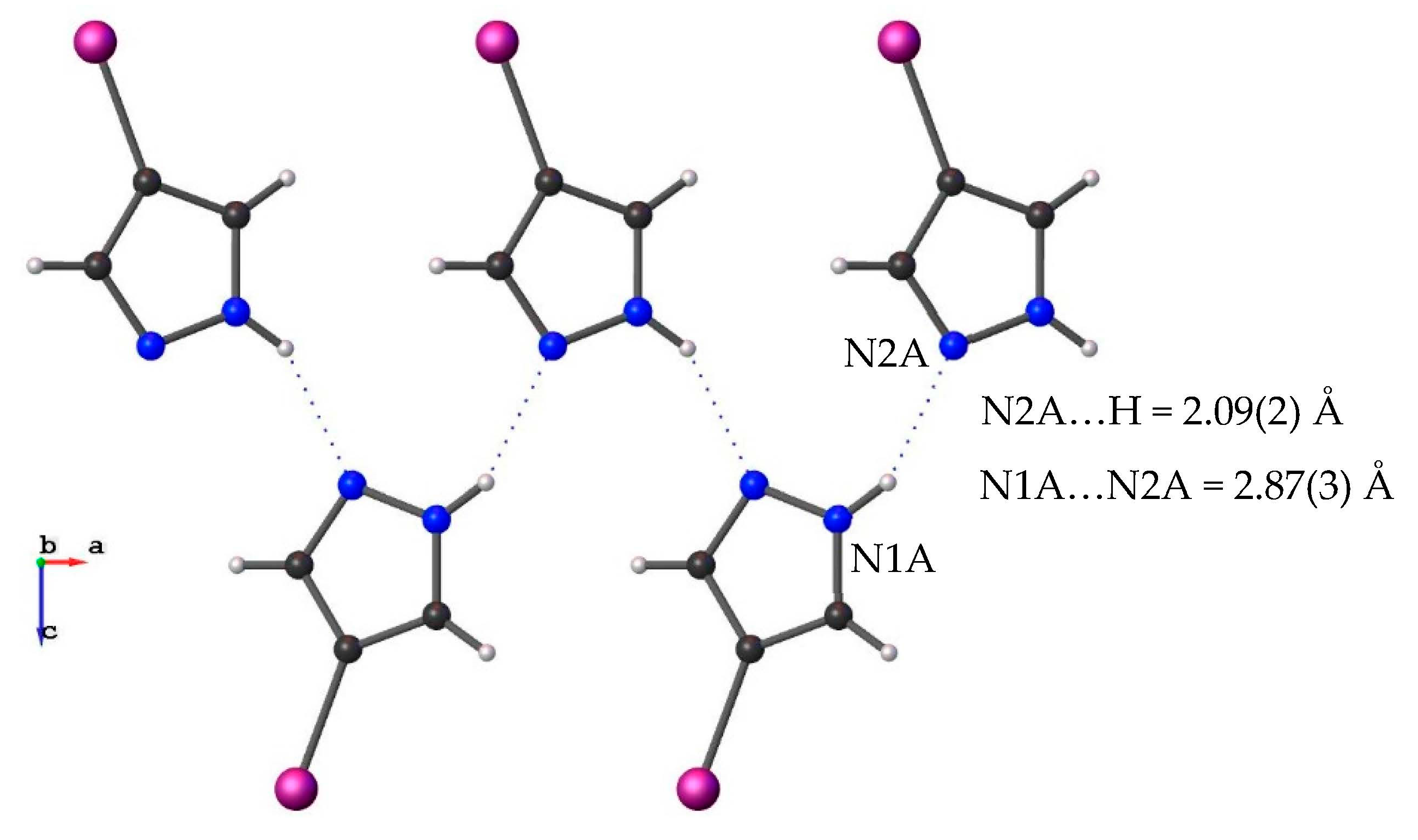
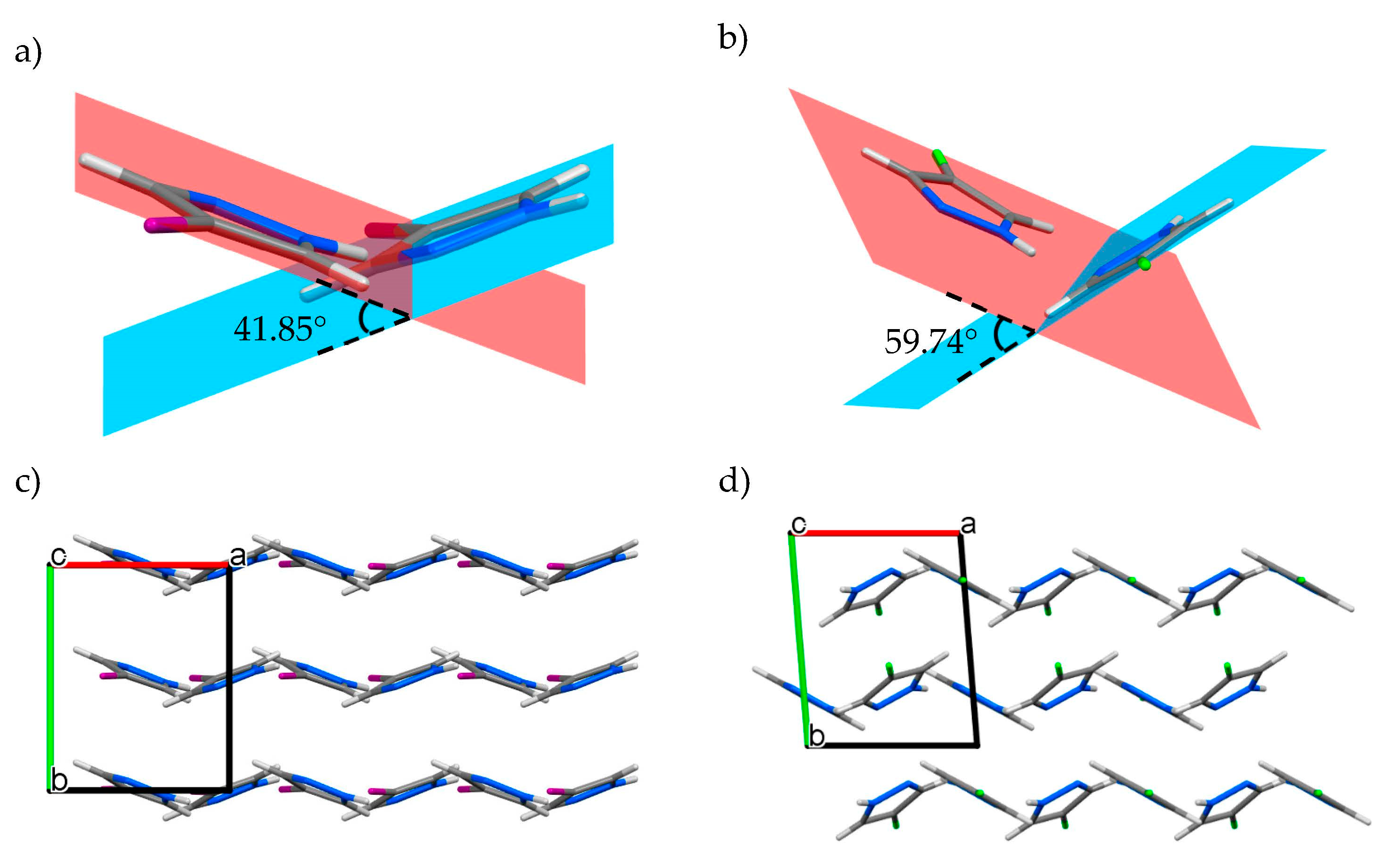
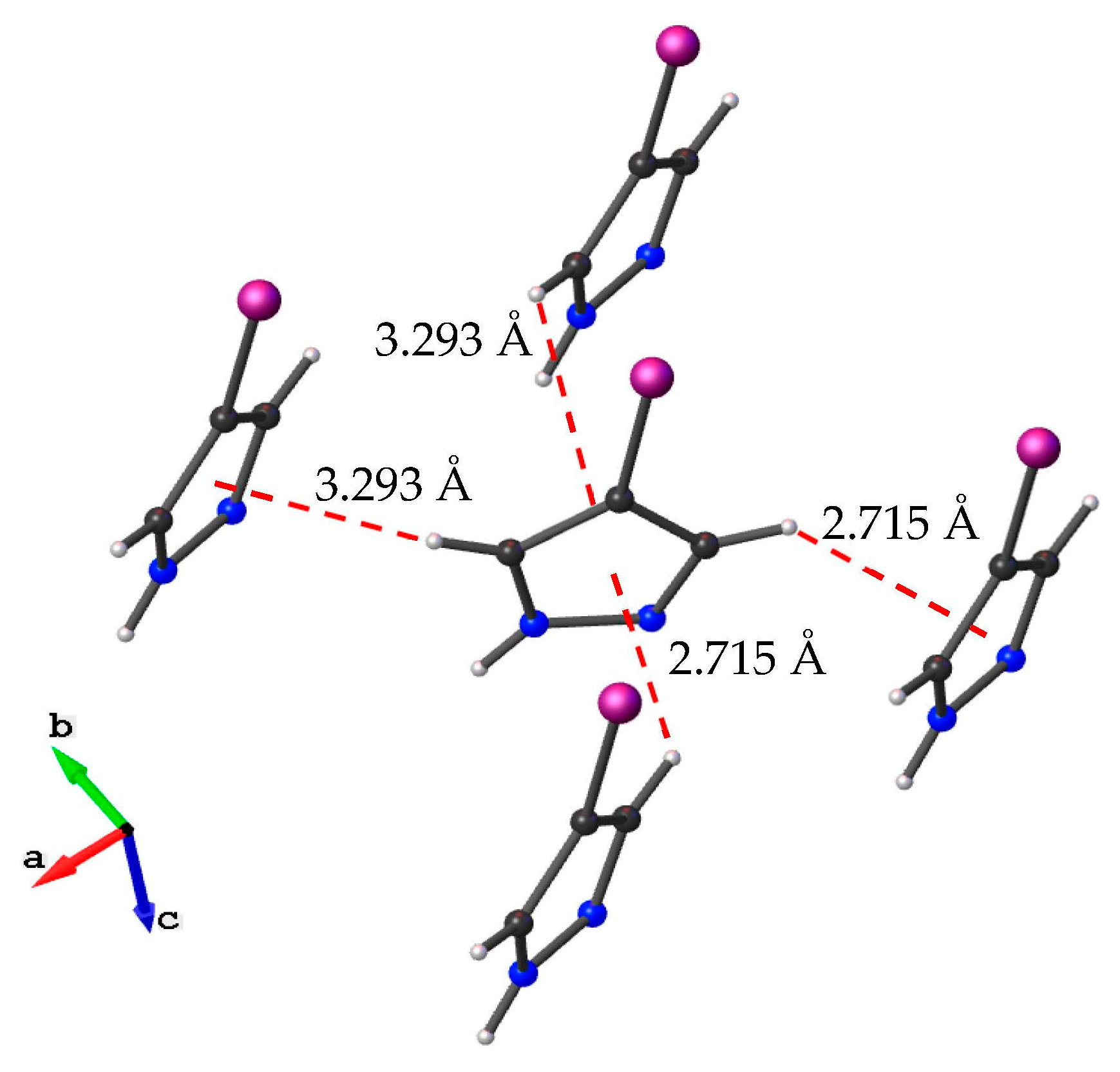
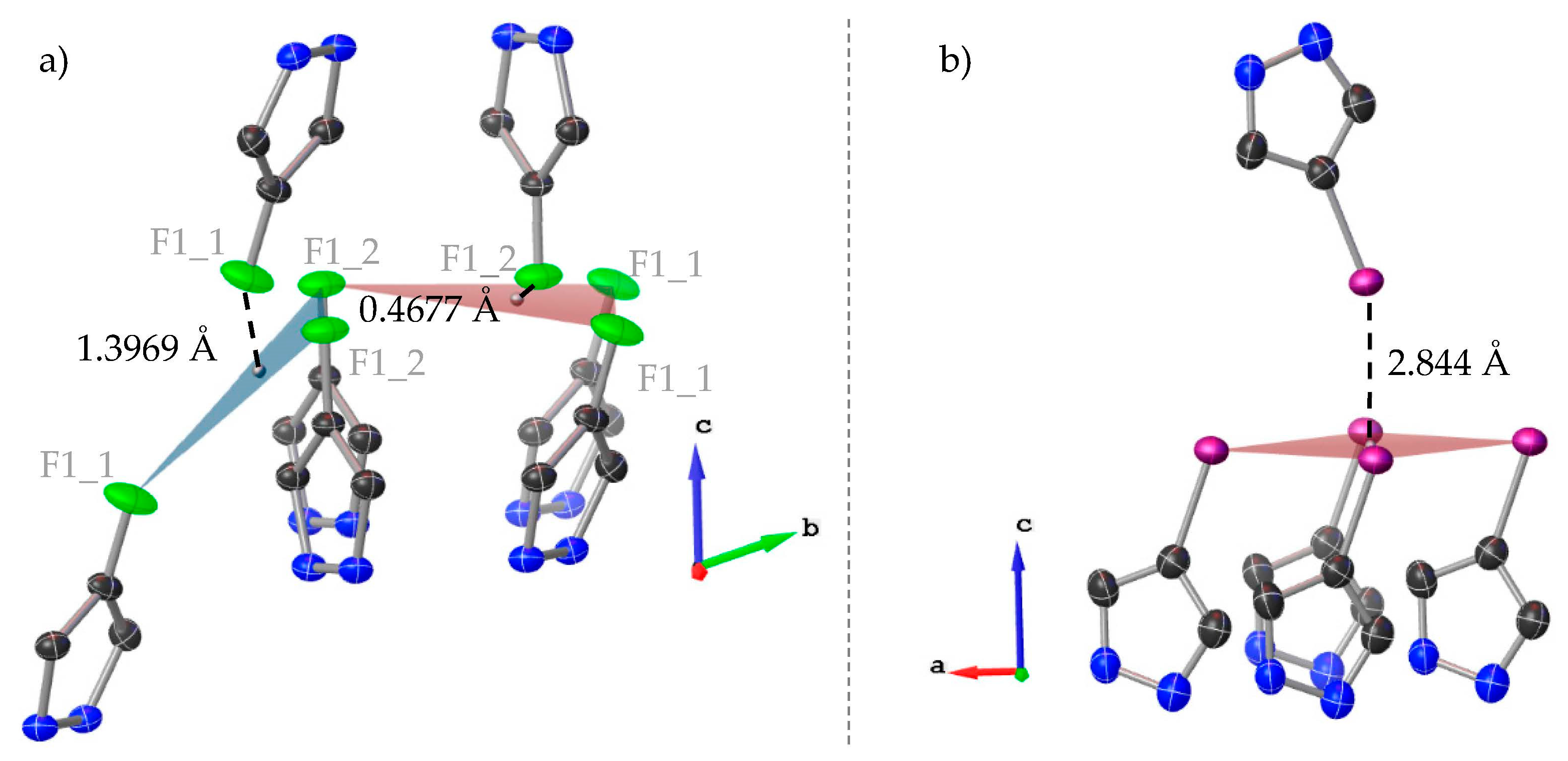
3.1. Crystal Structure Description
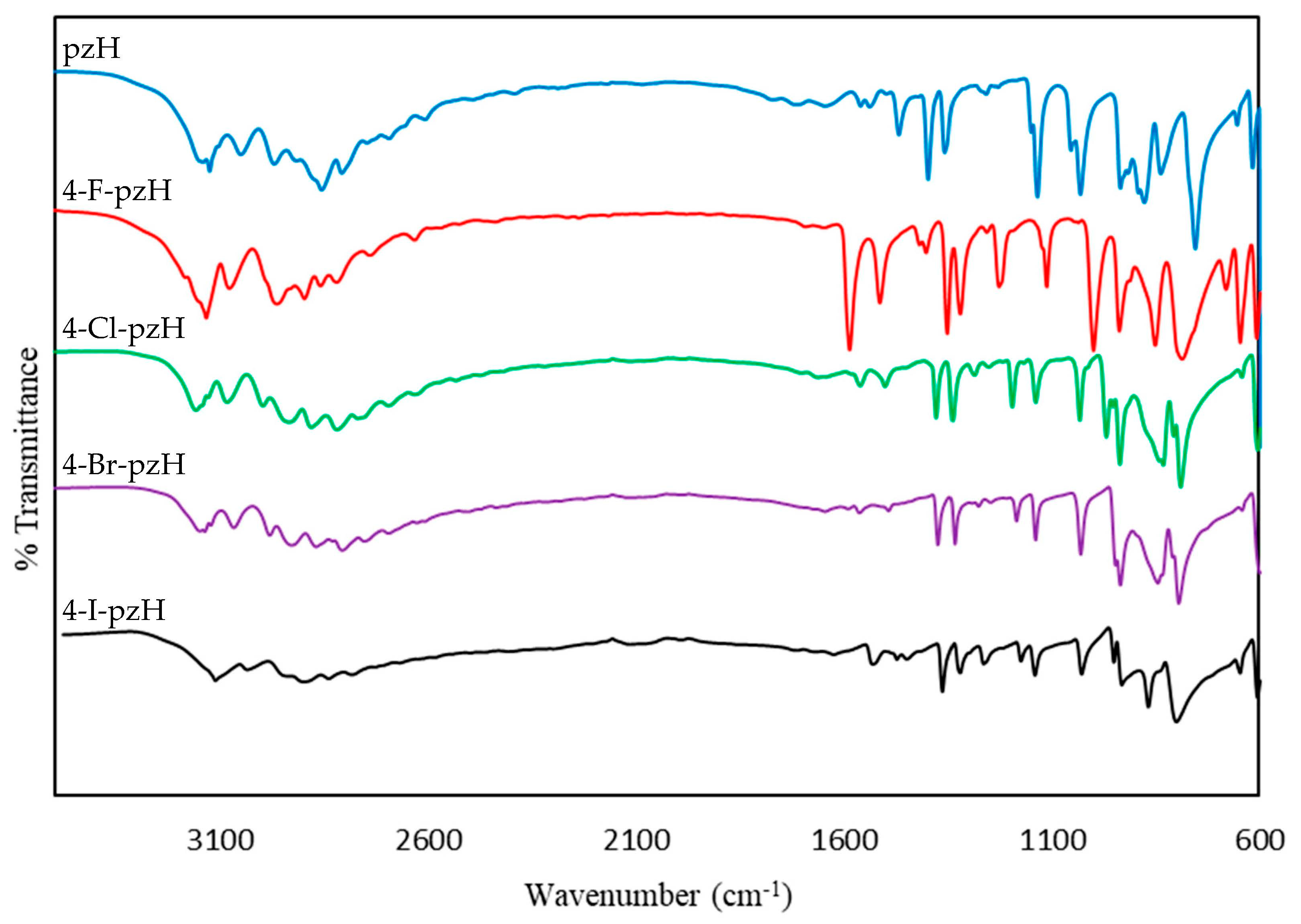
3.2. Spectroscopy and DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trofimenko, S. Coordination Chemistry of Pyrazole-Derived Ligands. Chem. Rev. 1972, 72, 497–509. [Google Scholar] [CrossRef]
- Viciano-Chumillas, M.; Tanase, S.; de Jongh, L.J.; Reedijk, J. Coordination Versatility of Pyrazole-Based Ligands towards High-Nuclearity Transition-Metal and Rare-Earth Clusters. Eur. J. Inorg. Chem. 2010, 2010, 3403–3418. [Google Scholar] [CrossRef]
- La Monica, G.; Ardizzoia, G.A. The Role of the Pyrazolate Ligand in Building Polynuclear Transition Metal Systems. In Progress in Inorganic Chemistry; Karlin, K.D., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 151–238. [Google Scholar] [CrossRef] [Green Version]
- Pérez, J.; Riera, L. Pyrazole Complexes and Supramolecular Chemistry. Eur. J. Inorg. Chem. 2009, 2009, 4913–4925. [Google Scholar] [CrossRef]
- Song, X.; Wang, Y.; Wang, C.; Wang, D.; Zhuang, G.; Kirlikovali, K.O.; Li, P.; Farha, O.K. Design Rules of Hydrogen-Bonded Organic Frameworks with High Chemical and Thermal Stabilities. J. Am. Chem. Soc. 2022, 144, 10663–10687. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, L.-R.; Tong, J.; Yu, Y.-M.; Wang, X.-Y.; Yu, S.-Y. Supramolecular Hydrogen Bonding Assembly from Non-Coplanar Aromatic Tetra-1H-Pyrazoles with Crystallization-Induced Emission (CIE). Int. J. Mol. Sci. 2022, 23, 4206. [Google Scholar] [CrossRef]
- Brewer, G.; Butcher, R.J.; Zavalij, P. Use of Pyrazole Hydrogen Bonding in Tripodal Complexes to Form Self Assembled Homochiral Dimers. Materials 2020, 13, 1595. [Google Scholar] [CrossRef] [Green Version]
- Moyano, S.; Diosdado, B.; Felices, L.S.; Elduque, A.; Giménez, R. Structural Diversity of Hydrogen-Bonded 4-Aryl-3,5-Dimethylpyrazoles for Supramolecular Materials. Materials 2021, 14, 4550. [Google Scholar] [CrossRef]
- Infantes, L.; Motherwell, S. Prediction of H-Bonding Motifs for Pyrazoles and Oximes Using the Cambridge Structural Database. Struct. Chem. 2004, 15, 173–184. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Foces-Foces, C.; Infantes, L. Classification of hydrogen-bond motives in crystals of NH-pyrazoles: A mixed empirical and theoretical approach. Arkivoc 2005, 2006, 15–30. [Google Scholar] [CrossRef] [Green Version]
- FFoces-Foces, C.; Alkorta, I.; Elguero, J. Supramolecular Structure of 1H-Pyrazoles in the Solid State: A Crystallographic and Ab Initio Study. Acta Crystallogr. Sect. B Struct. Sci. 2000, 56, 1018–1028. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [Green Version]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Biologically Active Pyrazole Derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Li, G.; Cheng, Y.; Han, C.; Song, C.; Huang, N.; Du, Y. Pyrazole-Containing Pharmaceuticals: Target, Pharmacological Activity, and Their SAR Studies. RSC Med. Chem. 2022, 13, 1300–1321. [Google Scholar] [CrossRef]
- Elkanzi, N.A.A.; Zahou, F.M. Short Review on Pharmacological Characteristics and Synthesis of Pyrazole. Heterocycl. Lett. 2021, 11, 111–130. [Google Scholar]
- Foces-Foces, C.; Llamas-Saiz, A.L.; Elguero, J. Crystal Structures of Two 4-Bromopyrazole Derivatives. Z. Kristallogr. Cryst. Mater. 1999, 214, 237–241. [Google Scholar] [CrossRef]
- Rue, K.; Raptis, R.G. Low-Temperature Crystal Structure of 4-Chloro-1 H -Pyrazole. Acta Crystallogr. E Cryst. Commun. 2021, 77, 955–957. [Google Scholar] [CrossRef]
- Ahmed, B.M.; Zeller, M.; Mezei, G. Crystal and Molecular Structure of 4-Fluoro-1 H -Pyrazole at 150 K. Acta Crystallogr. E Cryst. Commun. 2023, 79, 428–431. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GAUSSIAN 16, Revision C.1; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Basis Set Exchange Library. Available online: https://www.basissetexchange.org/ (accessed on 12 June 2023).
- CCheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruker. APEX3; Bruker AXS LLC: Madison, WI, USA, 2020. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, H.W. The Crystal and Molecular Structure of Pyrazole. Acta Crystallogr. 1960, 13, 946–952. [Google Scholar] [CrossRef]
- Sikora, M.; Katrusiak, A. Pressure-Controlled Neutral–Ionic Transition and Disordering of NH···N Hydrogen Bonds in Pyrazole. J. Phys. Chem. C 2013, 117, 10661–10668. [Google Scholar] [CrossRef]
- Larsen, F.K.; Lehmann, M.S.; Søtofte, I.; Rasmussen, S.E. A Neutron Diffraction Study of the Crystal and Molecular Structure of Pyrazole, C3H4N2. Acta Chem. Scand. 1970, 24, 3248–3258. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.K.; Veluthaparambath, R.V.; Krishna, G.V. Halogen⋯Halogen Interactions: Nature, Directionality and Applications. Chem. Asian J. 2023, 18, e202300067. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. [Google Scholar] [CrossRef]
- Secrieru, A.; O’neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2020, 25, 42. [Google Scholar] [CrossRef] [Green Version]
- Notario, R.; Herreros, M.; El Hammadi, A.; Homan, H.; Abboud, J.-L.M.; Forfar, I.; Claramunt, R.M.; Elguero, J. Similarity in Physical and Organic Chemistry: Substituent Effects on the Intrinsic Basicity of 4-Substituted Pyrazoles. J. Phys. Org. Chem. 1994, 7, 657–662. [Google Scholar] [CrossRef]
- Catalan, J.; Elguero, J. Basicity and Acidity of Azoles. In Advances in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 1987; Volume 41, pp. 187–274. [Google Scholar] [CrossRef]
- Gümüş, S.; Türker, L. Substituent effect on the aromaticity of 1,3-azole systems. Heterocycl. Commun. 2012, 18, 11–16. [Google Scholar] [CrossRef]
- Smith, W.B.; Ihrig, A.M.; Roark, J.L. Substituent Effects on Aromatic Proton Chemical Shifts. VII. Further Examples Drawn from Disubstituted Benzenes. J. Phys. Chem. 1970, 74, 74812–74821. [Google Scholar] [CrossRef]
- Scheiner, S.; Hunter, S. Influence of Substituents in the Benzene Ring on the Halogen Bond of Iodobenzene with Ammonia. ChemPhysChem 2022, 23, e202200011. [Google Scholar] [CrossRef]
- Baranac-Stojanović, M. Substituent Effect on the Triplet State Aromaticity of Benzene. J. Org. Chem. 2020, 85, 4289–4297. [Google Scholar] [CrossRef]
- Palusiak, M.; Domagała, M.; Dominikowska, J.; Bickelhaupt, F.M. The Substiuent Effect on Benzene Dications. Phys. Chem. Chem. Phys. 2014, 16, 4752–4763. [Google Scholar] [CrossRef] [Green Version]
- Abedini, N.; Kassaee, M.Z. The Effects of Halogen Substituents on Structure, Stability, and Electronic Properties of bicyclo[1.1.1]pentanylene at Density Functional Theory. J. Phys. Org. Chem. 2022, 35, e4304. [Google Scholar] [CrossRef]
- Wassermann, T.N.; Rice, C.A.; Suhm, M.A.; Luckhaus, D. Hydrogen Bonding Lights up Overtones in Pyrazoles. J. Chem. Phys. 2007, 127, 234309. [Google Scholar] [CrossRef] [PubMed]
- Wolff, H.; Müller, H. Structure of the NH Stretching Vibrational Band of Pyrazole. Multiple Resonance of Substances Forming Strong H or D Bonds. Spectrochim. Acta Part A Mol. Spectrosc. 1976, 32, 581–585. [Google Scholar] [CrossRef]
- Rice, C.A.; Borho, N.; Suhm, M.A. Dimerization of Pyrazole in Slit Jet Expansions. Z. Phys. Chem. 2005, 219, 379–388. [Google Scholar] [CrossRef]
- Hanamoto, T.; Koga, Y.; Kido, E.; Kawanami, T.; Furuno, H.; Inanaga, J. Palladium catalyzed cross-coupling reaction of 5-tributylstannyl-4-fluoropyrazole. Chem. Commun. 2005, 15, 2041–2043. [Google Scholar] [CrossRef]
- Zhao, Z.-G.; Wang, Z.-X. Halogenation of Pyrazoles using N-Halosuccinimides in CCl4 and in Water. Synth. Commun. 2007, 37, 137–147. [Google Scholar] [CrossRef]
- Mezei, G.; Raptis, R.G. Effect of Pyrazole-Substitution on the Structure and Nuclearity of Cu(II)-Pyrazolato Complexes. Inorg. Chim. Acta 2004, 357, 3279–3288. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4-I-pzH | |
|---|---|
| Formula | C3H3IN2 |
| Dcalc./g cm−3 | 2.571 |
| μ/mm−1 | 6.23 |
| Formula Weight | 193.97 |
| T/K | 172 |
| Crystal System | Orthorhombic |
| Space group | Cmme (No. 67) |
| a/Å | 6.9383 (6) |
| b/Å | 5.5231 (5) |
| c/Å | 13.077 (2) |
| α, β, γ/° | 90, 90, 90 |
| V/Å3 | 501.13 (8) |
| Z | 4 |
| Wavelength/Å | 0.71073 |
| Radiation Type | Mo Kα |
| θmin, θmax/° | 3.1, 31.1 |
| Measured Refl. | 5294 |
| Independent Refl. | 483 |
| Rint | 0.039 |
| Parameters | 55 |
| a GooF, b wR2, c R1 | 1.20, 0.068, 0.030 |
| N(H)…N | Dipole Moment | Motif | Reference | |
|---|---|---|---|---|
| pzH | 2.908 (2) | 2.2923 | Catemer | [33] |
| 4-F-pzH | 2.889 (1) | 2.3995 | Catemer | [18] |
| 4-Cl-pzH | 2.867 (3) | 2.5421 | Trimer | [17] |
| 4-Br-pzH | 2.89 (2) | 2.5158 | Trimer | [16] |
| 4-I-pzH | 2.87 (3) | 2.4155 | Catemer | This work |
| C-X Bond Length (Å) | Sum of C-X Covalent Radii (Å) [37] | Difference (Experimental—cov. Radii, Å) | |
|---|---|---|---|
| 4-F-pzH | 1.341(2) | 1.30(4) | 0.04 |
| 4-Cl-pzH | 1.717(3) | 1.75(4) | −0.02 |
| 4-Br-pzH | 1.874(11) | 1.93(4) | −0.06 |
| 4-I-pzH | 2.039(9) | 2.12(4) | −0.08 |
| Experimental N-H Stretching Modes (cm−1) | Calculated N-H Stretching Modes (cm−1) | ||||
|---|---|---|---|---|---|
| pzH | 3153.04 | 3126.05 | 3104.83 | 3286.3 | 3265.4 |
| 4-F-pzH | 3187.76 | 3154.97 | 3133.76 | 3291.2 | 3270.3 |
| 4-Cl-pzH | 3156.90 | 3143.40 | 3127.97 | 3286.7 | 3266.9 |
| 4-Br-pzH | 3153.04 | 3137.62 | 3122.19 | 3284.9 | 3265.4 |
| 4-I-pzH | - | - | 3110.62 | 3283.2 | 3264.2 |
| N-H | H3,5 a | H4 | ||
|---|---|---|---|---|
| pzH | Chemical Shift (ppm) | 11.087 (9.89) | 7.631 (7.74) | 6.358 (6.56) |
| Multiplicity | s | d | t | |
| J (Hz) | - | 2.1 | 2.1 | |
| Integration | - b | 2H | 1H | |
| 4-F-pzH | Chemical Shift (ppm) | 11.137 (9.47) | 7.469 (7.52) | - |
| Multiplicity | s | d | - | |
| J (Hz) | - | 4.5 | - | |
| Integration | 1H | 2H | - | |
| 4-Cl-pzH | Chemical Shift (ppm) | 11.570 (9.78) | 7.581 (7.63) | - |
| Multiplicity | s | s | - | |
| J (Hz) | - | - | - | |
| Integration | 1H | 2H | - | |
| 4-Br-pzH | Chemical Shift (ppm) | 11.530 (9.88) | 7.615 (7.64) | - |
| Multiplicity | s | s | - | |
| J (Hz) | - | - | - | |
| Integration | 1H | 2H | - | |
| 4-I-pzH | Chemical Shift (ppm) | 11.755 (10.00) | 7.658 (7.64) | - |
| Multiplicity | s | s | - | |
| J (Hz) | - | - | - | |
| Integration | 1H | 2H | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rue, K.L.; Herrera, S.; Chakraborty, I.; Mebel, A.M.; Raptis, R.G. Completion of Crystallographic Data for the Series of 4-Halogenated-1H-pyrazoles: Crystal Structure Determination of 4-Iodo-1H-pyrazole and Spectroscopic Comparison. Crystals 2023, 13, 1101. https://doi.org/10.3390/cryst13071101
Rue KL, Herrera S, Chakraborty I, Mebel AM, Raptis RG. Completion of Crystallographic Data for the Series of 4-Halogenated-1H-pyrazoles: Crystal Structure Determination of 4-Iodo-1H-pyrazole and Spectroscopic Comparison. Crystals. 2023; 13(7):1101. https://doi.org/10.3390/cryst13071101
Chicago/Turabian StyleRue, Kelly L., Susana Herrera, Indranil Chakraborty, Alexander M. Mebel, and Raphael G. Raptis. 2023. "Completion of Crystallographic Data for the Series of 4-Halogenated-1H-pyrazoles: Crystal Structure Determination of 4-Iodo-1H-pyrazole and Spectroscopic Comparison" Crystals 13, no. 7: 1101. https://doi.org/10.3390/cryst13071101