One Step Further in the Characterization of Synthetic Polymers by Ion Mobility Mass Spectrometry: Evaluating the Contribution of End-groups

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
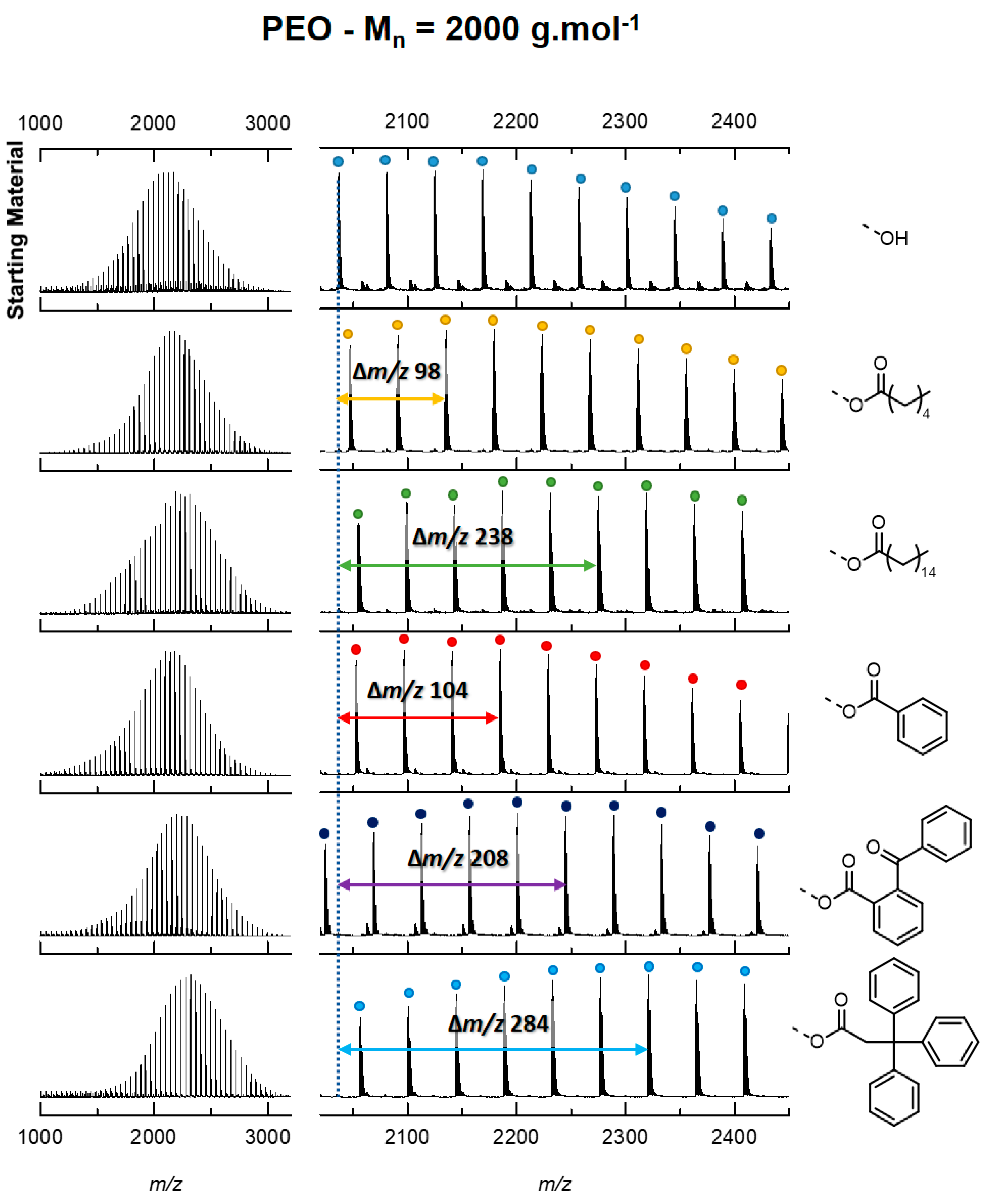
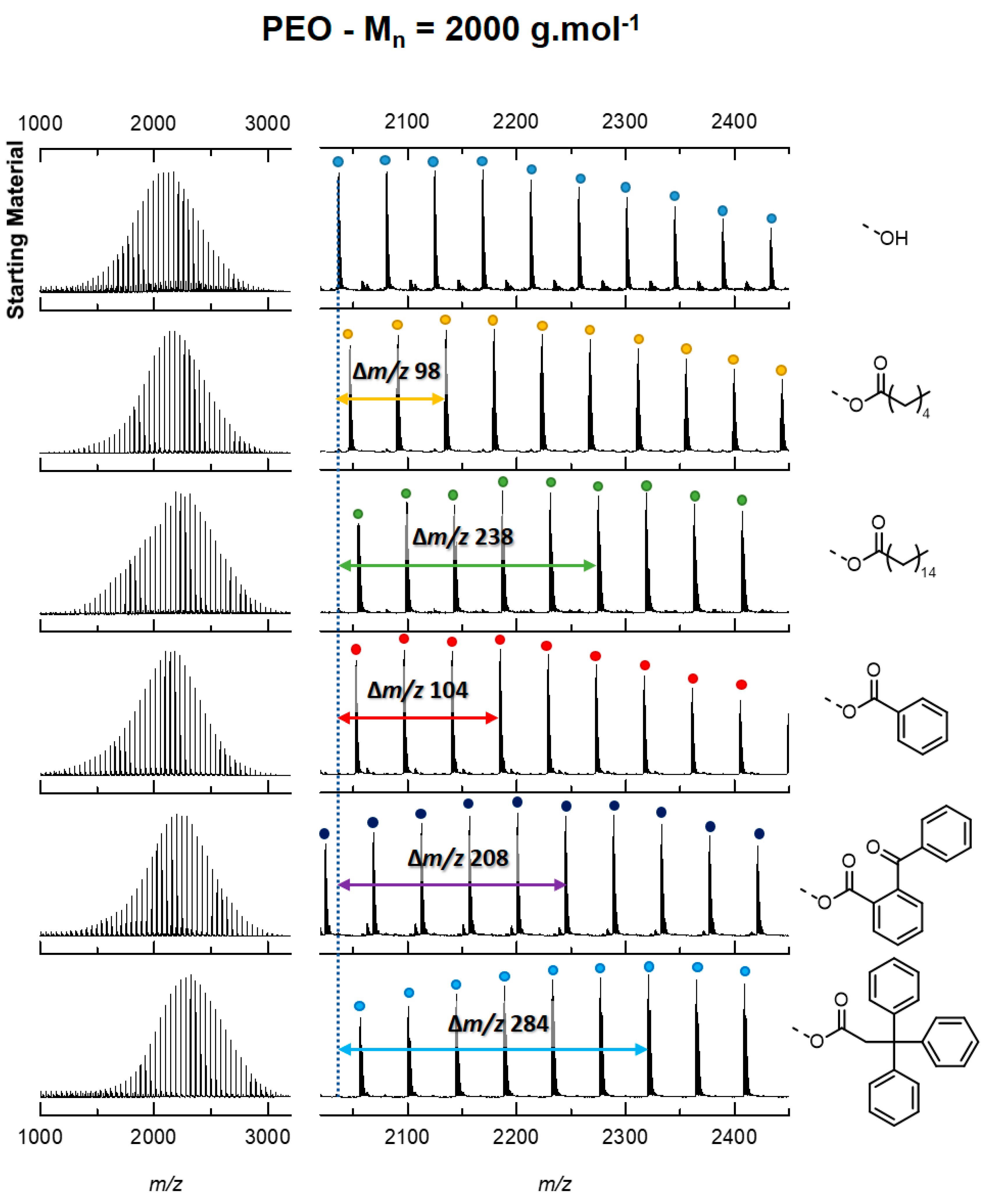
3. Results
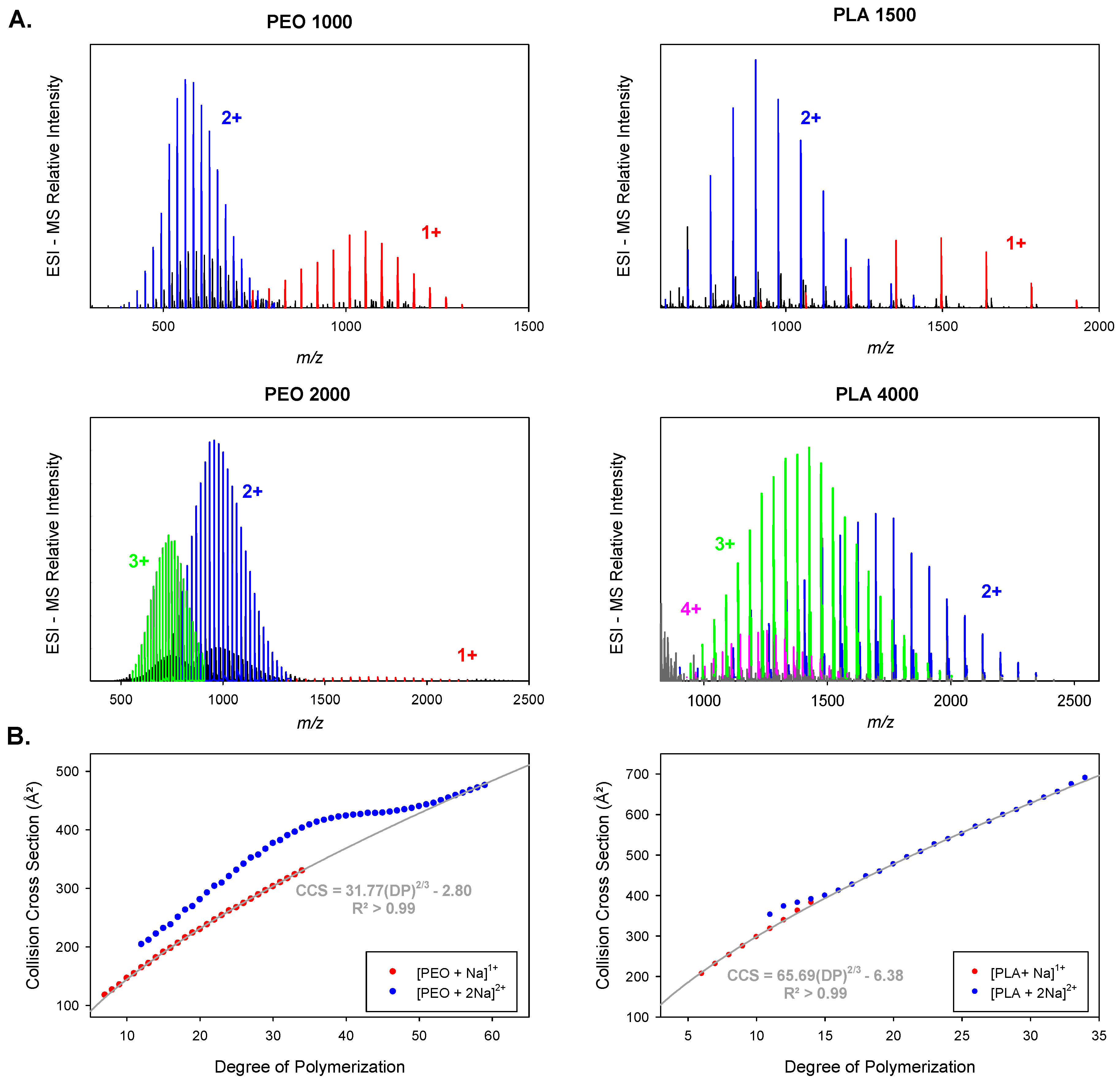
3.1. Model Description
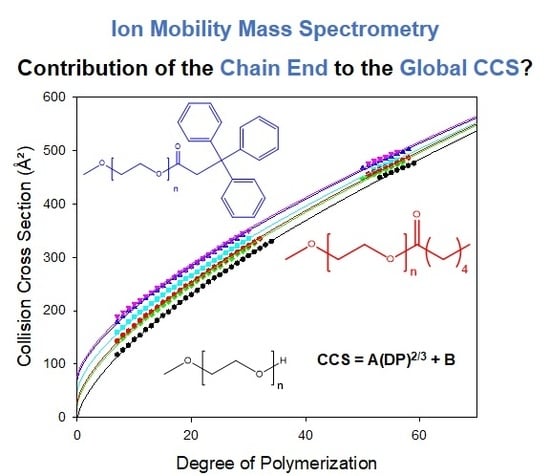
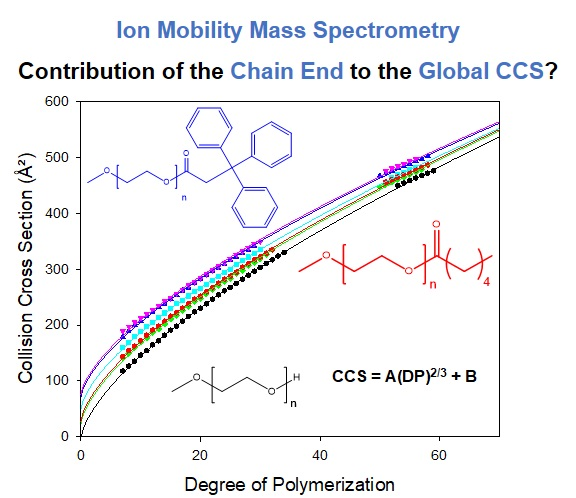
3.2. Contribution of the Chain Ends to the Collision Cross Section
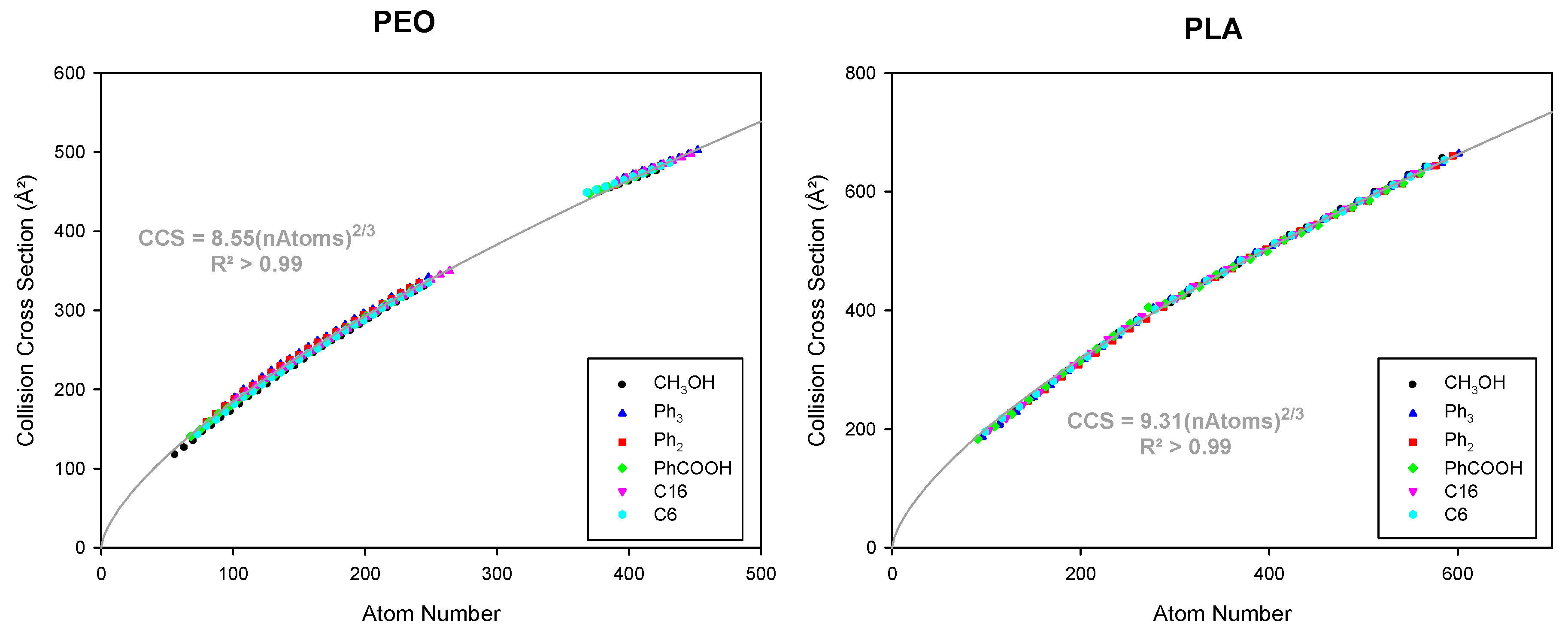
3.3. Considering Different Chain Ends and Polymer Backbones by Counting Atoms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Smith, D.P.; Knapman, T.W.; Campuzano, I.; Malham, R.W.; Berryman, J.T.; Radford, S.E.; Ashcroft, A.E. Deciphering drift time measurements from travelling wave ion mobility spectrometry-mass spectrometry studies. Eur. J. Mass Spectrom. 2009, 15, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Mason, E.A.; McDaniel, E.W. Transport Properties of Ions in Gases; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Gabelica, V.; Marklund, E. Fundamentals of Ion Mobility Spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 51–59. [Google Scholar] [CrossRef]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef]
- Sun, Y.; Vahidi, S.; Sowole, M.A.; Konermann, L. Protein Structural Studies by Traveling Wave Ion Mobility Spectrometry: A Critical Look at Electrospray Sources and Calibration Issues. J. Am. Soc. Mass. Spectrom. 2015, 27, 31–40. [Google Scholar] [CrossRef]
- Shi, L.; Holliday, A.E.; Glover, M.S.; Ewing, M.A.; Russell, D.H.; Clemmer, D.E. Ion Mobility-Mass Spectrometry Reveals the Energetics of Intermediates that Guide Polyproline Folding. J. Am. Soc. Mass Spectrom. 2015, 27, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroy, G.; Lemaur, V.; Henoumont, C.; Laurent, S.; De Winter, J.; De Pauw, E.; Cornil, J.; Gerbaux, P. Flying Cages in Traveling Wave Ion Mobility: Influence of the Instrumental Parameters on the Topology of the Host-Guest Complexes. J. Am. Soc. Mass Spectrom. 2018, 29, 121–132. [Google Scholar] [CrossRef]
- De Winter, J.; Lemaur, V.; Ballivian, R.; Chirot, F.; Coulembier, O.; Antoine, R.; Lemoine, J.; Cornil, J.; Dubois, P.; Dugourd, P.; et al. Size dependence of the folding of multiply charged sodium cationized polylactides revealed by ion mobility mass spectrometry and molecular modelling. Chem. Eur. J. 2011, 17, 9738–9745. [Google Scholar] [CrossRef]
- Haler, J.R.N.; Far, J.; Aqil, A.; Claereboudt, J.; Tomczyk, N.; Giles, K.; Jerome, C.; De Pauw, E. Multiple Gas-Phase Conformations of a Synthetic Linear Poly(acrylamide) Polymer Observed Using Ion Mobility-Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 2492–2499. [Google Scholar] [CrossRef]
- D’Atri, V.; Porrini, M.; Rosu, F.; Gabelica, V. Linking molecular models with ion mobility experiments. Illustration with a rigid nucleic acid structure. J. Mass Spectrom. 2015, 50, 711–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larriba, C.; de la Mora, J.F. The gas phase structure of coulombically stretched polyethylene glycol ions. J. Phys. Chem. B 2012, 116, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Larriba, C.; Fernandez de la Mora, J.; Clemmer, D.E. Electrospray ionization mechanisms for large polyethylene glycol chains studied through tandem ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25, 1332–1345. [Google Scholar] [CrossRef]
- Gidden, J.; Wyttenbach, T.; Jackson, A.T.; Scrivens, J.H.; Bowers, M.T. Gas-Phase Conformations of Synthetic Polymers: Poly(ethylene glycol), Poly(propylene glycol), and Poly(tetramethylene glycol). J. Am. Chem. Soc. 2000, 122, 4692–4699. [Google Scholar] [CrossRef]
- Trimpin, S.; Clemmer, D.E. Ion mobility spectrometry/mass spectrometry snapshots for assessing the molecular compositions of complex polymeric systems. Anal. Chem. 2008, 80, 9073–9083. [Google Scholar] [CrossRef]
- Duez, Q.; Metwally, H.; Konermann, L. Electrospray Ionization of Polypropylene Glycol: Rayleigh-Charged Droplets, Competing Pathways, and Charge State-Dependent Conformations. Anal. Chem. 2018, 90, 9912–9920. [Google Scholar] [CrossRef] [PubMed]
- Gidden, J.; Wyttenbach, T.; Batka, J.J.; Weis, P.; Jackson, A.T.; Scrivens, J.H.; Bowers, M.T. Folding energetics and dynamics of macromolecules in the gas phase: Alkali ion-cationized poly(ethylene terephthalate) oligomers. J. Am. Chem. Soc. 1999, 121, 1421–1422. [Google Scholar] [CrossRef]
- Gidden, J.; Bowers, M.T.; Jackson, A.T.; Scrivens, J.H. Gas-phase conformations of cationized poly(styrene) oligomers. J. Am. Soc. Mass Spectrom. 2002, 13, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Duez, Q.; Josse, T.; Lemaur, V.; Chirot, F.; Choi, C.M.; Dubois, P.; Dugourd, P.; Cornil, J.; Gerbaux, P.; De Winter, J. Correlation between the shape of the ion mobility signals and the stepwise folding process of polylactide ions. J. Mass Spectrom. 2017, 52, 133–138. [Google Scholar] [CrossRef]
- Hoskins, J.N.; Trimpin, S.; Grayson, S.M. Architectural Differentiation of Linear and Cyclic Polymeric Isomers by Ion Mobility Spectrometry-Mass Spectrometry. Macromolecules 2011, 44, 6915–6918. [Google Scholar] [CrossRef]
- Foley, C.D.; Zhang, B.; Alb, A.M.; Trimpin, S.; Grayson, S.M. Use of Ion Mobility Spectrometry–Mass Spectrometry to Elucidate Architectural Dispersity within Star Polymers. ACS Macro Lett. 2015, 4, 778–782. [Google Scholar] [CrossRef]
- Yol, A.M.; Wesdemiotis, C. Multidimensional mass spectrometry methods for the structural characterization of cyclic polymers. React. Funct. Polym. 2014, 80, 95–108. [Google Scholar] [CrossRef]
- Morsa, D.; Defize, T.; Dehareng, D.; Jerome, C.; De Pauw, E. Polymer topology revealed by ion mobility coupled with mass spectrometry. Anal. Chem. 2014, 86, 9693–9700. [Google Scholar] [CrossRef]
- Kokubo, S.; Vana, P. Easy Access to the Characteristic Ratio of Polymers Using Ion-Mobility Mass Spectrometry. Macromol. Chem. Phys. 2016, 2017, 1600373. [Google Scholar] [CrossRef]
- Kokubo, S.; Vana, P. Obtaining the Dielectric Constant of Polymers from Doubly Charged Species in Ion-Mobility Mass Spectrometry. Macromol. Chem. Phys. 2017, 218, 1700126. [Google Scholar] [CrossRef]
- Haler, J.R.N.; Morsa, D.; Lecomte, P.; Jerome, C.; Far, J.; De Pauw, E. Predicting Ion Mobility-Mass Spectrometry trends of polymers using the concept of apparent densities. Methods 2018, 144, 125–133. [Google Scholar] [CrossRef]
- Ruotolo, B.T.; Benesch, J.L.; Sandercock, A.M.; Hyung, S.J.; Robinson, C.V. Ion mobility-mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008, 3, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Duez, Q.; Chirot, F.; Liénard, R.; Josse, T.; Choi, C.M.; Coulembier, O.; Dugourd, P.; Cornil, J.; Gerbaux, P.; De Winter, J. Polymers for Traveling Wave Ion Mobility Spectrometry Calibration. J. Am. Soc. Mass Spectrom. 2017, 28, 2483–2491. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Johnson, N.R.; Contreras, S.; Wyttenbach, T.; Bowers, M.T. Molecular Structures and Ion Mobility Cross Sections: Analysis of the Effects of He and N2 Buffer Gas. Anal. Chem. 2015, 87, 7196–7203. [Google Scholar] [CrossRef] [PubMed]
- Trimpin, S.; Plasencia, M.; Isailovic, D.; Clemmer, D.E. Resolving oligomers from fully grown polymers with IMS-MS. Anal. Chem. 2007, 79, 7965–7974. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| End-Groups | Naming | Number of Atoms * |
|---|---|---|
| CH3O/H | CH3OH | 6 |
| CH3O/C(=O)C5H11 | C6 | 23 |
| CH3O/C(=O)C15H31 | C16 | 53 |
| CH3O/C(=O)Ph | PhCOOH | 18 |
| CH3O/C(=O)Ph(C=O)Ph | Ph2 | 30 |
| CH3O/C(=O)CH2CPh3 | Ph3 | 44 |
| PEO | PLA | |||
|---|---|---|---|---|
| End-Groups | A | B | A | B |
| CH3O/H | 31.77 | −2.80 | 65.69 | −6.38 |
| CH3O/C(=O)C5H11 | 30.77 | 27.41 | 63.90 | 22.93 |
| CH3O/C(=O)C15H31 | 28.80 | 75.24 | 60.96 | 67.60 |
| CH3O/C(=O)Ph | 30.91 | 22.65 | 62.85 | 24.00 |
| CH3O/C(=O)Ph(C=O)Ph | 29.63 | 49.12 | 62.78 | 38.13 |
| CH3O/C(=O)CH2CPh3 | 28.89 | 70.88 | 61.21 | 60.77 |
| PEO | PLA | ||
|---|---|---|---|
| End-Groups | Atom Number in the End-Groups * | A′ | A′ |
| CH3O/H | 6 | 8.42 | 9.26 |
| CH3O/C(=O)Ph | 18 | 8.55 | 9.28 |
| CH3O/C(=O)C5H11 | 23 | 8.54 | 9.31 |
| CH3O/C(=O)Ph(C=O)Ph | 30 | 8.62 | 9.26 |
| CH3O/C(=O)CH2CPh3 | 44 | 8.63 | 9.31 |
| CH3O/C(=O)C15H31 | 53 | 8.55 | 9.33 |
| Average | 8.55 ± 0.07 | 9.29 ± 0.03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duez, Q.; Liénard, R.; Moins, S.; Lemaur, V.; Coulembier, O.; Cornil, J.; Gerbaux, P.; De Winter, J. One Step Further in the Characterization of Synthetic Polymers by Ion Mobility Mass Spectrometry: Evaluating the Contribution of End-groups. Polymers 2019, 11, 688. https://doi.org/10.3390/polym11040688
Duez Q, Liénard R, Moins S, Lemaur V, Coulembier O, Cornil J, Gerbaux P, De Winter J. One Step Further in the Characterization of Synthetic Polymers by Ion Mobility Mass Spectrometry: Evaluating the Contribution of End-groups. Polymers. 2019; 11(4):688. https://doi.org/10.3390/polym11040688
Chicago/Turabian StyleDuez, Quentin, Romain Liénard, Sébastien Moins, Vincent Lemaur, Olivier Coulembier, Jérôme Cornil, Pascal Gerbaux, and Julien De Winter. 2019. "One Step Further in the Characterization of Synthetic Polymers by Ion Mobility Mass Spectrometry: Evaluating the Contribution of End-groups" Polymers 11, no. 4: 688. https://doi.org/10.3390/polym11040688