An Examination of the Electron Densities in a Series of Tripodal Cobalt Complexes Bridged by Magnesium, Calcium, Strontium, and Barium †



 and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
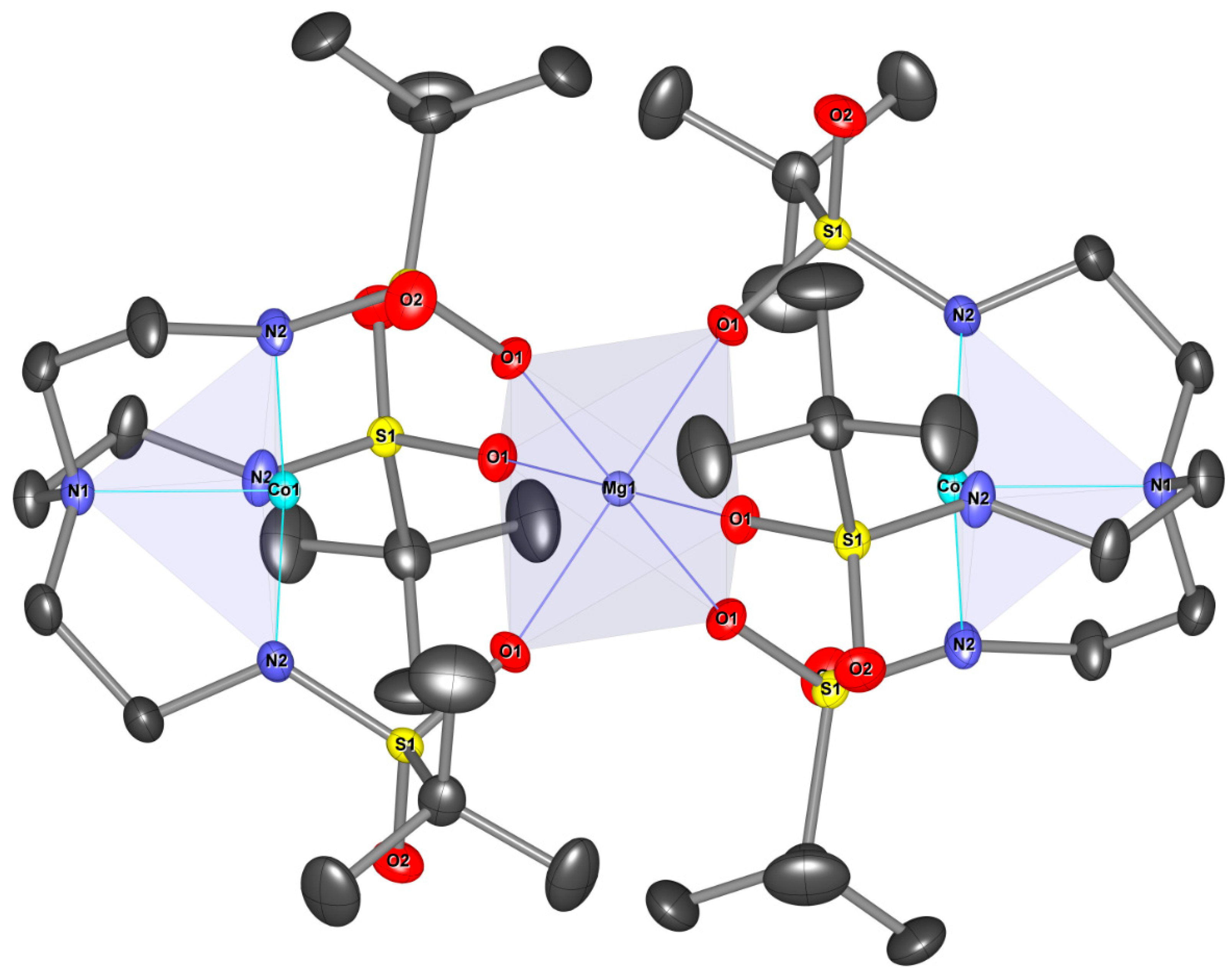
3.1. The Crystal Structures of [(Co(Ts3tren))M(Co(Ts3tren))]
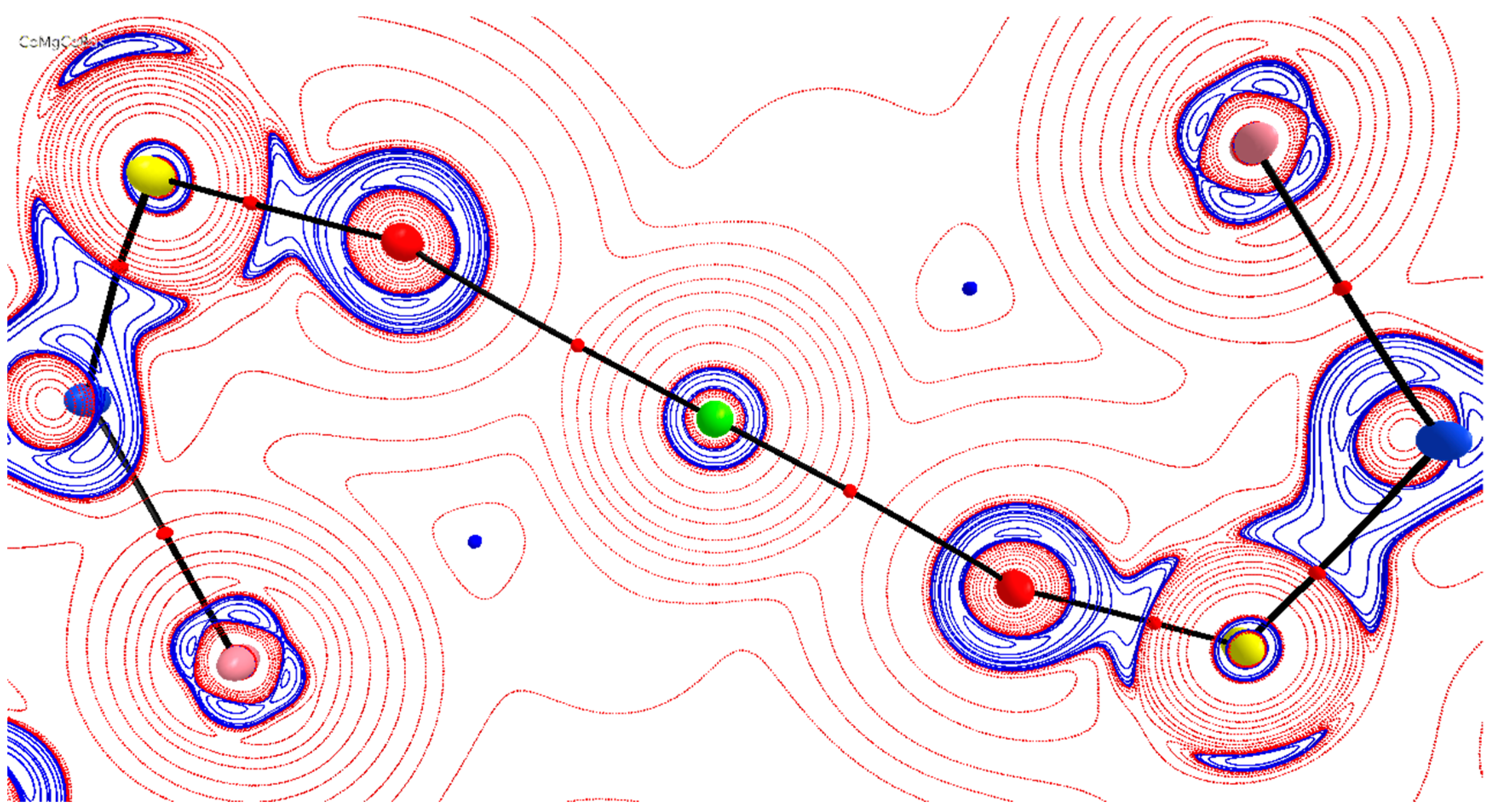
3.2. Theoretical Charge-Density Analysis of [C36H78MCo2N8O12S6]
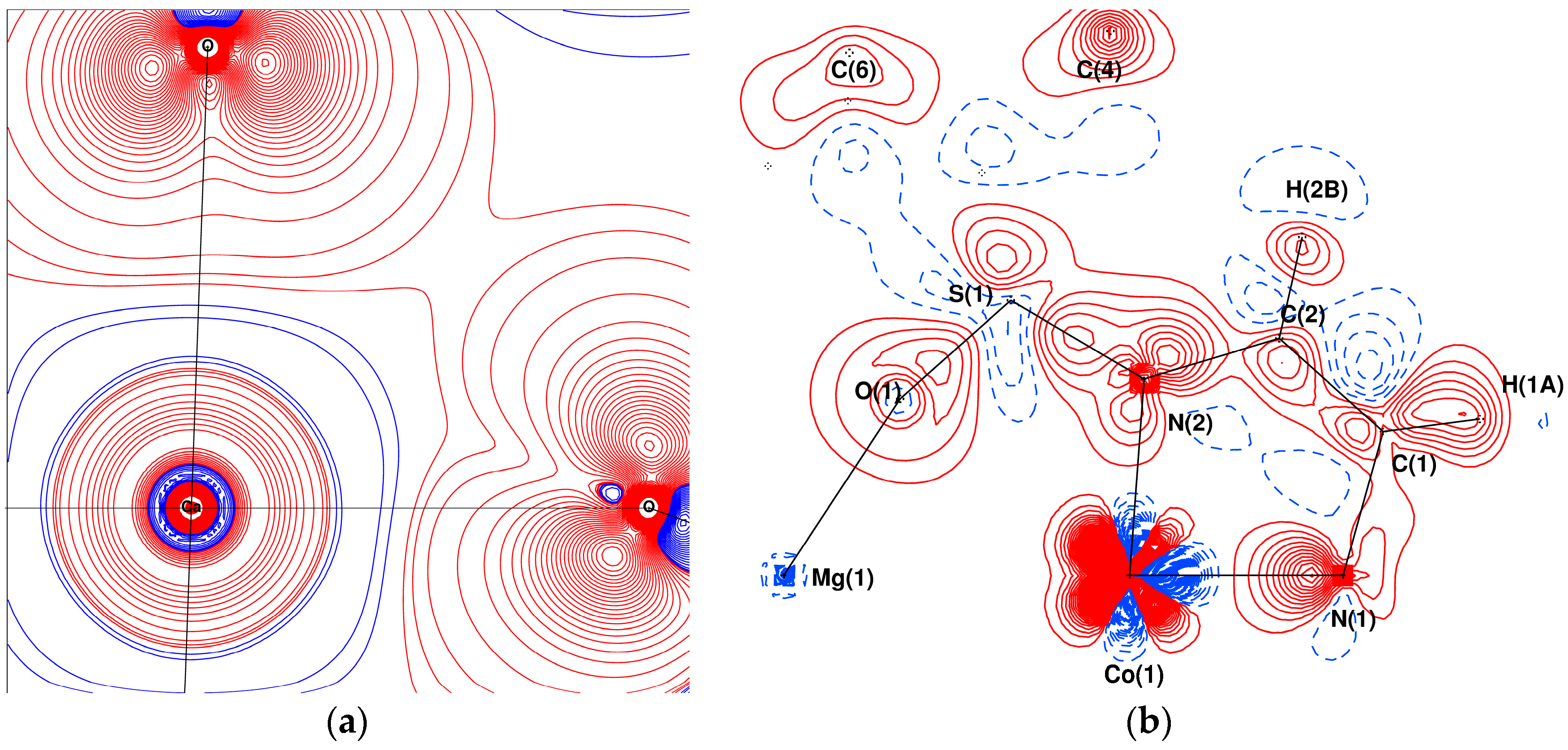
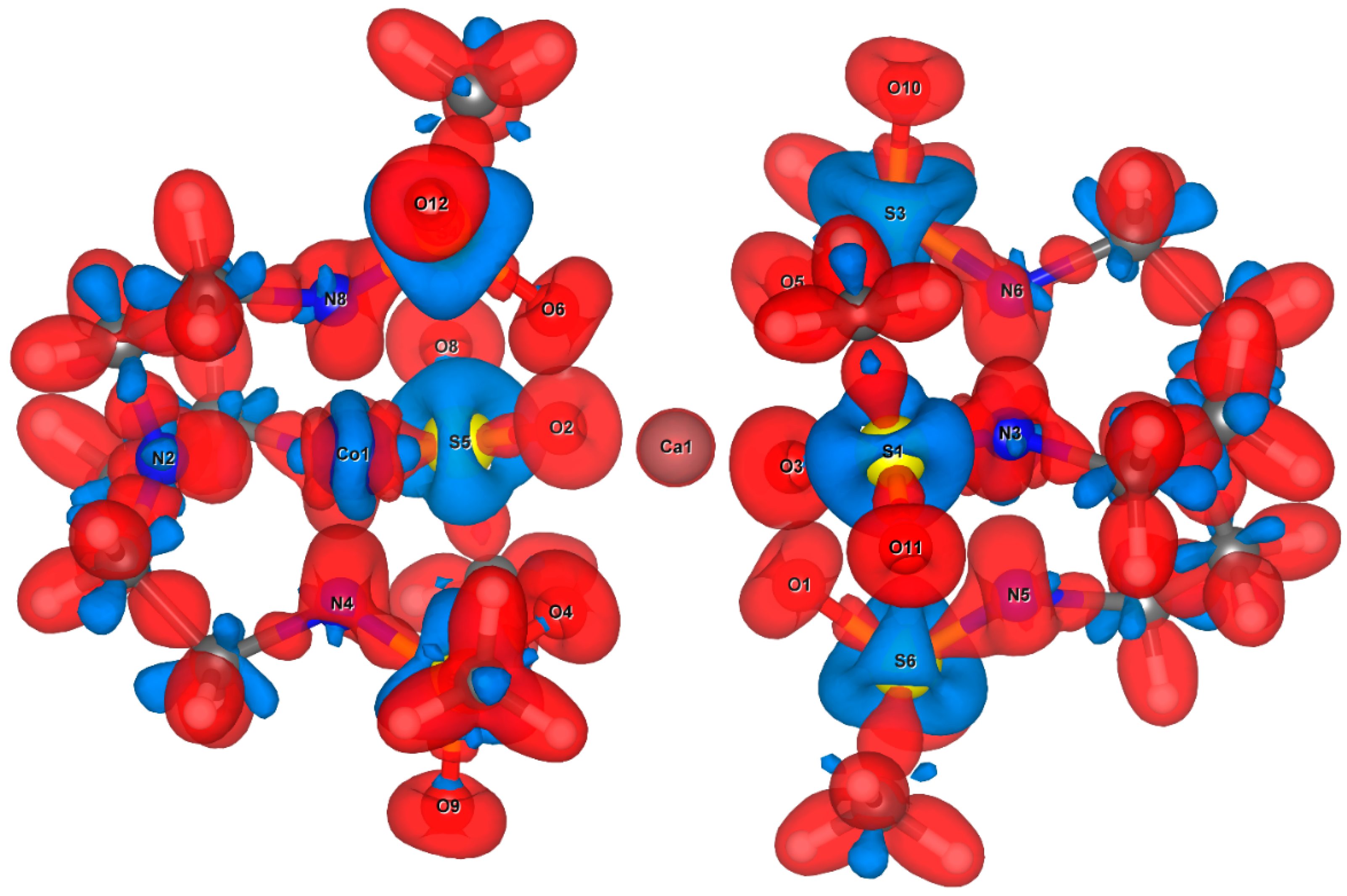
3.3. Visualisation and Qualitative Analysis of the Deformation Density of the [(Co(Ts3tren))M(Co(Ts3tren))] Complexes
3.4. Comparison of Some Experimentally and Theoretically Derived Parameters
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Wallen, C.M.; Wielizcko, M.; Bacsa, J.; Scarborough, C.C. Heterotrimetallic sandwich complexes supported by sulfonamido ligands. Inorg. Chem. Front. 2016, 3, 142–149. [Google Scholar] [CrossRef]
- Wallen, C.M.; Bacsa, J.; Scarborough, C.C. Coordination of Hydrogen Peroxide with Late-Transition-Metal Sulfonamido Complexes. Inorg. Chem. 2018, 57, 4841–4848. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Lee, Y.-M.; Hong, S.; Cho, K.-B.; Nishida, Y.; Seo, M.S.; Sarangi, R.; Fukuzumi, S.; Nam, W. Redox-inactive metal ions modulate the reactivity and oxygen release of mononuclear non-haem iron(III)–peroxo complexes. Nat. Chem. 2014, 6, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Pfaff, F.F.; Kwon, E.; Wang, Y.; Seo, M.-S.; Bill, E.; Ray, K.; Nam, W. Spectroscopic Capture and Reactivity of a Low-Spin Cobalt(IV)-Oxo Complex Stabilized by Binding Redox-Inactive Metal Ions. Angew. Chem. Int. Ed. 2014, 53, 10403–10407. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-M.; Bang, S.; Kim, Y.M.; Cho, J.; Hong, S.; Nomura, T.; Ogura, T.; Troeppner, O.; Ivanović-Burmazović, I.; Sarangi, R.; et al. A mononuclear nonheme iron(iii)-peroxo complex binding redox-inactive metal ions. Chem. Sci. 2013, 4, 3917–3923. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Gordon-Wylie, S.W.; Horwitz, C.P.; Strazisar, S.A.; Peraino, D.K.; Clark, G.R.; Weintraub, S.T.; Collins, T.J. A Method for Driving O-Atom Transfer: Secondary Ion Binding to a Tetraamide Macrocyclic Ligand. J. Am. Chem. Soc. 1998, 120, 11540–11541. [Google Scholar] [CrossRef]
- Monte-Pérez, I.; Kundu, S.; Ray, K. An Open-Shell Spin Singlet Copper-Nitrene Intermediate Binding Redox-innocent Metal Ions: Influence of the Lewis Acidity of the Metal Ions on Spectroscopic and Reactivity Properties. Z. Anorg. Allg. Chem. 2015, 641, 78–82. [Google Scholar] [CrossRef]
- Park, Y.J.; Cook, S.A.; Sickerman, N.S.; Sano, Y.; Ziller, J.W.; Borovik, A.S. Heterobimetallic complexes with MIII-([small mu]-OH)-MII cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): Structural, kinetic, and redox properties. Chem. Sci. 2013, 4, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Xiong, Y.; Vogt, M.; Grützmacher, H.; Herwig, C.; Limberg, C.; Driess, M. O–O Bond Activation in Heterobimetallic Peroxides: Synthesis of the Peroxide [LNi(μ,η2:η2-O2)K] and its Conversion into a Bis(μ-Hydroxo) Nickel Zinc Complex. Angew. Chem. Int. Ed. 2009, 48, 8107–8110. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Wotton-under-Edge, UK, 1990. [Google Scholar]
- Sun, Z.; Launder, A.M.; Schaefer, H.F. Prediction and Characterization of Alkaline-Earth (M = Be, Mg, Ca, Sr, and Ba) Metallacyclopentadienes and Relevant Derivatives. ChemistrySelect 2017, 2, 1442–1453. [Google Scholar] [CrossRef]
- Jorge, F.E.; Neto, A.C.; Camiletti, G.G.; Machado, S.F. Contracted Gaussian basis sets for Douglas–Kroll–Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.S.C.; Jorge, F.E.; Franco, M.L.; Ferreira, I.B. All-electron Gaussian basis sets of double zeta quality for the actinides. J. Chem. Phys. 2016, 145, 244113. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01.; Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, T.A. AIMAll, 17.11.14 ed.; TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. Sect. A 1978, 34, 909–921. [Google Scholar] [CrossRef]
- Koritsanszky, T.H.; Su, Z.; Mallinson, P.R.; Richter, T.; Hansen, N.K. XD2006—A Computer Program Package for Multipole Refinement and Analysis of Electron Densities from Diffraction Data; 2006. [Google Scholar]
- Bacsa, J.; Briones, J. Determination of the electron density in methyl (±)-(1S,2S,3R)-2-methyl-1,3-diphenylcyclopropanecarboxylate using refinements with X-ray scattering factors from wavefunction calculations of the whole molecule. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2013, 69, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Jayatilaka, D.; Grimwood, D.J. (Eds.) Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998, 27, 163–170. [Google Scholar] [CrossRef]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 238–239, 383–412. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Robinson, E.A. Models of molecular geometry. Chem. Soc. Rev. 2005, 34, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Guzmán, F.; Bader, R.F.W. Complementarity of QTAIM and MO theory in the study of bonding in donor–acceptor complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar] [CrossRef]
- Holladay, A.; Leung, P.; Coppens, P. Generalized relations between d-orbital occupancies of transition-metal atoms and electron-density multipole population parameters from X-ray diffraction data. Acta Crystallogr. Sect. A 1983, 39, 377–387. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Full Molecular Graph | Partial Molecular Graph (Zoom) |
|---|---|---|
| Mg |  |  |
| Ca |  |  |
| Sr |  |  |
| Ba |  |  |
| M2+ | ρbcp | ∇2ρbcp | ε | Hbcp | DI (M|O) |
|---|---|---|---|---|---|
| M–O | |||||
| Mg | 0.0282 | 0.2330 | 0.0016 | 0.0103 | 0.0793 |
| Ca | 0.0326 | 0.2170 | 0.0158 | 0.0065 | 0.1293 |
| Sr | 0.0330 | 0.1799 | 0.0259 | 0.0045 | 0.1676 |
| Ba | 0.0305 | 0.1707 | 0.0279 | 0.0047 | 0.1630 |
| Co–N | |||||
| Mg | 0.0976 0.0697 | 0.4739 0.3618 | 0.2631 0.0005 | −0.0233 −0.0108 | 0.5127 0.3350 |
| Ca | 0.0991 0.0710 | 0.4851 0.3725 | 0.2683 0.0005 | −0.0237 −0.0112 | 0.5232 0.3375 |
| Sr | 0.0924 0.0725 | 0.4469 0.3775 | 0.2688 0.0009 | −0.0203 −0.0118 | 0.5074 0.3463 |
| Ba | 0.0889 0.0972 | 0.4419 0.4740 | 0.1913 0.2859 | −0.0189 −0.0225 | 0.4634 0.5228 |
| Metal | Full Molecular Graph | Partial Molecular Graph (Zoom) |
|---|---|---|
| Mg |  |  |
| Ca |  |  |
| Sr |  |  |
| Ba |  |  |
| Atom | q(A) | N(A) | LI(A) | DI | DIB | DINB | E(A) | |
|---|---|---|---|---|---|---|---|---|
| M2+ | Mg | 1.8168 | 10.1832 | 9.924 | 0.2591 | 0.2378 | 0.0213 | −199.2018 |
| Ca | 1.8158 | 18.1842 | 17.7603 | 0.4239 | 0.4058 | 0.0181 | −676.1906 | |
| Sr | 1.7665 | 36.2335 | 35.6785 | 0.5550 | 0.5027 | 0.0523 | −3108.0289 | |
| Ba | 1.9283 | 54.0717 | 53.5193 | 0.5523 | 0.4892 | 0.0631 | −8363.3441 | |
| Co | Mg | 1.3174 | 25.6826 | 24.5528 | 1.1297 | 0.9365 | 0.1933 | −1379.8770 |
| Ca | 1.3133 | 25.6867 | 24.5496 | 1.1372 | 0.9535 | 0.1836 | −1380.2187 | |
| Sr | 1.3118 | 25.6882 | 24.5805 | 1.1077 | 0.9342 | 0.1735 | −1381.6302 | |
| Ba | 1.3098 | 25.6902 | 24.5639 | 1.1263 | 0.9565 | 0.1699 | −1242.0423 |
| 1 | 2 | |
|---|---|---|
| Pval | 7.675(58) | 7.077 |
| Kappa | 0.996(6) | 1.003 |
| Net charge | −0.435 | −0.037 |
| D10 | −0.218(22) | 0.004 |
| Q20 | 0.227(30) | 0.176 |
| Q30 | −0.102(19) | 0.001 |
| Q40 | −0.371(30) | −0.387 |
| O32- | 0.05(2) | −0.001 |
| H40 | 0.27(3) | 0.235 |
| H43+ | −0.087(27) | −0.045 |
| H43− | 0.068(27) | −0.108 |
| 1 | 2 (Mg) | 2 (Ca) | |
|---|---|---|---|
| Orbital | |||
| z2 | 16.2 | 15.0 | 16.6 |
| xz | 26.2 | 26.4 | 25.4 |
| yz | 26.2 | 26.4 | 24.2 |
| x2-y2 | 15.7 | 16.1 | 17.0 |
| xy | 15.7 | 16.2 | 16.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacsa, J.; Ramírez-Palma, L.G.; Cortés-Guzmán, F.; Wallen, C.M.; Scarborough, C.C. An Examination of the Electron Densities in a Series of Tripodal Cobalt Complexes Bridged by Magnesium, Calcium, Strontium, and Barium †. Crystals 2018, 8, 234. https://doi.org/10.3390/cryst8060234
Bacsa J, Ramírez-Palma LG, Cortés-Guzmán F, Wallen CM, Scarborough CC. An Examination of the Electron Densities in a Series of Tripodal Cobalt Complexes Bridged by Magnesium, Calcium, Strontium, and Barium †. Crystals. 2018; 8(6):234. https://doi.org/10.3390/cryst8060234
Chicago/Turabian StyleBacsa, John, Lillian G. Ramírez-Palma, Fernando Cortés-Guzmán, Christian M. Wallen, and Christopher C. Scarborough. 2018. "An Examination of the Electron Densities in a Series of Tripodal Cobalt Complexes Bridged by Magnesium, Calcium, Strontium, and Barium †" Crystals 8, no. 6: 234. https://doi.org/10.3390/cryst8060234
APA StyleBacsa, J., Ramírez-Palma, L. G., Cortés-Guzmán, F., Wallen, C. M., & Scarborough, C. C. (2018). An Examination of the Electron Densities in a Series of Tripodal Cobalt Complexes Bridged by Magnesium, Calcium, Strontium, and Barium †. Crystals, 8(6), 234. https://doi.org/10.3390/cryst8060234