
Toward Direct Exploration of the Few-Femtosecond Dynamics of Electronic Coherence and Correlation in Quantum Materials Using Time- and Angle-Resolved Photoemission Spectroscopy


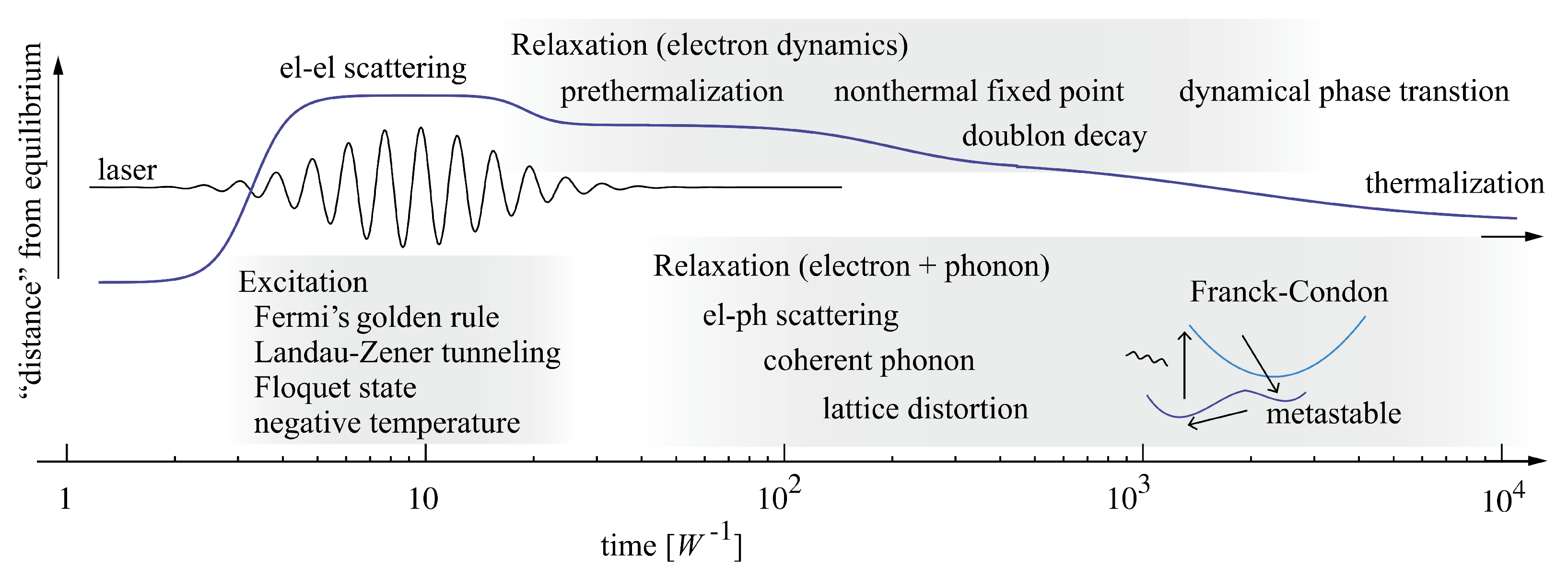{kind=link}
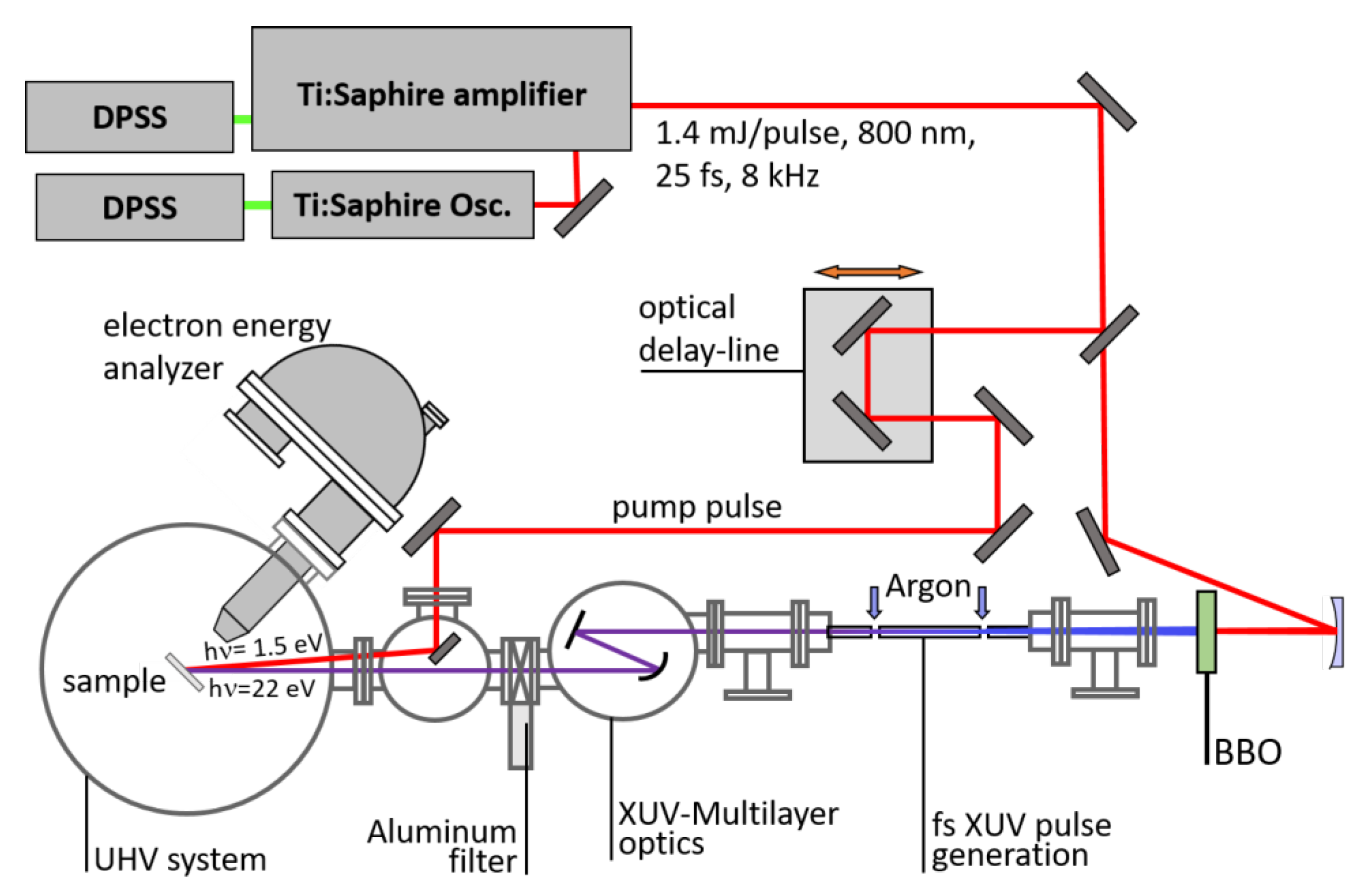{kind=link}
{kind=link}
{kind=link}
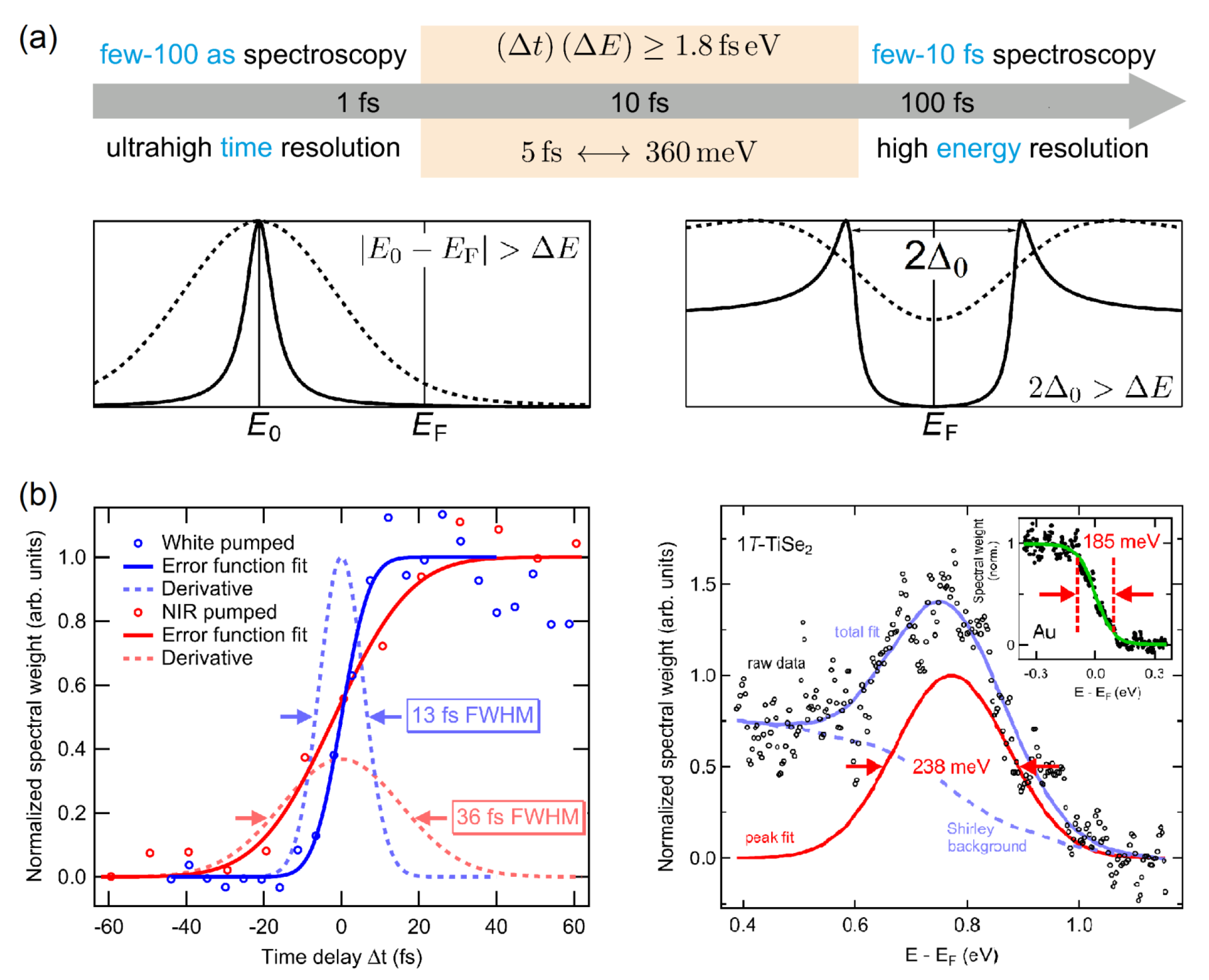{kind=link}
{kind=link}
{kind=link}
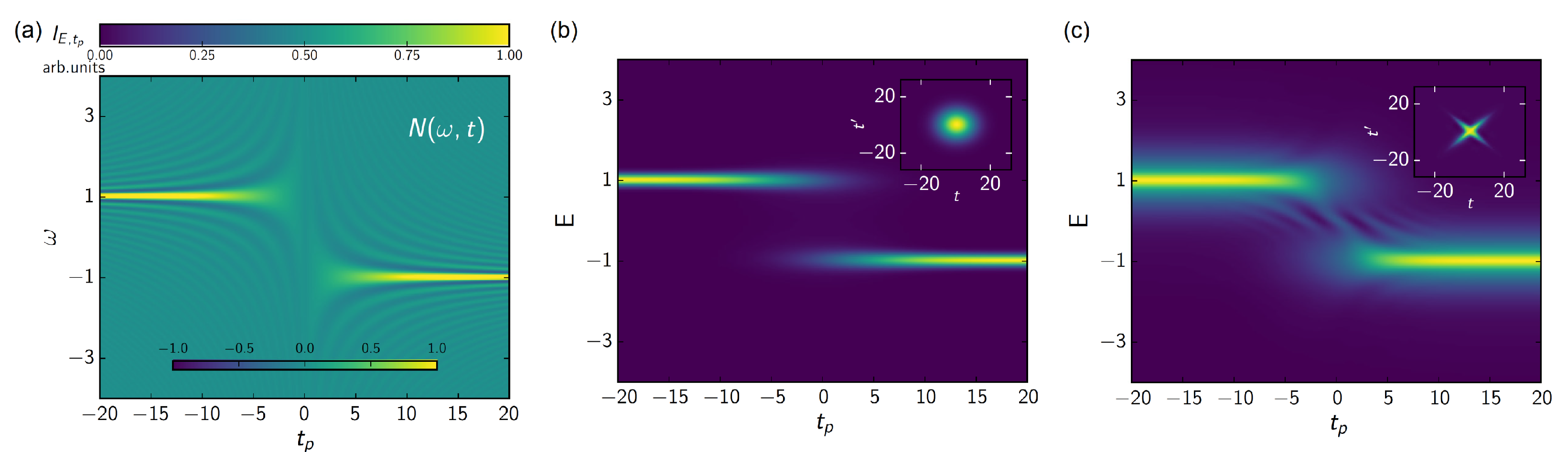{kind=link}
Abstract
:1. Introduction
2. Current Temporal Resolution Limits of trARPES: Few-Femtosecond Dynamics in Two Prominent Layered Quantum Materials
2.1. Photoexcitation of a (Possible) Mott-Insulator Phase in 1T-TaS2
2.2. Ultrafast Suppression of a (Possible) Excitonic Insulator Phase in 1T-TiSe2
3. Pushing the Temporal Resolution Limit of trARPES: Entering the Sub-10 fs Time Regime


4. Future Perspectives of Few-Femtosecond trARPES: Exploring Quantum Interference and Overcoming the Time–Bandwidth Product
4.1. Few-Cycle Strong-Field ARPES at PHz Frequencies
4.2. Coherent Multidimensional ARPES
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARPES | angle-resolved photoemission spectroscopy |
| BZ | Brillouin zone |
| CCDW | commensurate charge-density wave |
| CDW | charge-density wave |
| CEP | carrier–envelope phase |
| EDC | energy distribution curve |
| FWHM | full width at half maximum |
| LAPE | laser-assisted photoemission |
| LHB | lower Hubbard band |
| NIR | near-infrared |
| RABBIT | reconstruction of attosecond beating by interference of two-photon transitions |
| TMDC | transition-metal dichalcogenide |
| trARPES | time- and angle-resolved photoemission spectroscopy |
| UHB | upper Hubbard band |
| XUV | extreme ultraviolet |
References
- Aoki, H.; Tsuji, N.; Eckstein, M.; Kollar, M.; Oka, T.; Werner, P. Nonequilibrium dynamical mean-field theory and its applications. Rev. Mod. Phys. 2014, 86, 779–837. [Google Scholar] [CrossRef]
- Zhang, J.; Averitt, R.D. Dynamics and control in complex transition metal oxides. Annu. Rev. Mater. Res. 2014, 44, 19–43. [Google Scholar] [CrossRef]
- De La Torre, A.; Kennes, D.M.; Claassen, M.; Gerber, S.; McIver, J.W.; Sentef, M.A. Colloquium: Nonthermal pathways to ultrafast control in quantum materials. Rev. Mod. Phys. 2021, 93, 41002. [Google Scholar] [CrossRef]
- Gauthier, A.; Sobota, J.A.; Gauthier, N.; Xu, K.J.; Pfau, H.; Rotundu, C.R.; Shen, Z.X.; Kirchmann, P.S. Tuning time and energy resolution in time-resolved photoemission spectroscopy with nonlinear crystals. J. Appl. Phys. 2020, 128, 093101. [Google Scholar] [CrossRef]
- Zhang, H.; Pincelli, T.; Jozwiak, C.; Kondo, T.; Ernstorfer, R.; Sato, T.; Zhou, S. Angle-resolved photoemission spectroscopy. Nat. Rev. Methods Prim. 2022, 2, 54. [Google Scholar] [CrossRef]
- Na, M.X.; Mills, A.K.; Jones, D.J. Advancing time- and angle-resolved photoemission spectroscopy: The role of ultrafast laser development. Phys. Rep. 2023, 1036, 1–47. [Google Scholar] [CrossRef]
- Boschini, F.; Zonno, M.; Damascelli, A. Time-resolved ARPES studies of quantum materials. Rev. Mod. Phys. 2024, 96, 015003. [Google Scholar] [CrossRef]
- Damascelli, A.; Hussain, Z.; Shen, Z.X. Angle-resolved photoemission studies of the cuprate superconductors. Rev. Mod. Phys. 2003, 75, 473–541. [Google Scholar] [CrossRef]
- Sobota, J.A.; He, Y.; Shen, Z.X. Angle-resolved photoemission studies of quantum materials. Rev. Mod. Phys. 2021, 93, 1–72. [Google Scholar] [CrossRef]
- Schmitt, F.; Kirchmann, P.S.; Bovensiepen, U.; Moore, R.G.; Chu, J.H.; Lu, D.H.; Rettig, L.; Wolf, M.; Fisher, I.R.; Shen, Z.X. Ultrafast electron dynamics in the charge density wave material TbTe3. New J. Phys. 2011, 13, 063022. [Google Scholar] [CrossRef]
- Wallauer, R.; Reimann, J.; Armbrust, N.; Güdde, J.; Höfer, U. Intervalley scattering in MoS2 imaged by two-photon photoemission with a high-harmonic probe. Appl. Phys. Lett. 2016, 109, 162102. [Google Scholar] [CrossRef]
- Gierz, I.; Calegari, F.; Aeschlimann, S.; Chávez Cervantes, M.; Cacho, C.; Chapman, R.; Springate, E.; Link, S.; Starke, U.; Ast, C.; et al. Tracking Primary Thermalization Events in Graphene with Photoemission at Extreme Time Scales. Phys. Rev. Lett. 2015, 115, 086803. [Google Scholar] [CrossRef]
- Gerber, S.; Yang, S.; Zhu, D.; Soifer, H.; Sobota, J.A.; Rebec, S.; Lee, J.J.; Jia, T.; Moritz, B.; Jia, C.; et al. Femtosecond electron-phonon lock-in by photoemission and X-ray free-electron laser. Science 2017, 357, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Hein, P.; Jauernik, S.; Erk, H.; Yang, L.; Qi, Y.; Sun, Y.; Felser, C.; Bauer, M. Mode-resolved reciprocal space mapping of electron-phonon interaction in the Weyl semimetal candidate Td-WTe2. Nat. Commun. 2020, 11, 2613. [Google Scholar] [CrossRef]
- Perfetti, L.; Loukakos, P.A.; Lisowski, M.; Bovensiepen, U.; Berger, H.; Biermann, S.; Cornaglia, P.S.; Georges, A.; Wolf, M. Time Evolution of the Electronic Structure of 1T-TaS2 through the Insulator-Metal Transition. Phys. Rev. Lett. 2006, 97, 067402. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.; Kirchmann, P.S.; Bovensiepen, U.; Moore, R.G.; Rettig, L.; Krenz, M.; Chu, J.H.; Ru, N.; Perfetti, L.; Lu, D.H.; et al. Transient electronic structure and melting of a charge density wave in TbTe3. Science 2008, 321, 1649–1652. [Google Scholar] [CrossRef]
- Petersen, J.; Kaiser, S.; Dean, N.; Simoncig, A.; Liu, H.; Cavalieri, A.; Cacho, C.; Turcu, I.; Springate, E.; Frassetto, F.; et al. Clocking the Melting Transition of Charge and Lattice Order in 1T-TaS2 with Ultrafast Extreme-Ultraviolet Angle-Resolved Photoemission Spectroscopy. Phys. Rev. Lett. 2011, 107, 177402. [Google Scholar] [CrossRef]
- Smallwood, C.L.; Hinton, J.P.; Jozwiak, C.; Zhang, W.; Koralek, J.D.; Eisaki, H.; Lee, D.H.; Orenstein, J.; Lanzara, A. Tracking Cooper pairs in a cuprate superconductor by ultrafast angle-resolved photoemission. Science 2012, 336, 1137. [Google Scholar] [CrossRef] [PubMed]
- Cortés, R.; Rettig, L.; Yoshida, Y.; Eisaki, H.; Wolf, M.; Bovensiepen, U. Momentum-Resolved Ultrafast Electron Dynamics in Superconducting Bi2Sr2CaCu2O8+δ. Phys. Rev. Lett. 2011, 107, 097002. [Google Scholar] [CrossRef]
- Beaulieu, S.; Dong, S.; Tancogne-Dejean, N.; Dendzik, M.; Pincelli, T.; Maklar, J.; Patrick Xian, R.; Sentef, M.A.; Wolf, M.; Rubio, A.; et al. Ultrafast dynamical Lifshitz transition. Sci. Adv. 2021, 7, eabd9275. [Google Scholar] [CrossRef]
- Reimann, J.; Schlauderer, S.; Schmid, C.P.; Langer, F.; Baierl, S.; Kokh, K.A.; Tereshchenko, O.E.; Kimura, A.; Lange, C.; Güdde, J.; et al. Subcycle observation of lightwave-driven Dirac currents in a topological surface band. Nature 2018, 562, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, R.; Nicholson, C.W.; Waldecker, L.; Hübener, H.; Monney, C.; De Giovannini, U.; Puppin, M.; Hoesch, M.; Springate, E.; Chapman, R.T.; et al. Generation and Evolution of Spin-, Valley-, and Layer-Polarized Excited Carriers in Inversion-Symmetric WS2. Phys. Rev. Lett. 2016, 117, 277201. [Google Scholar] [CrossRef] [PubMed]
- Beyer, H.; Rohde, G.; Grubišić Čabo, A.; Stange, A.; Jacobsen, T.; Bignardi, L.; Lizzit, D.; Lacovig, P.; Sanders, C.; Lizzit, S.; et al. 80% Valley Polarization of Free Carriers in Singly Oriented Single-Layer WS2 on Au(111). Phys. Rev. Lett. 2019, 123, 236802. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ziffer, M.E.; Hansen, K.R.; Wang, J.; Zhu, X. Direct Determination of Band-Gap Renormalization in the Photoexcited Monolayer MoS2. Phys. Rev. Lett. 2019, 122, 246803. [Google Scholar] [CrossRef] [PubMed]
- Karni, O.; Barré, E.; Pareek, V.; Georgaras, J.D.; Man, M.K.; Sahoo, C.; Bacon, D.R.; Zhu, X.; Ribeiro, H.B.; O’Beirne, A.L.; et al. Structure of the moiré exciton captured by imaging its electron and hole. Nature 2022, 603, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, D.; Bange, J.P.; Bennecke, W.; AlMutairi, A.A.; Meneghini, G.; Watanabe, K.; Taniguchi, T.; Steil, D.; Luke, D.R.; Weitz, R.T.; et al. Formation of moiré interlayer excitons in space and time. Nature 2022, 608, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Steinberg, H.; Jarillo-Herrero, P.; Gedik, N. Observation of Floquet-Bloch states on the surface of a topological insulator. Science 2013, 342, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Carley, R.; Döbrich, K.; Frietsch, B.; Gahl, C.; Teichmann, M.; Schwarzkopf, O.; Wernet, P.; Weinelt, M. Femtosecond Laser Excitation Drives Ferromagnetic Gadolinium out of Magnetic Equilibrium. Phys. Rev. Lett. 2012, 109, 057401. [Google Scholar] [CrossRef] [PubMed]
- Eich, S.; Plötzing, M.; Rollinger, M.; Emmerich, S.; Adam, R.; Chen, C.; Kapteyn, H.C.; Murnane, M.M.; Plucinski, L.; Steil, D.; et al. Band structure evolution during the ultrafast ferromagnetic-paramagnetic phase transition in cobalt. Sci. Adv. 2017, 3, e1602094. [Google Scholar] [CrossRef] [PubMed]
- Gort, R.; Bühlmann, K.; Däster, S.; Salvatella, G.; Hartmann, N.; Zemp, Y.; Holenstein, S.; Stieger, C.; Fognini, A.; Michlmayr, T.; et al. Early Stages of Ultrafast Spin Dynamics in a in a 3d Ferromagnet. Phys. Rev. Lett. 2018, 121, 087206. [Google Scholar] [CrossRef]
- Moritz, B.; Kemper, F.; Sentef, M.; Devereaux, T.P.; Freericks, J.K. Electron-Mediated Relaxation Following Ultrafast Pumping of Strongly Correlated Materials: Model Evidence of a Correlation-Tuned Crossover between Thermal and Nonthermal States. Phys. Rev. Lett. 2013, 111, 077401. [Google Scholar] [CrossRef] [PubMed]
- Golež, D.; Eckstein, M.; Werner, P. Dynamics of screening in photodoped Mott insulators. Phys. Rev. B 2015, 92, 195123. [Google Scholar] [CrossRef]
- Wang, Y.; Claassen, M.; Moritz, B.; Devereaux, T.P. Producing coherent excitations in pumped Mott antiferromagnetic insulators. Phys. Rev. B 2017, 96, 235142. [Google Scholar] [CrossRef]
- Shen, W.; Ge, Y.; Liu, A.Y.; Krishnamurthy, H.R.; Devereaux, T.P.; Freericks, J.K. Nonequilibrium “Melting” of a Charge Density Wave Insulator via an Ultrafast Laser Pulse. Phys. Rev. Lett. 2014, 112, 176404. [Google Scholar] [CrossRef]
- Hellmann, S.; Rohwer, T.; Kalläne, M.; Hanff, K.; Sohrt, C.; Stange, A.; Carr, A.; Murnane, M.M.; Kapteyn, H.C.; Kipp, L.; et al. Time-domain classification of charge-density-wave insulators. Nat. Commun. 2012, 3, 1069. [Google Scholar] [CrossRef] [PubMed]
- Ligges, M.; Avigo, I.; Golež, D.; Strand, H.; Beyazit, Y.; Hanff, K.; Diekmann, F.; Stojchevska, L.; Kalläne, M.; Zhou, P.; et al. Ultrafast Doublon Dynamics in Photoexcited 1T-TaS2. Phys. Rev. Lett. 2018, 120, 166401. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, T.; Hellmann, S.; Wiesenmayer, M.; Sohrt, C.; Stange, A.; Slomski, B.; Carr, A.; Liu, Y.; Avila, L.; Kalläsignne, M.; et al. Collapse of long-range charge order tracked by time-resolved photoemission at high momenta. Nature 2011, 471, 490–494. [Google Scholar] [CrossRef]
- Burian, M.; Porer, M.; Mardegan, J.R.; Esposito, V.; Parchenko, S.; Burganov, B.; Gurung, N.; Ramakrishnan, M.; Scagnoli, V.; Ueda, H.; et al. Structural involvement in the melting of the charge density wave in 1T-TiSe2. Phys. Rev. Res. 2021, 3, 13128. [Google Scholar] [CrossRef]
- Dardel, B.; Grioni, M.; Malterre, D.; Weibel, P.; Baer, Y.; Lévy, F. Temperature-dependent pseudogap and electron localization in 1T-TaS2. Phys. Rev. B 1992, 45, 1462–1465. [Google Scholar] [CrossRef]
- Perfetti, L.; Gloor, T.A.; Mila, F.; Berger, H.; Grioni, M. Unexpected periodicity in the quasi-two-dimensional Mott insulator 1T-TaS2 revealed by angle-resolved photoemission. Phys. Rev. B 2005, 71, 153101. [Google Scholar] [CrossRef]
- Rossnagel, K.; Smith, N.V. Spin-orbit coupling in the band structure of reconstructed 1T-TaS2. Phys. Rev. B 2006, 73, 073106. [Google Scholar] [CrossRef]
- Freericks, J.K.; Krishnamurthy, H.R.; Ge, Y.; Liu, A.; Pruschke, T. Theoretical description f time-resolved pump/probe photoemission in TaS2: A single-band DFT+DMFT(NRG)study within the quasiequilibrium approximation. Phys. Status Solidi B 2009, 246, 948–954. [Google Scholar] [CrossRef]
- Rossnagel, K.; Kipp, L.; Skibowski, M. Charge-density-wave phase transition in 1T-TiSe2: Excitonic insulator versus band-type Jahn-Teller mechanism. Phys. Rev. B 2002, 65, 235101. [Google Scholar] [CrossRef]
- Kidd, T.; Miller, T.; Chou, M.; Chiang, T.C. Electron-Hole Coupling and the Charge Density Wave Transition in TiSe2. Phys. Rev. Lett. 2002, 88, 226402. [Google Scholar] [CrossRef] [PubMed]
- Cercellier, H.; Monney, C.; Clerc, F.; Battaglia, C.; Despont, L.; Garnier, M.; Beck, H.; Aebi, P.; Patthey, L.; Berger, H.; et al. Evidence for an Excitonic Insulator Phase in 1T-TiSe2. Phys. Rev. Lett. 2007, 99, 146403. [Google Scholar] [CrossRef] [PubMed]
- Rossnagel, K. On the origin of charge-density waves in select layered transition-metal dichalcogenides. J. Phys. Condens. Matter 2011, 23, 213001. [Google Scholar] [CrossRef]
- Perfetti, L.; Loukakos, P.A.; Lisowski, M.; Bovensiepen, U.; Wolf, M.; Berger, H.; Biermann, S.; Georges, A. Femtosecond dynamics of electronic states in the Mott insulator 1T-TaS2 by time resolved photoelectron spectroscopy. New J. Phys. 2008, 10, 053019. [Google Scholar] [CrossRef]
- Fazekas, P.; Tosatti, E. Electrical, structural and magnetic properties of pure and doped 1T-TaS2. Philos. Mag. B 1979, 39, 229–244. [Google Scholar] [CrossRef]
- Bayliss, S.C.; Ghorayeb, A.M.; Guy, D.R. Thermal and transport evidence for a phase transition in 1T-TaS2 observed at 282K upon warming. J. Phys. C Solid State Phys. 1984, 17. [Google Scholar] [CrossRef]
- Ritschel, T.; Trinckauf, J.; Koepernik, K.; Büchner, B.; Zimmermann, M.V.; Berger, H.; Joe, Y.I.; Abbamonte, P.; Geck, J. Orbital textures and charge density waves in transition metal dichalcogenides. Nat. Phys. 2015, 11, 328–331. [Google Scholar] [CrossRef]
- Petocchi, F.; Nicholson, C.W.; Salzmann, B.; Pasquier, D.; Yazyev, O.V.; Monney, C.; Werner, P. Mott versus Hybridization Gap in the Low-Temperature Phase of 1T-TaS2. Phys. Rev. Lett. 2022, 129, 16402. [Google Scholar] [CrossRef] [PubMed]
- Moritz, B.; Devereaux, T.P.; Freericks, J.K. Time-resolved photoemission of correlated electrons driven out of equilibrium. Phys. Rev. B 2010, 81, 1–5. [Google Scholar] [CrossRef]
- Wall, S.; Brida, D.; Clark, S.R.; Ehrke, H.P.; Jaksch, D.; Ardavan, A.; Bonora, S.; Uemura, H.; Takahashi, Y.; Hasegawa, T.; et al. Quantum interference between charge excitation paths in a solid-state Mott insulator. Nat. Phys. 2011, 7, 114–118. [Google Scholar] [CrossRef]
- Di Salvo, F.J.; Moncton, D.E.; Waszczak, J.V. Electronic properties and superlattice formation in the semimetal TiSe2. Phys. Rev. B 1976, 14, 4321–4328. [Google Scholar] [CrossRef]
- Woo, K.C.; Brown, F.; McMillan, W.L.; Miller, R.J.; Schaffman, M.J.; Sears, M.P. Superlattice formation in titanium diselenide. Phys. Rev. B 1976, 14, 3242–3248. [Google Scholar] [CrossRef]
- Monney, G.; Monney, C.; Hildebrand, B.; Aebi, P.; Beck, H. Impact of electron-hole correlations on the 1T-TiSe2 electronic structure. Phys. Rev. Lett. 2015, 114, 086402. [Google Scholar] [CrossRef]
- Kogar, A.; Rak, M.S.; Vig, S.; Husain, A.A.; Flicker, F.; Joe, Y.I.; Venema, L.; MacDougall, G.J.; Chiang, T.C.; Fradkin, E.; et al. Signatures of exciton condensation in a transition metal dichalcogenide. Science 2017, 358, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Van Zandt, L.L.; Honig, J.M. Theories Pertaining to the Semiconductor-Metal Transition in Crystals. Annu. Rev. Mater. Sci. 1974, 4, 191–220. [Google Scholar] [CrossRef]
- Mathias, S.; Eich, S.; Urbancic, J.; Michael, S.; Carr, A.V.; Emmerich, S.; Stange, A.; Popmintchev, T.; Rohwer, T.; Wiesenmayer, M.; et al. Self-amplified photo-induced gap quenching in a correlated electron material. Nat. Commun. 2016, 7, 12902. [Google Scholar] [CrossRef]
- Porer, M.; Leierseder, U.; Ménard, J.M.; Dachraoui, H.; Mouchliadis, L.; Perakis, I.E.; Heinzmann, U.; Demsar, J.; Rossnagel, K.; Huber, R. Non-thermal separation of electronic and structural orders in a persisting charge density wave. Nat. Mater. 2014, 13, 857–861. [Google Scholar] [CrossRef]
- Glover, T.; Schoenlein, R.; Chin, A.; Shank, C. Observation of laser assisted photoelectric effect and femtosecond high order harmonic radiation. Phys. Rev. Lett. 1996, 76, 2468–2471. [Google Scholar] [CrossRef]
- Bányai, L.; Vu, Q.; Mieck, B.; Haug, H. Ultrafast Quantum Kinetics of Time-Dependent RPA-Screened Coulomb Scattering. Phys. Rev. Lett. 1998, 81, 882–885. [Google Scholar] [CrossRef]
- Huber, R.; Tauser, F.; Brodschelm, A.; Bichler, M.; Abstreiter, G.; Leitenstorfer, A. How many-particle interactions develop after ultrafast excitation of an electron-hole plasma. Nature 2001, 414, 286–289. [Google Scholar] [CrossRef]
- Eich, S.; Stange, A.; Carr, A.; Urbancic, J.; Popmintchev, T.; Wiesenmayer, M.; Jansen, K.; Ruffing, A.; Jakobs, S.; Rohwer, T.; et al. Time- and angle-resolved photoemission spectroscopy with optimized high-harmonic pulses using frequency-doubled Ti:Sapphire lasers. J. Electron Spectrosc. Relat. Phenom. 2014, 195, 231–236. [Google Scholar] [CrossRef]
- Nisoli, M.; De Silvestri, S.; Svelto, O.; Szipöcs, R.; Ferencz, K.; Spielmann, C.; Sartania, S.; Krausz, F. Compression of high-energy laser pulses below 5 fs. Opt. Lett. 1997, 22, 522–524. [Google Scholar] [CrossRef]
- Rohde, G.; Hendel, A.; Stange, A.; Hanff, K.; Oloff, L.P.; Yang, L.X.; Rossnagel, K.; Bauer, M. Time-resolved ARPES with sub-15 fs temporal and near Fourier-limited spectral resolution. Rev. Sci. Instrum. 2016, 87, 103102. [Google Scholar] [CrossRef]
- Rohde, G.; Stange, A.; Müller, A.; Behrendt, M.; Oloff, L.P.; Hanff, K.; Albert, T.; Hein, P.; Rossnagel, K.; Bauer, M. Ultrafast Formation of a Fermi-Dirac Distributed Electron Gas. Phys. Rev. Lett. 2018, 121, 256401. [Google Scholar] [CrossRef] [PubMed]
- Breusing, M.; Ropers, C.; Elsaesser, T. Ultrafast Carrier Dynamics in Graphite. Phys. Rev. Lett. 2009, 102, 086809. [Google Scholar] [CrossRef] [PubMed]
- Malic, E.; Winzer, T.; Bobkin, E.; Knorr, A. Microscopic theory of absorption and ultrafast many-particle kinetics in graphene. Phys. Rev. B 2011, 84, 205406. [Google Scholar] [CrossRef]
- Malic, E.; Winzer, T.; Knorr, A. Efficient orientational carrier relaxation in optically excited graphene. Appl. Phys. Lett. 2012, 101, 213110. [Google Scholar] [CrossRef]
- Brida, D.; Tomadin, A.; Manzoni, C.; Kim, Y.J.; Lombardo, A.; Milana, S.; Nair, R.R.; Novoselov, K.S.; Ferrari, A.C.; Cerullo, G.; et al. Ultrafast collinear scattering and carrier multiplication in graphene. Nat. Commun. 2013, 4, 2987. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.F.; Ott, C.; Kraus, P.M.; Kaplan, C.J.; Pouse, W.; Marvel, R.E.; Haglund, R.F.; Neumark, D.M.; Leone, S.R. Tracking the insulator-to-metal phase transition in VO2 with few-femtosecond extreme UV transient absorption spectroscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 9558–9563. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Guggenmos, A.; Chen, C.T.; Oh, J.; Géneaux, R.; Chuang, Y.D.; Schwartzberg, A.M.; Aloni, S.; Neumark, D.M.; Leone, S.R. Coupled valence carrier and core-exciton dynamics in WS2 probed by few-femtosecond extreme ultraviolet transient absorption spectroscopy. Phys. Rev. B 2021, 104, 064309. [Google Scholar] [CrossRef]
- Sidiropoulos, T.P.; Di Palo, N.; Rivas, D.E.; Severino, S.; Reduzzi, M.; Nandy, B.; Bauerhenne, B.; Krylow, S.; Vasileiadis, T.; Danz, T.; et al. Probing the Energy Conversion Pathways between Light, Carriers, and Lattice in Real Time with Attosecond Core-Level Spectroscopy. Phys. Rev. X 2021, 11, 41060. [Google Scholar] [CrossRef]
- Geneaux, R.; Marroux, H.J.; Guggenmos, A.; Neumark, D.M.; Leone, S.R. Transient absorption spectroscopy using high harmonic generation: A review of ultrafast X-ray dynamics in molecules and solids. Philos. Trans. R. Soc. A 2019, 377. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, A.L.; Müller, N.; Uphues, T.; Yakovlev, V.S.; Baltuska, A.; Horvath, B.; Schmidt, B.; Blümel, L.; Holzwarth, R.; Hendel, S.; et al. Attosecond spectroscopy in condensed matter. Nature 2007, 449, 1029–1032. [Google Scholar] [CrossRef]
- Neppl, S.; Ernstorfer, R.; Cavalieri, A.L.; Lemell, C.; Wachter, G.; Magerl, E.; Bothschafter, E.M.; Jobst, M.; Hofstetter, M.; Kleineberg, U.; et al. Direct observation of electron propagation and dielectric screening on the atomic length scale. Nature 2015, 517, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Siek, F.; Neb, S.; Bartz, P.; Hensen, M.; Strüber, C.; Fiechter, S.; Torrent-Sucarrat, M.; Silkin, V.M.; Krasovskii, E.E.; Kabachnik, N.M.; et al. Angular momentum–induced delays in solid-state photoemission enhanced by intra-atomic interactions. Science 2017, 357, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Locher, R.; Castiglioni, L.; Lucchini, M.; Greif, M.; Gallmann, L.; Osterwalder, J.; Hengsberger, M.; Keller, U. Energy-dependent photoemission delays from noble metal surfaces by attosecond interferometry. Optica 2015, 2, 405–410. [Google Scholar] [CrossRef]
- Tao, Z.; Chen, C.; Szilvási, T.; Keller, M.; Mavrikakis, M.; Kapteyn, H.; Murnane, M. Direct time-domain observation of attosecond final-state lifetimes in photoemission from solids. Science 2016, 353, aaf6793. [Google Scholar] [CrossRef]
- Krausz, F.; Stockman, M.I. Attosecond metrology: From electron capture to future signal processing. Nat. Photonics 2014, 8, 205–213. [Google Scholar] [CrossRef]
- Kruchinin, S.Y.; Krausz, F.; Yakovlev, V.S. Colloquium: Strong-field phenomena in periodic systems. Rev. Mod. Phys. 2018, 90, 21002. [Google Scholar] [CrossRef]
- Higuchi, T.; Heide, C.; Ullmann, K.; Weber, H.B.; Hommelhoff, P. Light-field-driven currents in graphene. Nature 2017, 550, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Heide, C.; Higuchi, T.; Weber, H.B.; Hommelhoff, P. Coherent Electron Trajectory Control in Graphene. Phys. Rev. Lett. 2018, 121, 207401. [Google Scholar] [CrossRef]
- Breusing, M.; Kuehn, S.; Winzer, T.; Malić, E.; Milde, F.; Severin, N.; Rabe, J.; Ropers, C.; Knorr, A.; Elsaesser, T. Ultrafast nonequilibrium carrier dynamics in a single graphene layer. Phys. Rev. B 2011, 83, 153410. [Google Scholar] [CrossRef]
- Oloff, L.P.; Hanff, K.; Stange, A.; Rohde, G.; Diekmann, F.; Bauer, M.; Rossnagel, K. Pump laser-induced space-charge effects in HHG-driven time- and angle-resolved photoelectron spectroscopy. J. Appl. Phys. 2016, 119, 225106. [Google Scholar] [CrossRef]
- Kuzmenko, A.B.; Van Heumen, E.; Carbone, F.; Van Der Marel, D. Universal optical conductance of graphite. Phys. Rev. Lett. 2008, 100, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Papoular, R.J.; Papoular, R. Some optical properties of graphite from IR to millimetric wavelengths. Mon. Not. R. Astron. Soc. 2014, 443, 2974–2982. [Google Scholar] [CrossRef]
- Kelardeh, H.K.; Apalkov, V.; Stockman, M.I. Attosecond strong-field interferometry in graphene: Chirality, singularity, and Berry phase. Phys. Rev. B 2016, 93, 155434. [Google Scholar] [CrossRef]
- Aeschlimann, M.; Brixner, T.; Fischer, A.; Kramer, C.; Melchior, P.; Pfeiffer, W.; Schneider, C.; Strüber, C.; Tuchscherer, P.; Voronine, D.V. Coherent Two-Dimensional Nanoscopy. Science 2011, 333, 1723–1726. [Google Scholar] [CrossRef]
- Reutzel, M.; Li, A.; Wang, Z.; Petek, H. Coherent multidimensional photoelectron spectroscopy of ultrafast quasiparticle dressing by light. Nat. Commun. 2020, 11, 2230. [Google Scholar] [CrossRef] [PubMed]
- Uhl, D.; Bangert, U.; Bruder, L.; Stienkemeier, F. Coherent optical 2D photoelectron spectroscopy. Optica 2021, 8, 1316–1324. [Google Scholar] [CrossRef]
- Ogawa, S.; Nagano, H.; Petek, H.; Heberle, A.P. Optical Dephasing in Cu(111) Measured by Interferometric Two-Photon Time-Resolved Photoemission. Phys. Rev. Lett. 1997, 78, 1339–1342. [Google Scholar] [CrossRef]
- Petek, H.; Nagano, H.; Ogawa, S. Hole decoherence of d bands in copper. Phys. Rev. Lett. 1999, 83, 832–835. [Google Scholar] [CrossRef]
- Cui, X.; Wang, C.; Argondizzo, A.; Garrett-Roe, S.; Gumhalter, B.; Petek, H. Transient excitons at metal surfaces. Nat. Phys. 2014, 10, 505–509. [Google Scholar] [CrossRef]
- Randi, F.; Fausti, D.; Eckstein, M. Bypassing the energy-time uncertainty in time-resolved photoemission. Phys. Rev. B 2017, 95, 115132. [Google Scholar] [CrossRef]
- Mukamel, S. Principles of Nonlinear Optics and Spectroscopy; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Mukamel, S. Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 2000, 51, 691–729. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.H.; Wang, H.; Xu, Y.; Maklar, J.; Joucken, F.; Zeng, L.; Stoll, S.; Jozwiak, C.; Pepper, J.; Chuang, Y.D.; et al. A setup for extreme-ultraviolet ultrafast angle-resolved photoelectron spectroscopy at 50-kHz repetition rate. Rev. Sci. Instrum. 2018, 90, 023105. [Google Scholar] [CrossRef] [PubMed]
- Puppin, M.; Deng, Y.; Nicholson, C.W.; Feldl, J.; Schroeter, N.; Vita, H.; Kirchmann, P.S.; Monney, C.; Rettig, L.; Wolf, M.; et al. Time- and angle-resolved photoemission spectroscopy of solids in the extreme ultraviolet at 500 kHz repetition rate. Rev. Sci. Instrum. 2018, 90, 023104. [Google Scholar] [CrossRef]
- Mills, A.K.; Zhdanovich, S.; Na, M.X.; Boschini, F.; Razzoli, E.; Michiardi, M.; Sheyerman, A.; Schneider, M.; Hammond, T.J.; Süss, V.; et al. Cavity-enhanced high harmonic generation for extreme ultraviolet time- and angle-resolved photoemission spectroscopy. Rev. Sci. Instrum. 2019, 90, 083001. [Google Scholar] [CrossRef]
- Chini, M.; Mashiko, H.; Wang, H.; Chen, S.; Yun, C.; Scott, S.; Gilbertson, S.; Chang, Z. Delay control in attosecond pump-probe experiments. Opt. Express 2009, 17, 21459–21464. [Google Scholar] [CrossRef] [PubMed]

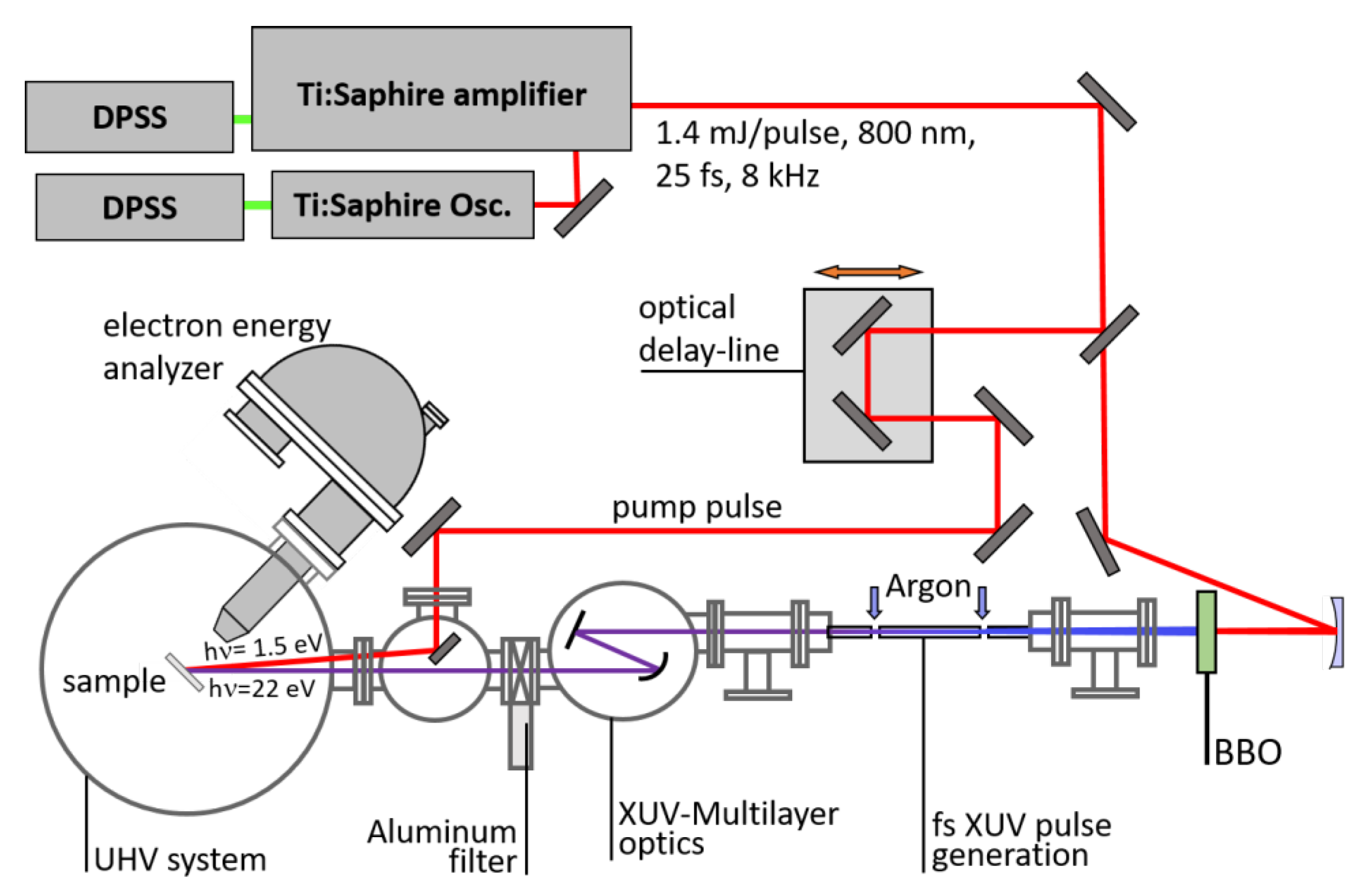

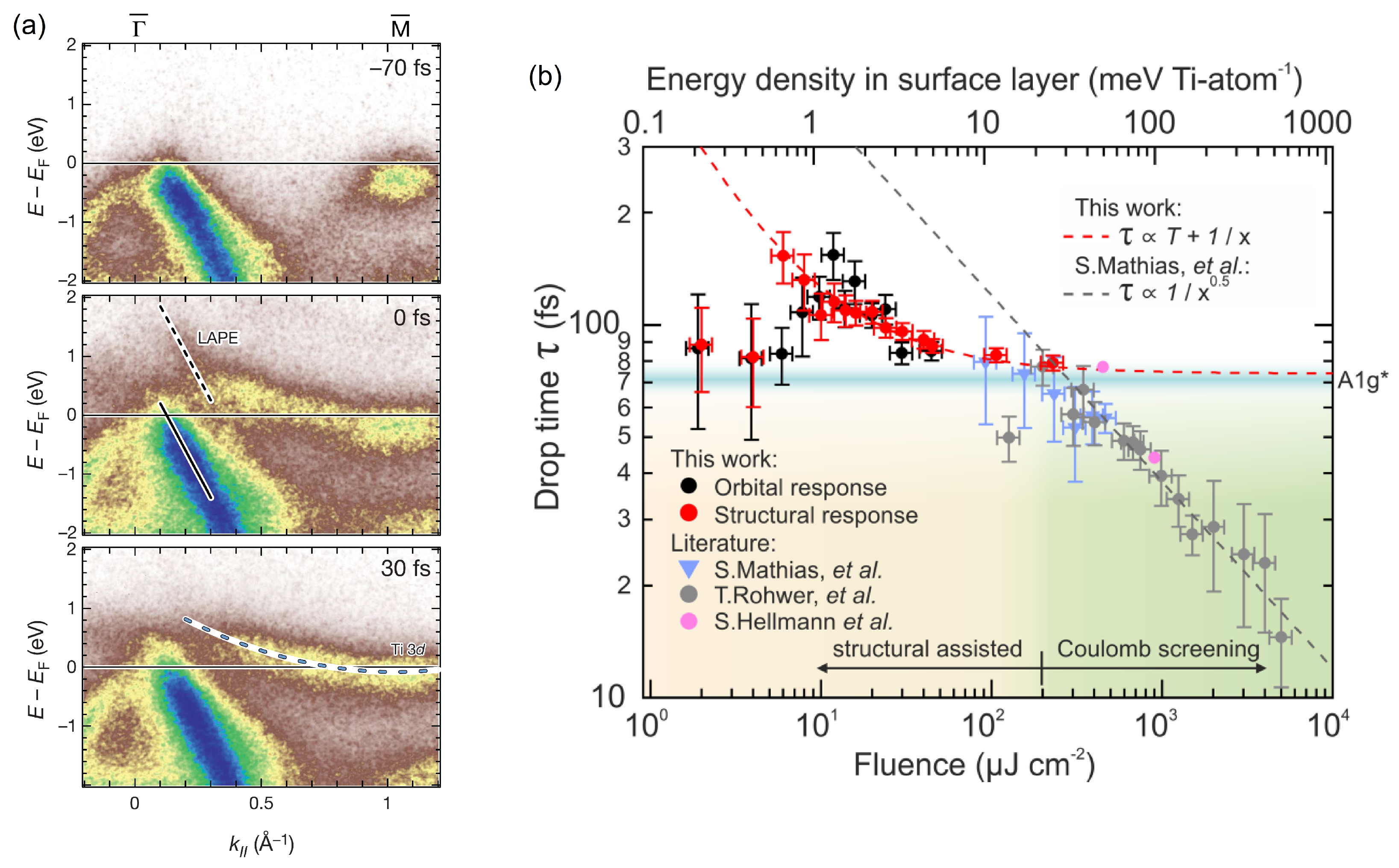
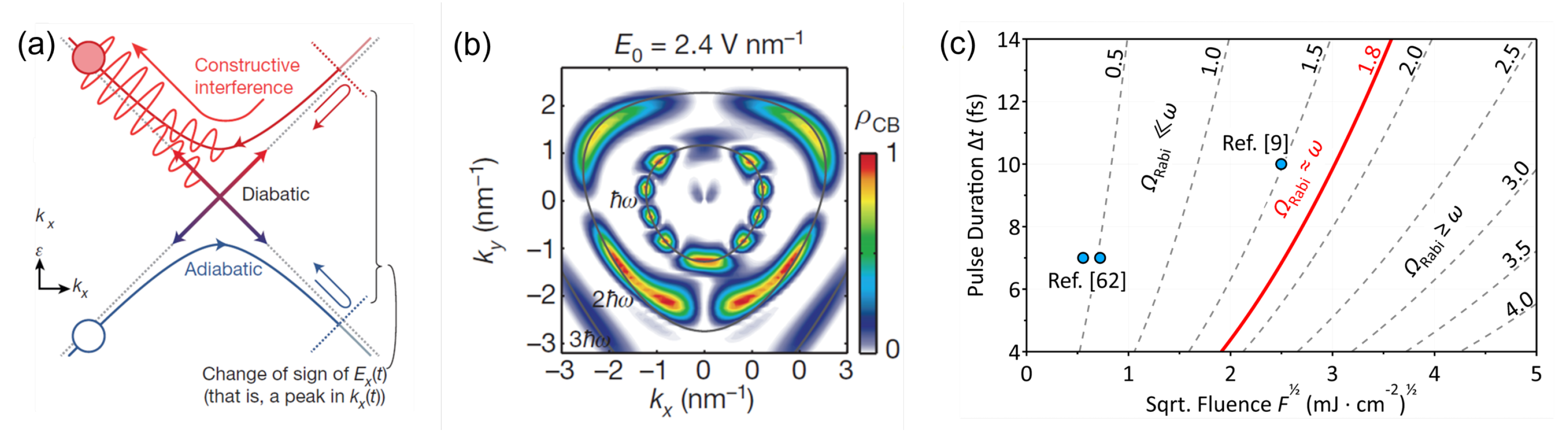

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossnagel, K.; Bauer, M. Toward Direct Exploration of the Few-Femtosecond Dynamics of Electronic Coherence and Correlation in Quantum Materials Using Time- and Angle-Resolved Photoemission Spectroscopy. Crystals 2024, 14, 404. https://doi.org/10.3390/cryst14050404
Rossnagel K, Bauer M. Toward Direct Exploration of the Few-Femtosecond Dynamics of Electronic Coherence and Correlation in Quantum Materials Using Time- and Angle-Resolved Photoemission Spectroscopy. Crystals. 2024; 14(5):404. https://doi.org/10.3390/cryst14050404
Chicago/Turabian StyleRossnagel, Kai, and Michael Bauer. 2024. "Toward Direct Exploration of the Few-Femtosecond Dynamics of Electronic Coherence and Correlation in Quantum Materials Using Time- and Angle-Resolved Photoemission Spectroscopy" Crystals 14, no. 5: 404. https://doi.org/10.3390/cryst14050404