1. Introduction
In the past decades enzymes have become part of the chemists’ toolbox and they have proven to be effective in different organic reactions. In addition to replacing traditional processes, biocatalysis opened new synthetic routes not available before. Enzymes perform their catalytic activity under mild reaction conditions with a high chemo-, regio- and stereospecificity. Therefore enzymes promise to be a green alternative for conventional processes in industry.
Oligomers produced by enzymatic oligomerizations include polysaccharides, polyesters and even vinyl oligomers [
1,
2,
3,
4]. Polyamides are versatile engineering plastics and excellent fiber materials due to their toughness over wide ranges of temperatures. While there are many reports on enzymatic polyester formation, there are, surprisingly, only few publications on enzyme-catalyzed synthesis of polyamides [
5,
6,
7,
8,
9] which were recently reviewed in an excellent review [
10].
The enzymatic oligomerization of α-amino acid esters was studied by many authors for a wide array of proteases and amino acid esters over the years. The first reports by Brenner
et al. in 1950 showed the oligomerization of threonine and methionine esters by α-chymotrypsin [
11,
12,
13]. Others used bovine lung proteinase [
14], ficin and Cathepsin-C [
15] a protease from
Streptomyces sp. was used for the oligomerization of diethyl L-aspartate by Matsumura
et al. [
16] and Soeda
et al. [
17].
One of the most versatile proteases is papain. It was used in the past to catalyze the oligomerization of leucine [
18,
19] methionine [
18,
20] glycine [
18], tyrosine [
18,
20] and amino acid ethyl esters [
21,
22,
23]. The oligomerizations were carried out in citrate buffer pH 5–6 or phosphate buffer pH 7 leading to oligo amino acids with a degree of oligomerization (DP) of 5–9 amino acids.
Co-oligomers of glutamic acid diethyl ester with ethyl- or methyl esters of alanine, leucine, phenylalanine, tyrosine and aspartic acid ethyl ester were reported by Uyama
et al. [
24]. The co-oligomers were exclusively α-linked and the yield of oligomeric product depended on the comonomer used. Co-oligomers with leucine were obtained in the highest yield, 70%, with DP 8 while a co-oligomer with alanine gave chains with DP 15 but in a lower 31% yield. Co-oligomers of L-leucine ethyl ester with diethyl-L-glutamate were studied by Li
et al. [
25].
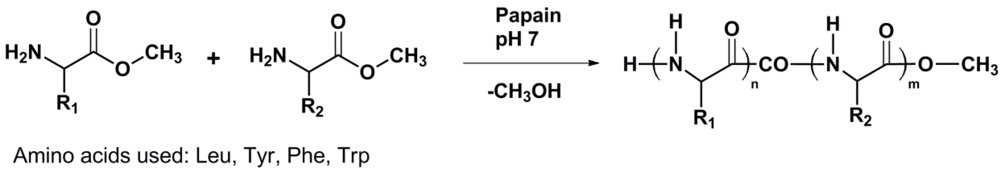
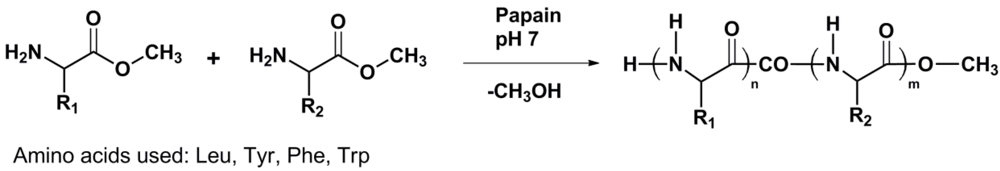
Papain has a known preference for hydrolysis of the peptide bond one amino acid away from hydrophobic aromatic residues in peptides. Therefore it is expected that hydrophobic amino acids are also oligomerized by papain. Here we add the papain catalyzed homo- and co-oligomerization of Leu, Tyr, Trp and Phe (see
Figure 1) to this field.
Figure 1.
Reaction scheme of enzyme catalyzed amino acid (co)oligomerization R1 = R2 and R1 ≠ R2.
Figure 1.
Reaction scheme of enzyme catalyzed amino acid (co)oligomerization R1 = R2 and R1 ≠ R2.
Oligo amino acids containing 1 to 4 of the listed amino acids were prepared in phosphate and tris(hydroxymethyl)aminomethane buffers and a suitable reaction medium was chosen from this.
Since a selectivity of papain is reported for hydrolysis and binding sites are identified in the active site, it is expected that the amino acids will not be polymerized equally well. MALDI-ToF mass spectrometry was used to determine the composition of the oligo amino acids. From the composition of the polymer chains, conclusions were drawn regarding the reactivity order in which the amino acids are oligomerized. The thermal stability of oligo amino acids was determined by thermogravimetric analysis.
2. Experimental Section
2.1. Materials
Papain was purchased from ACROS Organics as a lyophilized powder. The methyl ester hydrochlorides of l-leucine, l-phenylalanine, l-tryptophan and l-tyrosine, α-cyano-4-hydroxycinnamic acid, DMSO-d6, tris(hydroxymethyl)aminomethane and 2,2,2-trifluoroethanol (TFE) were obtained from Sigma-Aldrich. N-Carbobenzoxy-glycine (Z-Gly), trifluoroacetic acid (TFA), Na2CO3 and acetonitrile are used as received from Acros Organics. Hydrochloric acid (37%), NaH2PO4·H2O, Na2HPO4·7H2O, citric acid monohydrate and sodium citrate dihydrate were obtained from Merck. Water was purified by reverse osmosis from an in-house tap. Methanol was bought from Lab-Scan. The MALDI-ToF calibration mixture (bradykinin, angiotensin I and ACTH 18–39) was obtained from Sigma-Aldrich. Citrate buffer (pH 5.6; 2.0 M), phosphate buffer (pH 7; 1.0 M) and TRIS buffer (pH 7; 1.0 M) were prepared in the laboratory.
2.2. Methods
1H-NMR spectra were recorded on a Varian 400 or 300 MHz spectrometer. Either DMSO-d6 or a 9:1 mixture of DMSO-d6 with trifluoroacetic acid was used as a solvent.
MALDI-ToF-MS measurements were performed on a Biosystems Voyager-DE PRO spectrometer in reflector mode with α-cyano-4–hydroxycinnamic acid as the matrix.
Thermogravimetric analysis measurements were carried out using a Perkin-Elmer TGA7 with a heat rate of 10 °C∙min−1 and N2 flow from 18–800 °C. The samples were dried over P2O5 prior to the measurement.
2.3. Synthesis of Z-Gly-(Leu)5-OMe
Papain (38 mg) was dissolved in 10 mL buffer solution (citrate buffer pH 5.6) and to this Z-Gly-OH was added (140 mg; 6.69 × 10−4 mol), followed by Leu-OMe·HCl (1.229 g; 6.77 × 10−3·mol). The clear solution was stirred for 66 h at room temperature. After this a white precipitate was collected by filtration and washed twice with 2 mL of a Na2CO3 solution (5%) and twice with a 1 M solution of HCl in water, and finally with water. The product was dried overnight under reduced pressure at 46 °C and gave a crude yield of 49%. The crude product was purified by dissolving in hot methanol. Water was added dropwise to the hot solution until a precipitate formed. After cooling in an ice bath for 45 min the product was collected by filtration and dried in vacuum overnight at 46 °C (yield 36%).
1H-NMR (1 drop CF3COOH in CDCl3): δ = 7.8 (s, 1H, CONHCH (Gly)); 7.5 (s, 1H, CONHCH (Leu)); 7.3 (m, 5H, Ar); 5.1 (s, 2H, Ar-CH2O); 4.5 (s, 1H, NCHRCO); 4.0 (s, 2H, NCH2CO); 3.8 (s, 3H OCH3); 1.5 (m, 3H, CHCH2 CH(CH3)2); 0.8 (m, 6H, CH(CH3)2).
2.4. Oligomerization and Co-Oligomerization of α-Amino Acids
All of the homo-oligomerizations and co-oligomerizations where conducted according to the following experimental procedure.
A mixture of the amino acid ester hydrochloride (5 mmol), 25 mL phosphate buffer (pH 7.0; 1.0 M) and papain (300 mg) was placed in a 50 mL flask, equipped with a stirring egg. The mixture was kept at 40 °C and stirred for 24 h in air. Precipitates were collected by centrifugation (7500 rpm) and washed once with dilute HCl solution (0.1 M) and twice with water. The resulting white powder was dried in vacuo for three hours, recrystallized from 2,2,2-trifluoroethanol and subsequently dried to give 30 to 50% yield, depending on the nature of the amino acid ester. In the case of co-oligomerization the amino acid esters were added in equimolar amounts.
2.5. MALDI-ToF MS Sample Preparation
For MALDI-ToF analysis the samples were dissolved (3 mg∙mL−1) in TFE, acetonitrile/H2O/TFA (30/70/0.1) mixtures or pure TFA depending on the co-oligomer. Sometimes a 10-fold dilution of the sample is needed to obtain a decent mass spectrum. The spots were created by 2 μL of a mixture containing matrix solution (10 mg∙mL−1) and sample solution in a 1:1 ratio by volume. Calibration was performed with a mixture of matrix dimer, bradykinin, angiotensin I and ACTH 18–39, dissolved in a mixture of acetonitrile/H2O/TFA (30/70/0.1). Each spectrum was obtained by a minimum of 600 shots.
3. Results and Discussion
3.1. Homo-Oligomerization of α-Amino Acids
Homo-oligomers of leucine, tyrosine and tryptophan were synthesized with papain from the corresponding methyl ester hydrochlorides in phosphate buffer (pH 7; 1 M). After 24 h of reaction the precipitate was collected by centrifugation, while in the absence of papain no precipitation was formed. Yields depend on the amino acids used varying from 30 to 80% by weight (see
Table 1).
Table 1.
Yield and DPavg of the Leu, Phe, Tyr and Trp homo-oligomers.
Table 1.
Yield and DPavg of the Leu, Phe, Tyr and Trp homo-oligomers.
| Entry | Oligomer | Yield(wt%) | DP by 1H-NMR | DPavg by MALDI-ToF MS |
|---|
| 1 | Z-Gly(Leu)n | 31 | 5 | 6 a |
| 2 | (Leu)n | 55 | 12 | 6.86 |
| 3 | (Phe)n | 60 | 6 | 5.35 |
| 4 | (Tyr)n | 80 | 9 | 6.98 |
| 5 | (Trp)n | 30 | 5 | 4.85 |
The average chain lengths determined from
1H-NMR spectroscopy and MALDI-ToF MS are shown in
Table 1. Signals from the methyl ester end-groups are used to determine the DP from
1H-NMR spectra. The overestimation of the degree of oligomerization of polyleucine (DP 12) and polytyrosine (DP 9) by
1H-NMR spectroscopy can be explained by hydrolysis reactions creating acid end-groups. In MALDI-ToF spectra the resulting acid end groups are found in significant amounts as well.
To prove this, leucine was oligomerized starting from a N-carbobenzoxy-glycine (Z-Gly) molecule (see
Table 1 entry 1). The chain length (DP 5) can now be determined using the aromatic protons of the protecting group and the leucine main chain. Including the starting Z-Gly moiety, 6 amino acids are connected by papain in agreement with the DP
avg calculated from the MALDI-ToF mass spectra.
From the MALDI-ToF MS data DPavg was calculated using Equation (1) (see experimental section). Often the smallest chains detected are 4 or 5 amino acids. Chains with a lower DP are not detected because of overlapping peaks with matrix fragments and oligomer fragments.
From the results presented in
Table 1, the following order of reactivity was determined: Tyr > Leu > Phe > Trp. Polytyrosine is obtained in highest yield (80%) combined with the highest DP
avg (6.98). Polyleucine was obtained with a DP
avg 6.86 but in comparable yield (55%) as polyphenylalanine (60%) with DP
avg 5.35. Polytryptophan was the homo-oligomer obtained in the lowest yield of the series (30%) and formed the shortest chains with DP
avg 4.85.
3.2. Co-Oligomerization of α-Amino Acid Esters
Co-oligomers where synthesized by papain starting from a 1:1 molar ratio of the amino acids. After 24 h of reaction in the buffer the precipitate was collected by centrifugation in yields 28–60% in phosphate buffer and 13–35% in TRIS buffer. Since the yields are lower in TRIS buffer than in the phosphate buffer (see
Table 2) TRIS was discarded as a suitable reaction medium and all further experiments are conducted in phosphate buffer (pH 7; 1.0 M).
Table 2.
Co-oligomer yields in phosphate and TRIS buffer.
Table 2.
Co-oligomer yields in phosphate and TRIS buffer.
| entry | Co-oligomer | Yield in phosphate buffer (wt%) | Yield in TRIS buffer (wt%) |
|---|
| 1 | Tyr-Phe | 44 | 15 |
| 2 | Tyr-Leu | 40 | 19 |
| 3 | Tyr-Trp | 36 | 13 |
| 4 | Leu-Phe | 52 | 35 |
| 5 | Trp-Phe | 35 | 20 |
| 6 | Trp-Leu | 60 | 41 |
| 7 | Tyr-Phe-Trp | 41 | 15 |
| 8 | Phe-Tyr-Leu | 28 | 23 |
| 9 | Trp-Tyr-Leu | 35 | 16 |
| 10 | Phe-Leu-Trp | 39 | 28 |
| 11 | Phe-Leu-Trp-Tyr | 38 | 18 |
TRIS buffer is used for some protease mediated reactions [
28,
29,
30,
31] including papain [
31,
33], so it is not likely that the enzyme loses its activity in this buffer. However, in the oligomerization of amino acids [
16,
17,
18,
19,
20,
21,
22], TRIS was not reported; only citrate and phosphate buffers are mentioned.
3.2.1. Composition of Binary Co-Oligo Amino Acids
MALDI-ToF mass spectrometry provides detailed information about the composition and endgroups of the oligomer chains. Hydrolysis of the ester endgroups leads to acid endgroups and both are found in the MALDI-ToF mass spectra. The most abundant molecular ion is the sodium adduct [M-Na]+ but also some [M-H]+ ions are detected. At low m/z values (<700 m/z) the peaks overlap with matrix ions and oligomer fragments. Molecular ions with a lower m/z value can therefore not be identified.
In
Table 3 the average degree of oligomerization and the composition at DP
avg for all the binary co-oligomers is reported. From the number of each containing amino acid and the assumption that all the chains have an ester endgroup the mass of the average chains is calculated. All binary co-oligomers show an average composition of a random co-oligomer (a 1:1 ratio of the amino acids AA1 and AA2) in most of the chains at DP
avg.
Table 3.
Composition of binary co-oligomers composed of amino acids AA1 and AA2 at DPavg and mass at DPavg.
Table 3.
Composition of binary co-oligomers composed of amino acids AA1 and AA2 at DPavg and mass at DPavg.
| entry | Co-oligomer | DPavg | AA1 | AA2 | M at DPavg |
|---|
| 1 | Tyr-Phe | 6.9 | 4.14 | 2.74 | 1,110.5 |
| 2 | Tyr-Leu | 7.7 | 3.94 | 3.78 | 1,103.9 |
| 3 | Tyr-Trp | 5.9 | 2.50 | 3.35 | 1,063.1 |
| 4 | Leu-Phe | 6.7 | 3.57 | 3.12 | 895.8 |
| 5 | Trp-Phe | 4.3 | 2.13 | 2.18 | 749.6 |
| 6 | Trp-Leu | 7.2 | 3.74 | 3.42 | 1,114.8 |
From the homoligomerization it is expected that the amino acids do not have an equal reactivity in the co-oligomerization. By analyzing the composition of copolymers this can be verified.
Below, first the MALDI-ToF MS analysis of the copolymer poly(tyrosine-co-phenylalanine) is illustrated in detail. Second, a brief description of the other binary co-oligomers is given, accompanied by the resulting graphs illustrating the composition of the binary co-oligomers. Finally, the effect of combining amino acids with another is evaluated to rank the amino acids in order of reactivity.
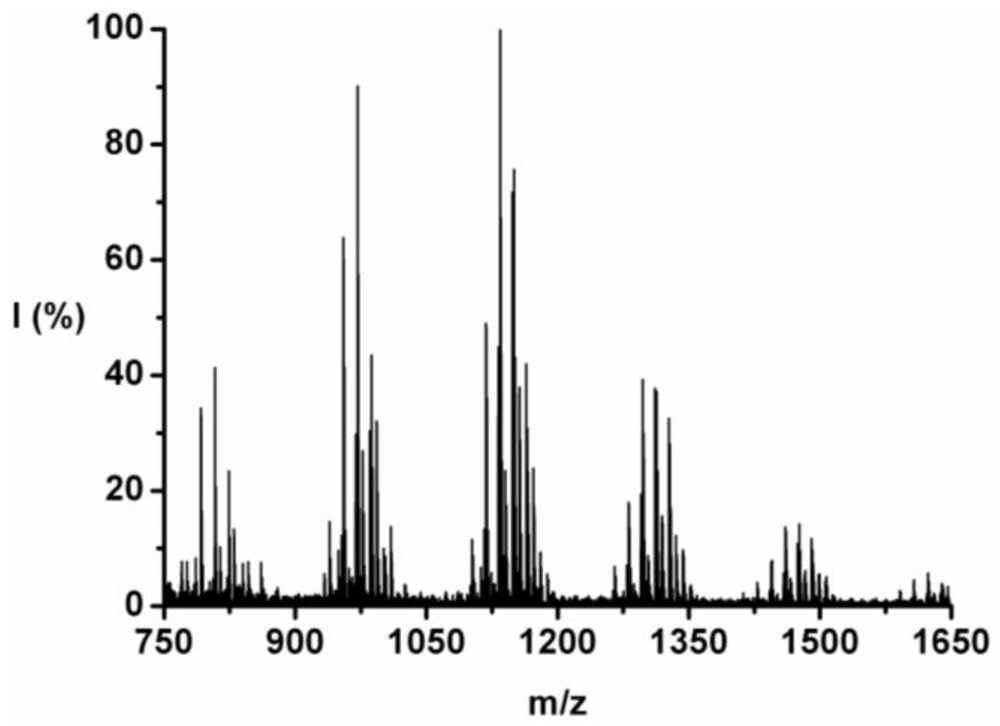
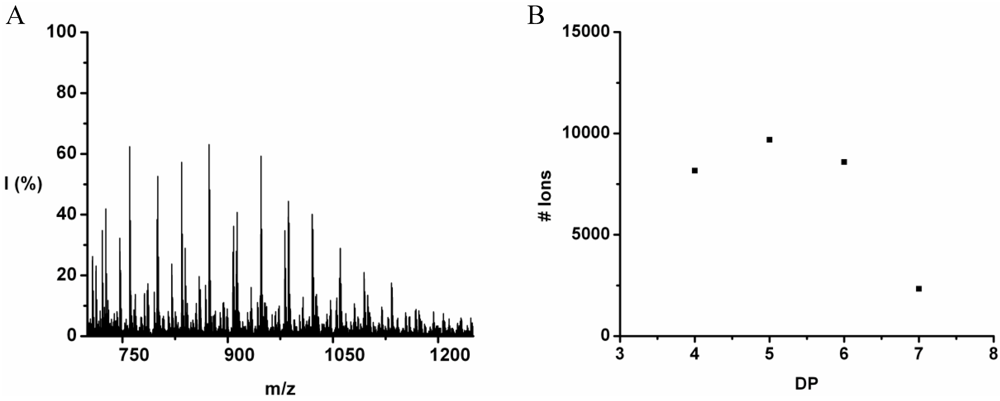
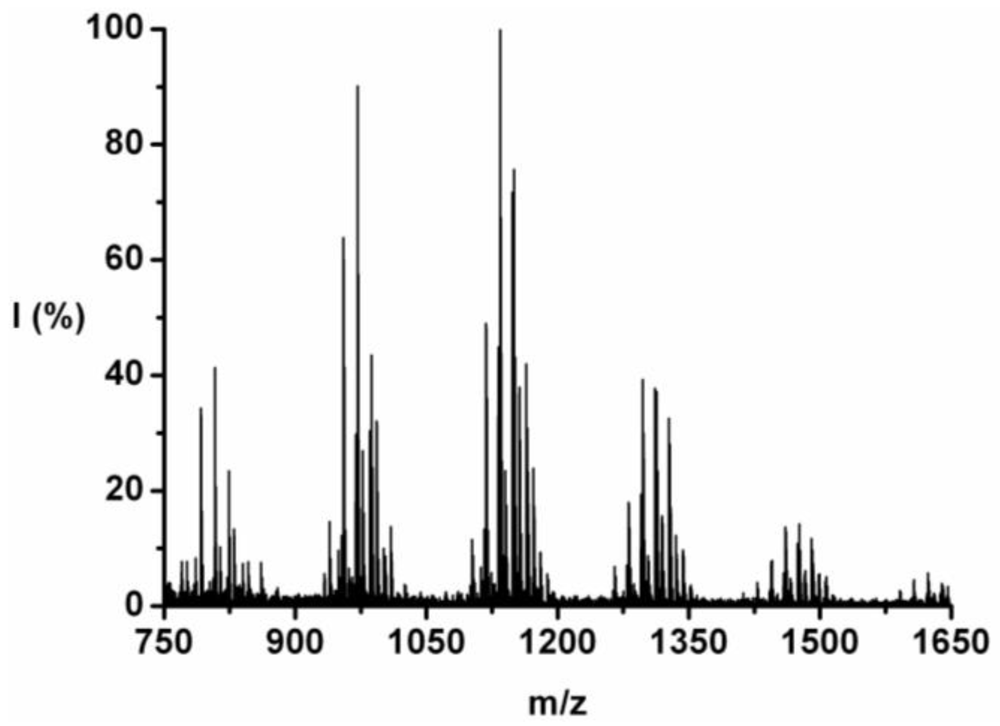
3.2.2. Poly(Tyrosine-Co-Phenylalanine)
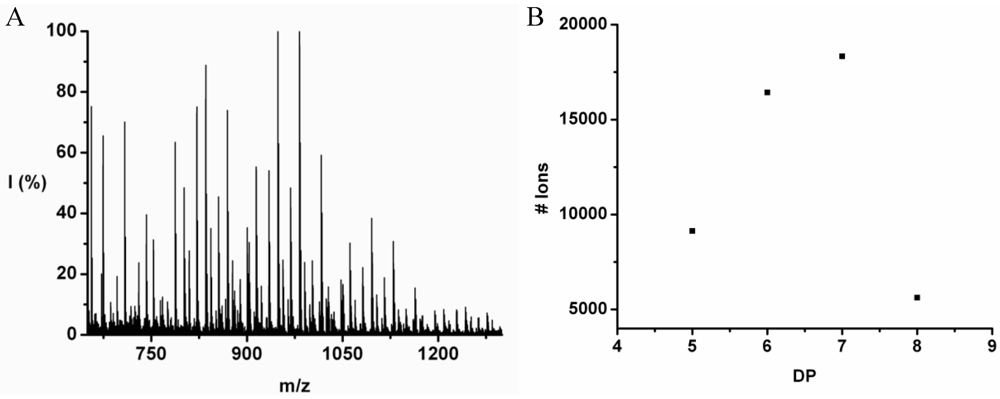
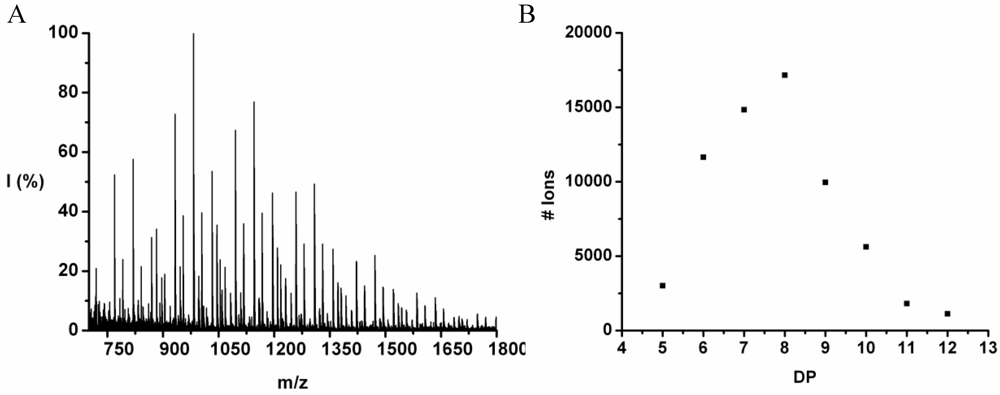
The co-oligomer of tyrosine and phenylalanine was obtained in 44 wt% yield. The mass difference between Tyr and Phe causes the mass peaks to be grouped per DP ranging from 5 to 10, see
Figure 2. Each peak can be explained as one of the four possible molecular ions, [M-H]
+ or [M-Na]
+ with either an ester or an acid endgroup.
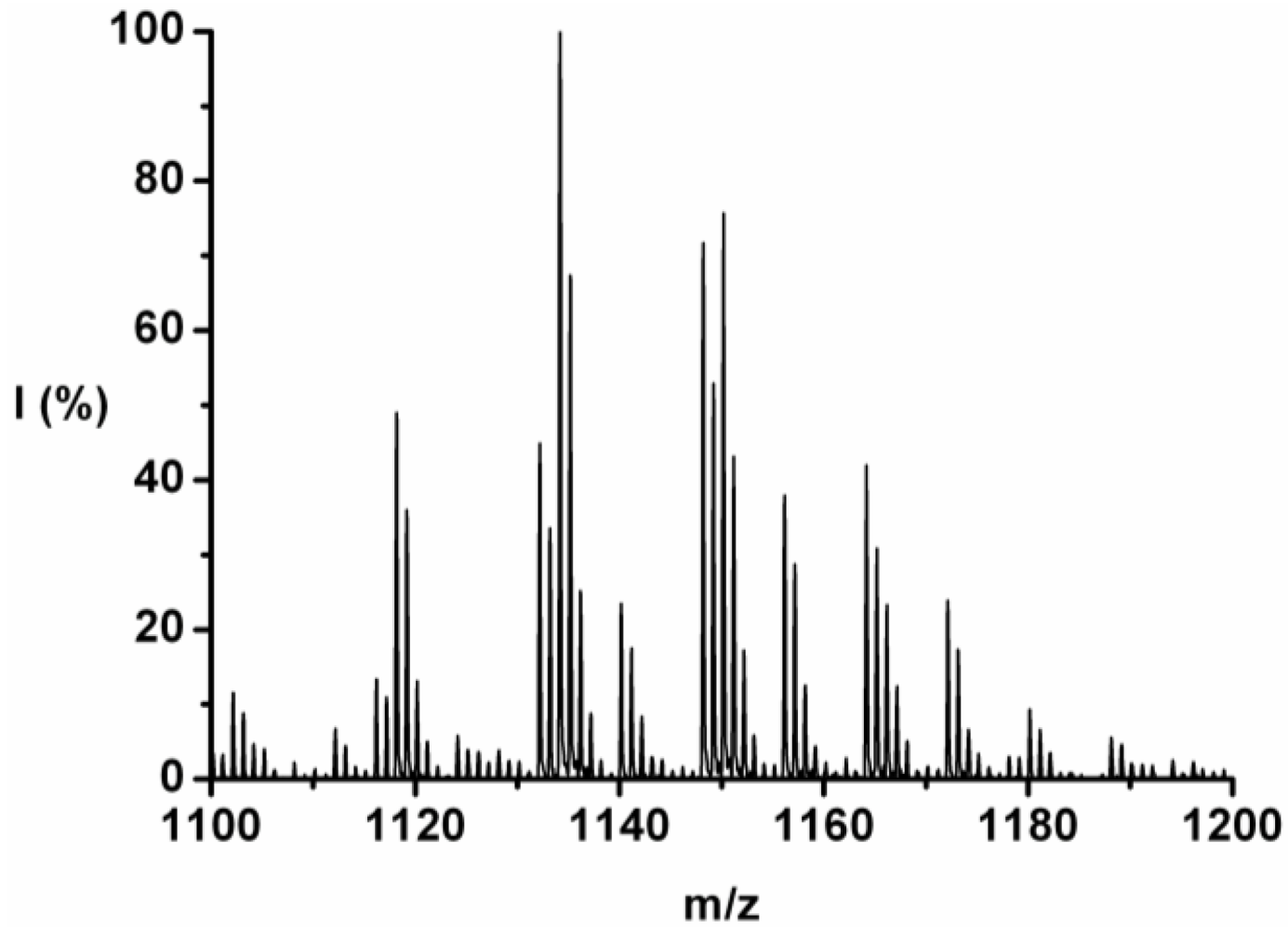
For each DP the ions are identified and counted. This is illustrated for the 7-mer.
Figure 3 zooms in on this fraction and its peaks are explained in
Table 4. Peaks of all the possible chains with seven amino acid residues were found, except the chains with a 1:6 and a 0:7 ratio of tyrosine to phenylalanine. Either they are not present or too small to distinguish from the noise.
Figure 2.
MALDI-ToF mass spectrum of poly(Tyr-co-Phe).
Figure 2.
MALDI-ToF mass spectrum of poly(Tyr-co-Phe).
Figure 3.
MALDI-ToF mass spectrum of poly(Tyr-co-Phe) zoomed in on the 7-mer.
Figure 3.
MALDI-ToF mass spectrum of poly(Tyr-co-Phe) zoomed in on the 7-mer.
The chains with an average composition of 4 plus 3 amino acids are the most abundant, they make up around 55% of the ion counts for this DP. In
Table 4 the peaks (m/z value) are listed from 1102 to 1180 and the composition Tyr_Phe, ion-type, end-group, absolute I (counts) and relative (I%; ion counts divided by total counts per DP) are listed. Most of the ions are [M-Na]
+ type with ester or acid endgroup.
Table 4.
Identification of the ions found in the poly(Tyr-co-Phe) 7-mer.
Table 4.
Identification of the ions found in the poly(Tyr-co-Phe) 7-mer.
| m/z | Tyr_Phe | Ion | Endgroup | I (counts) | I (%) |
|---|
| 1102 | 2_5 | [M-Na]+ | Acid | 597.77 | 2.14 |
| 1112 | 4_3 | [M-H]+ | Acid | 347.79 | 1.25 |
| 1116 | 2_5 | [M-Na]+ | Ester | 690.90 | 2.47 |
| 1118 | 3_4 | [M-Na]+ | Acid | 2533.93 | 9.07 |
| 1124 | 4_3 | [M-H]+ | Ester | 300.12 | 1.07 |
| 1128 | 5_2 | [M-H]+ | Acid | 200.40 | 0.72 |
| 1132 | 3_4 | [M-Na]+ | Ester | 2320.56 | 8.31 |
| 1134 | 4_3 | [M-Na]+ | Acid | 5170.25 | 18.51 |
| 1140 | 5_2 | [M-H]+ | Ester | 1213.88 | 4.35 |
| 1148 | 4_3 | [M-Na]+ | Ester | 3710.17 | 13.28 |
| 1150 | 5_2 | [M-Na]+ | Acid | 3915.33 | 14.02 |
| 1156 | 6_1 | [M-H]+ | Ester | 1965.10 | 7.03 |
| 1164 | 5_2 | [M-Na]+ | Ester | 2046.85 | 7.33 |
| 1166 | 6_1 | [M-Na]+ | Acid | 1205.64 | 4.32 |
| 1172 | 7_0 | [M-H]+ | Ester | 1233.70 | 4.42 |
| 1180 | 6_1 | [M-Na]+ | Ester | 481.27 | 1.72 |
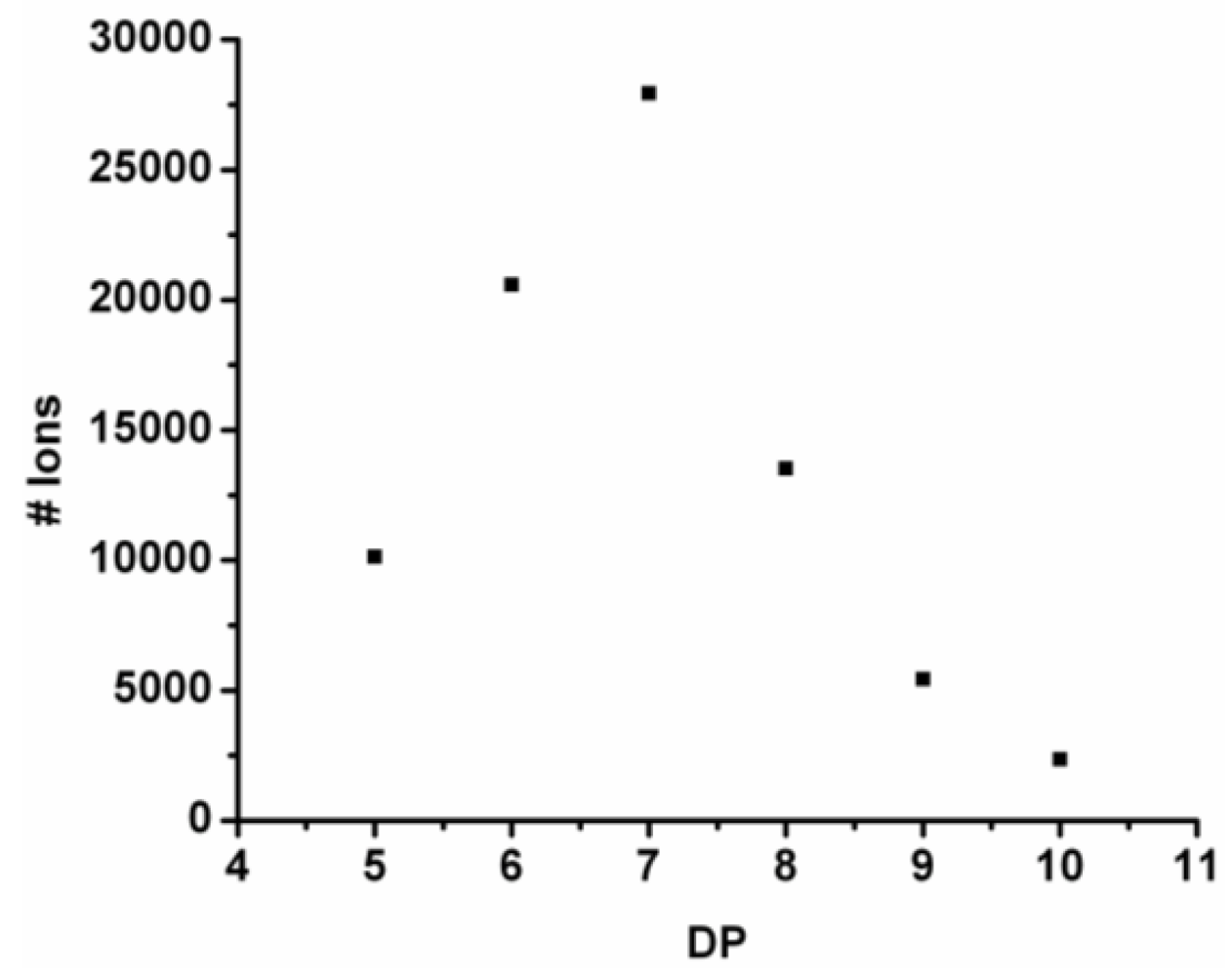
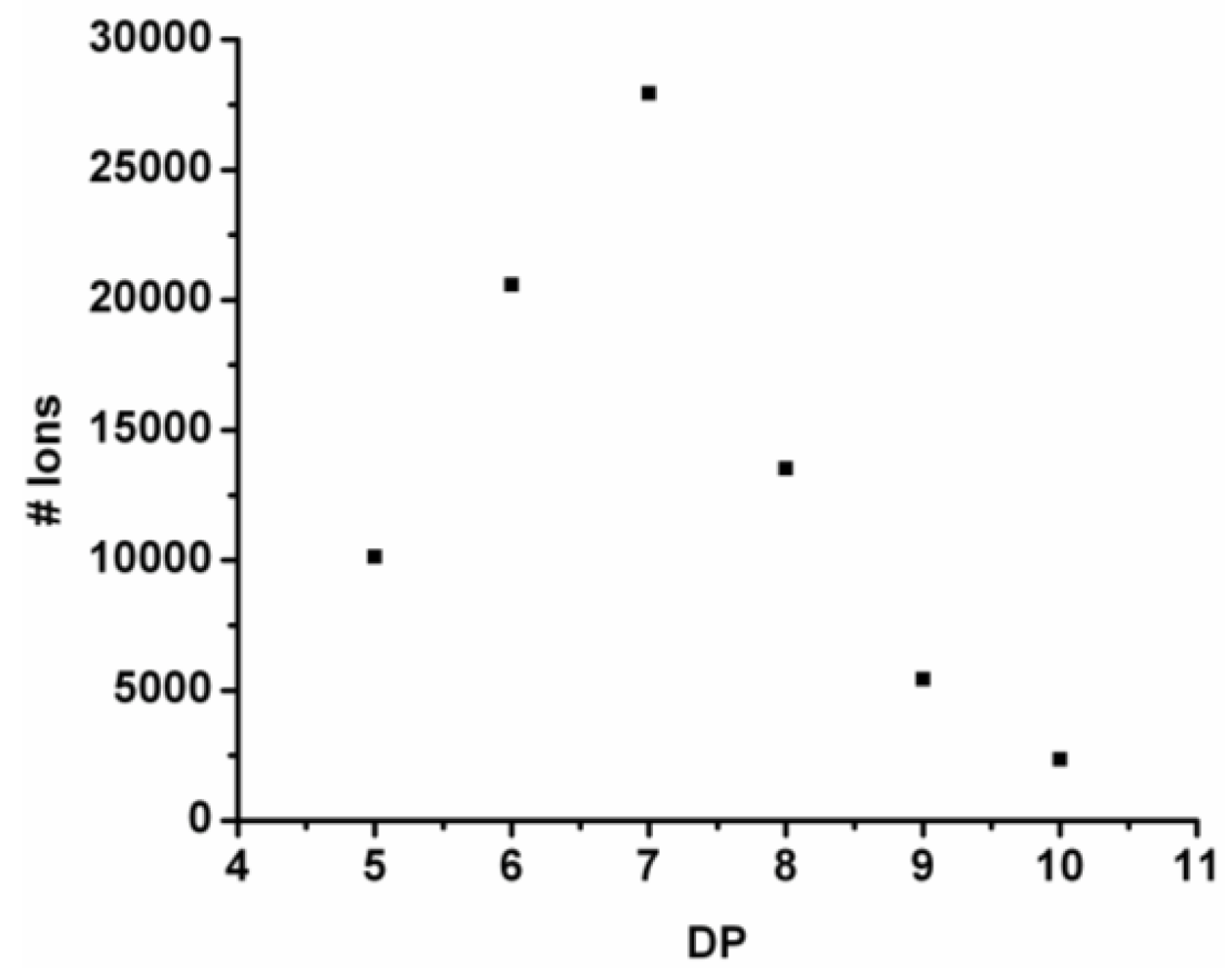
For every observed DP the ion counts are added and the resulting chain length distribution is plotted in
Figure 4. The DP
avg is calculated weighted by the number of ions using Equation (1) (see
Section 2) to be 6.9.
Figure 4.
Observed chain length distribution of poly(Tyr-co-Phe).
Figure 4.
Observed chain length distribution of poly(Tyr-co-Phe).
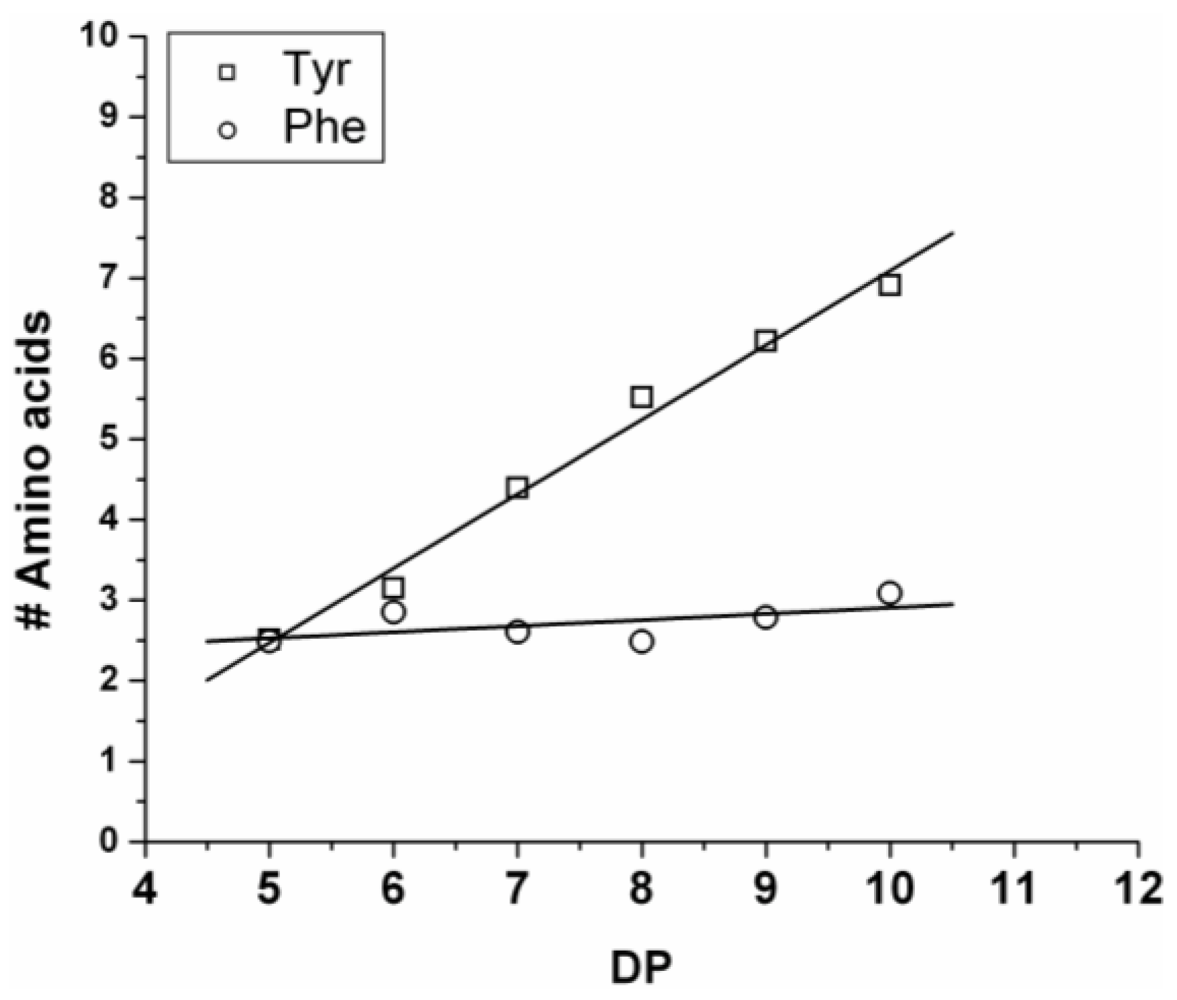
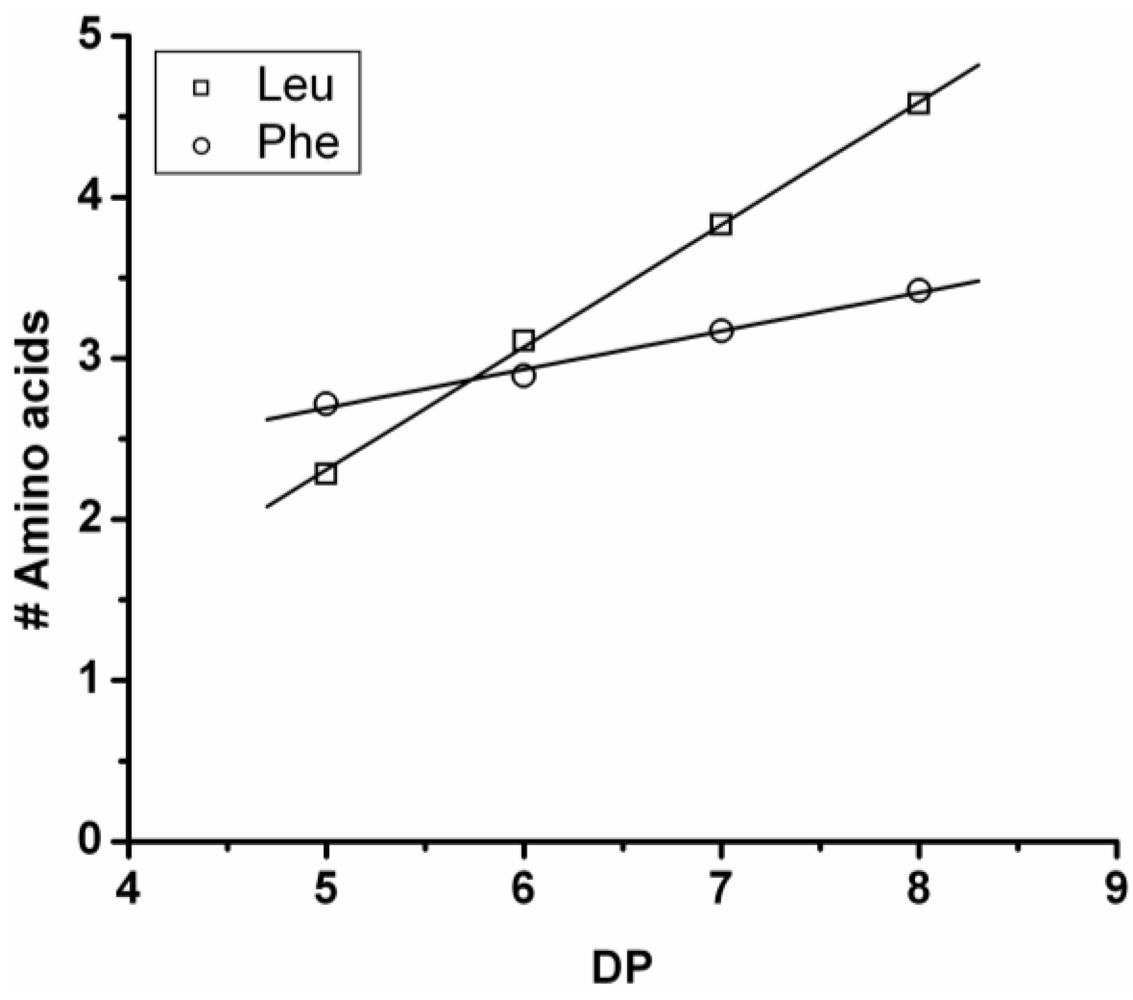
The average amount of the two amino acids was calculated from the average fraction (from Equation (2)—see
Section 2) and plotted in
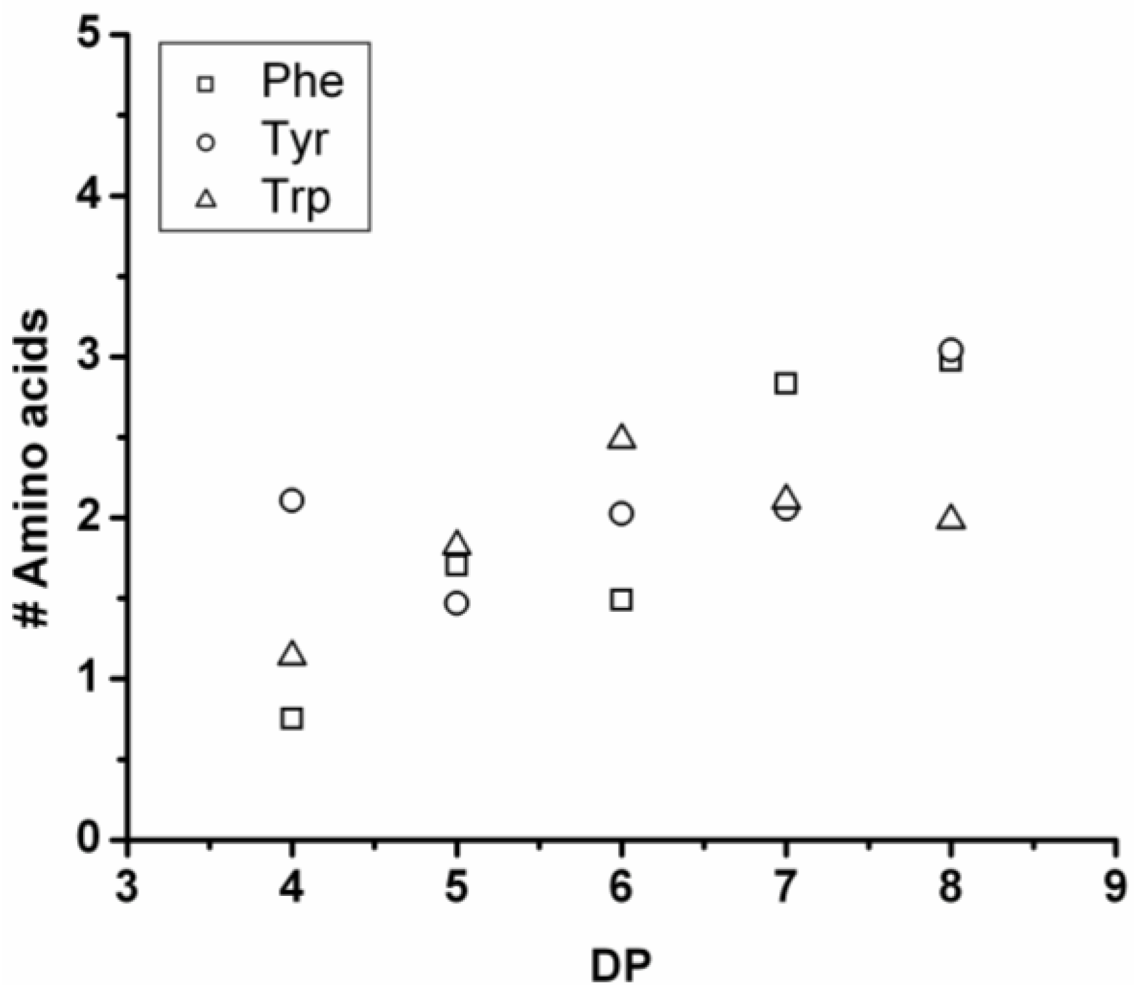
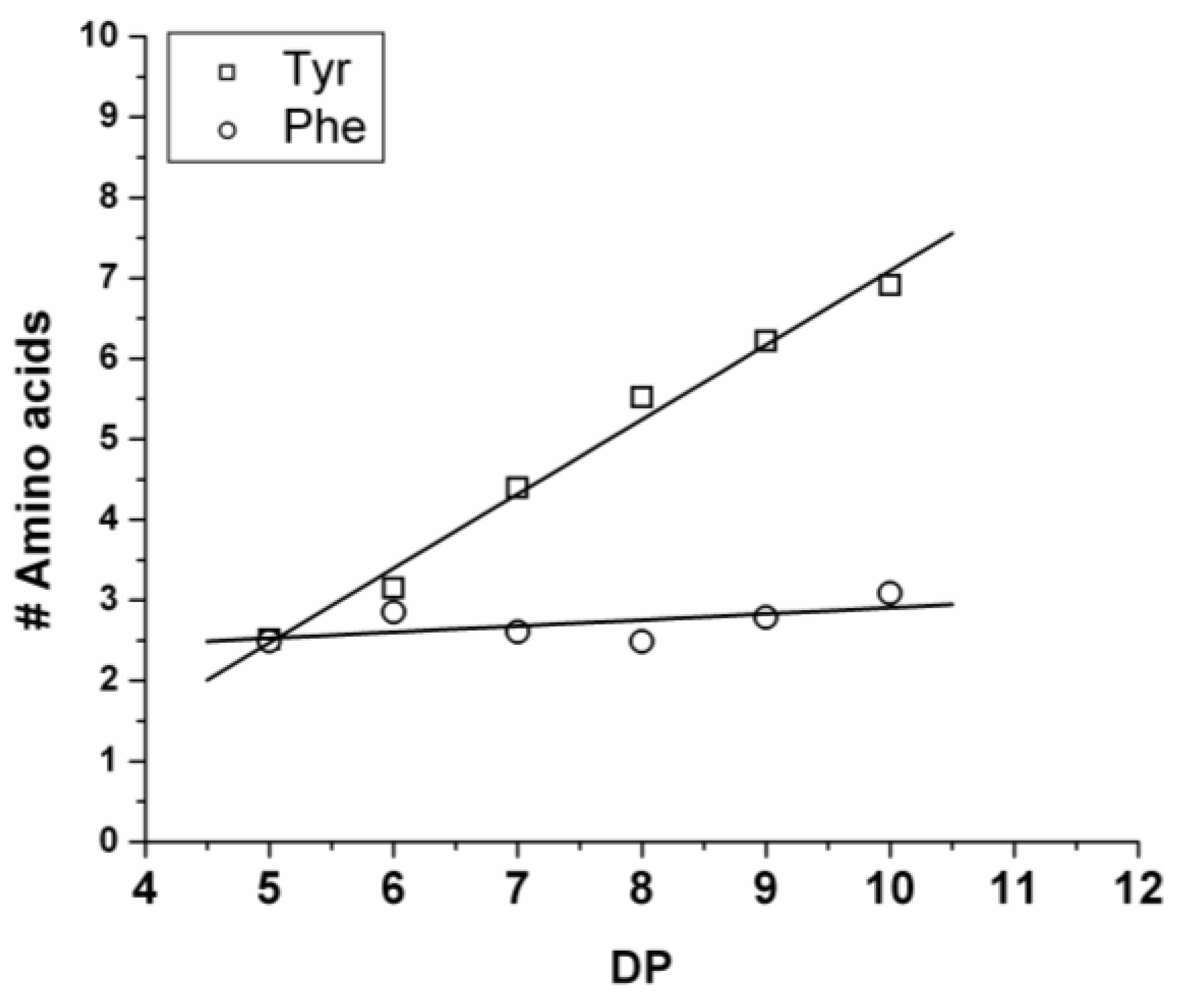
Figure 5. The chains grow by adding on average 0.92 units of Tyr and 0.08 units of Phe (slope of the lines). Showing the low reactivity of phenylalanine when co-oligomerized with a tyrosine. Tyrosine is preferred over phenylalanine as it was observed for the reactivity in the case of the homo-oligomers.
Figure 5.
Average number of amino acid residues in poly(Tyr-co-Phe) for every observed degree of oligomerization (DP).
Figure 5.
Average number of amino acid residues in poly(Tyr-co-Phe) for every observed degree of oligomerization (DP).
Poly(Tyrosine-Co-Leucine)
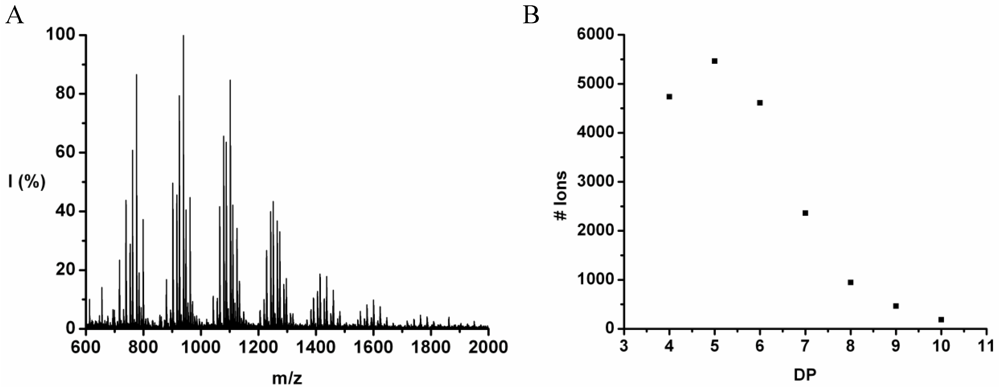
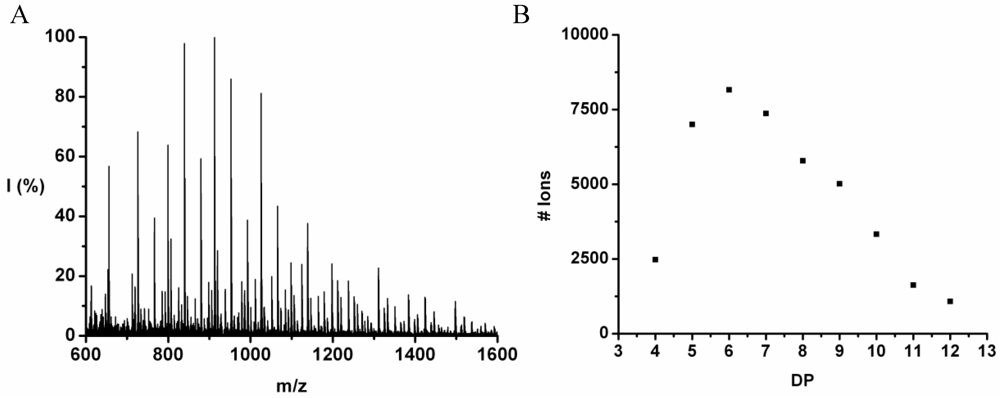
The co-oligomer of tyrosine and leucine was obtained in 40% yield. The MALDI-ToF MS spectrum shown in
Figure 6(A) shows peaks with m/z values 769–1656. The peaks belong to chains with a DP 6–12. The DPavg was calculated as 7.8 corresponding to the maximum of the distribution shown in
Figure 6(B) at DP 8.
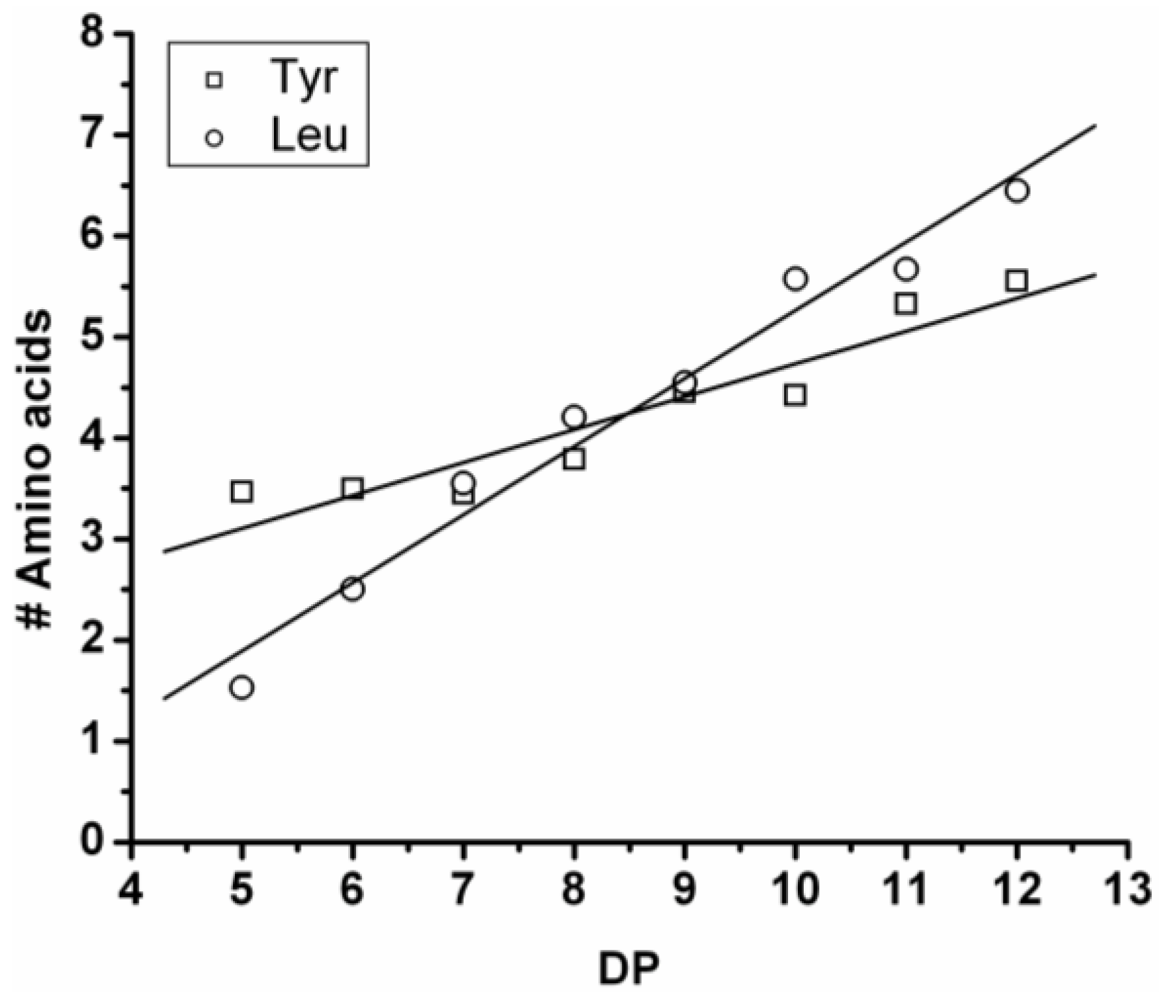
In
Figure 7 the average number of tyrosine and leucine residues for every DP are plotted. The chains grow by preferentially adding leucine. The slopes of the two amino acids (determined from a linear fit) show an increase of 0.37 per DP for Tyr and 0.72 for Leu. Leucine is therefore preferred over tyrosine in this reaction. This result is not in accordance with the reactivity found for homo‑oligomerization of leucine and tyrosine.
Figure 6.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of poly(Tyr-co-Leu).
Figure 6.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of poly(Tyr-co-Leu).
Figure 7.
Average number of amino acids in poly(Tyr-co-Leu) for every observed DP.
Figure 7.
Average number of amino acids in poly(Tyr-co-Leu) for every observed DP.
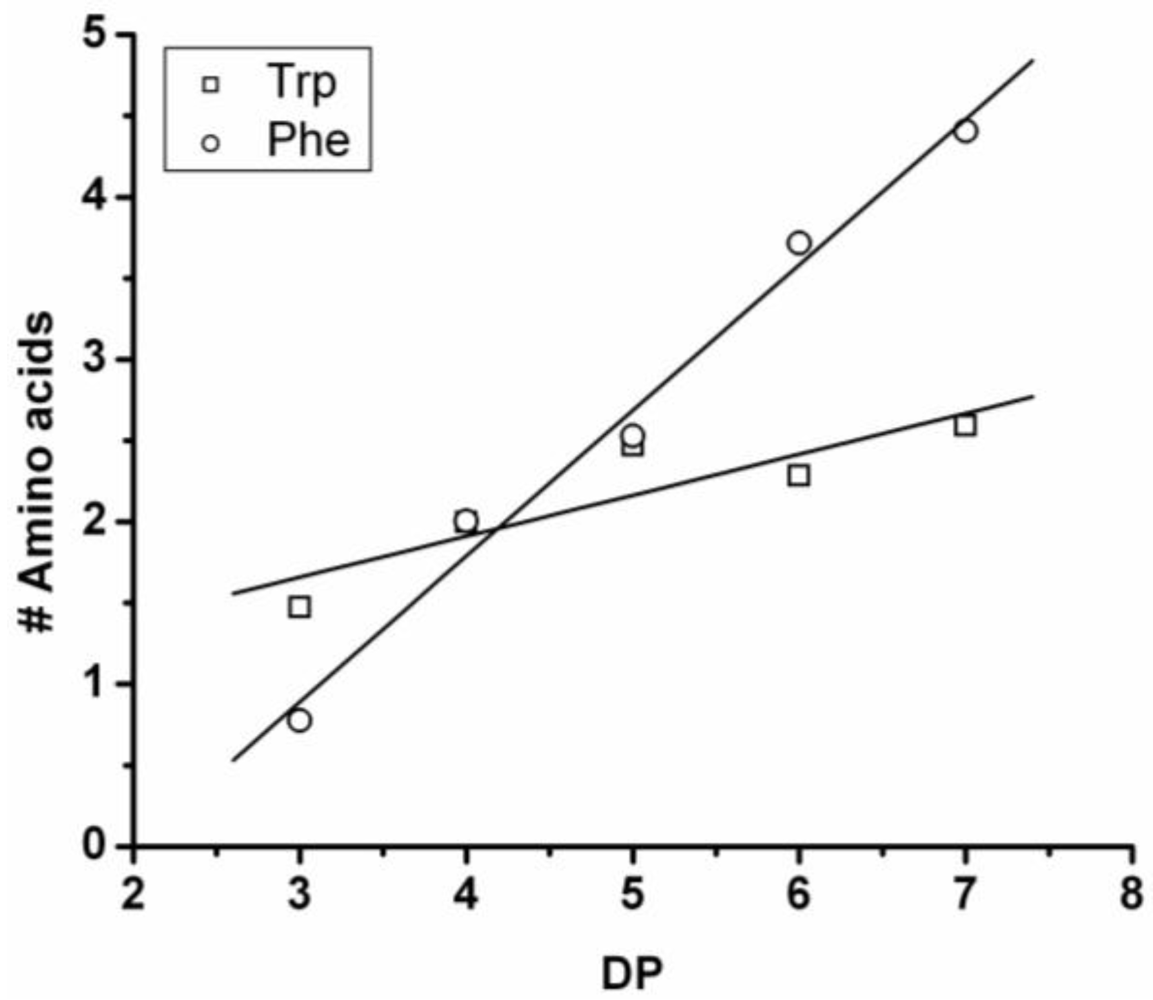
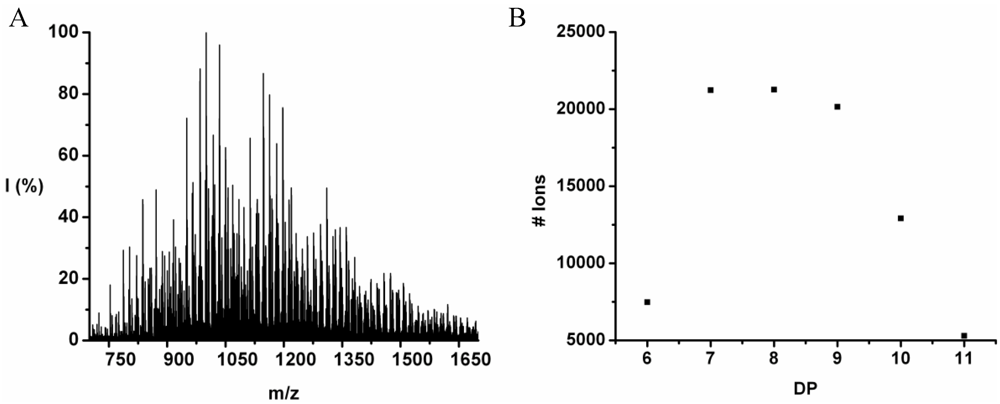
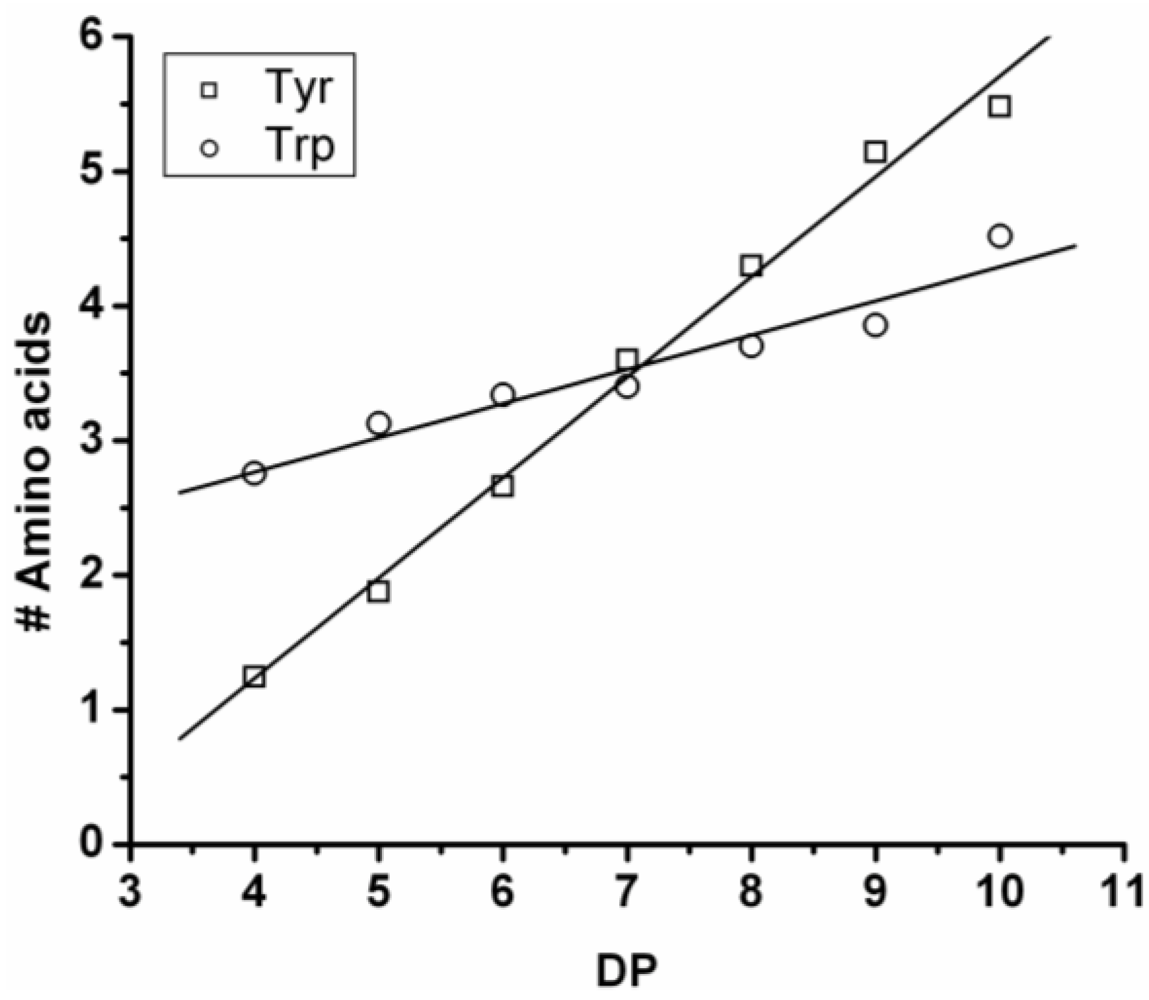
Poly(Tyrosine-Co-Tryptophan)
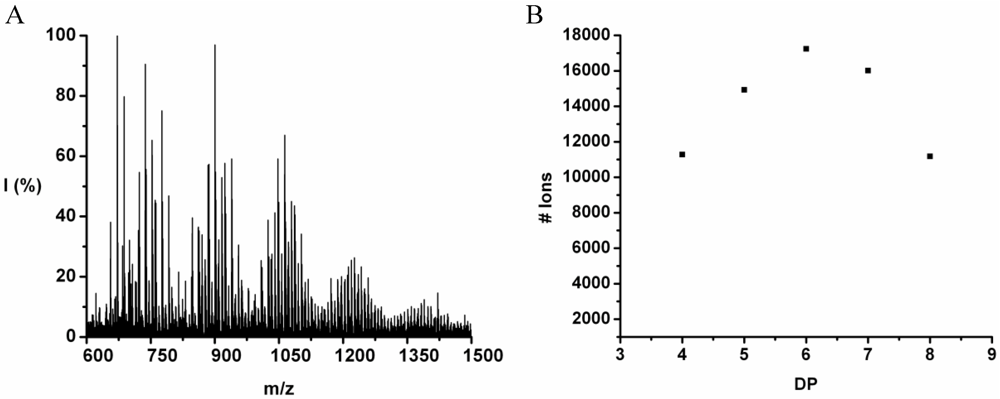
The copolymer of tyrosine and tryptophan was produced in 36% yield. The MALDI-ToF MS spectrum of this copolymer is shown in
Figure 8(A). The peaks with values 799–1,717 m/z were analyzed and belong to chains with a DP 4–10. The most abundant ion is the 939 m/z, 2_3 [M-Na]
+ ester terminated molecular ion.
In
Figure 8(B) the chain length distribution of the poly(Tyr-co-Trp) is plotted. The DP
avg of the oligomer is calculated at 5.5. This is a slight overestimation (DP 1,2 and 3 are missing) caused by the lack of MALDI-ToF peaks below 700 m/z that could not be distinguished from oligomer fragments and noise.
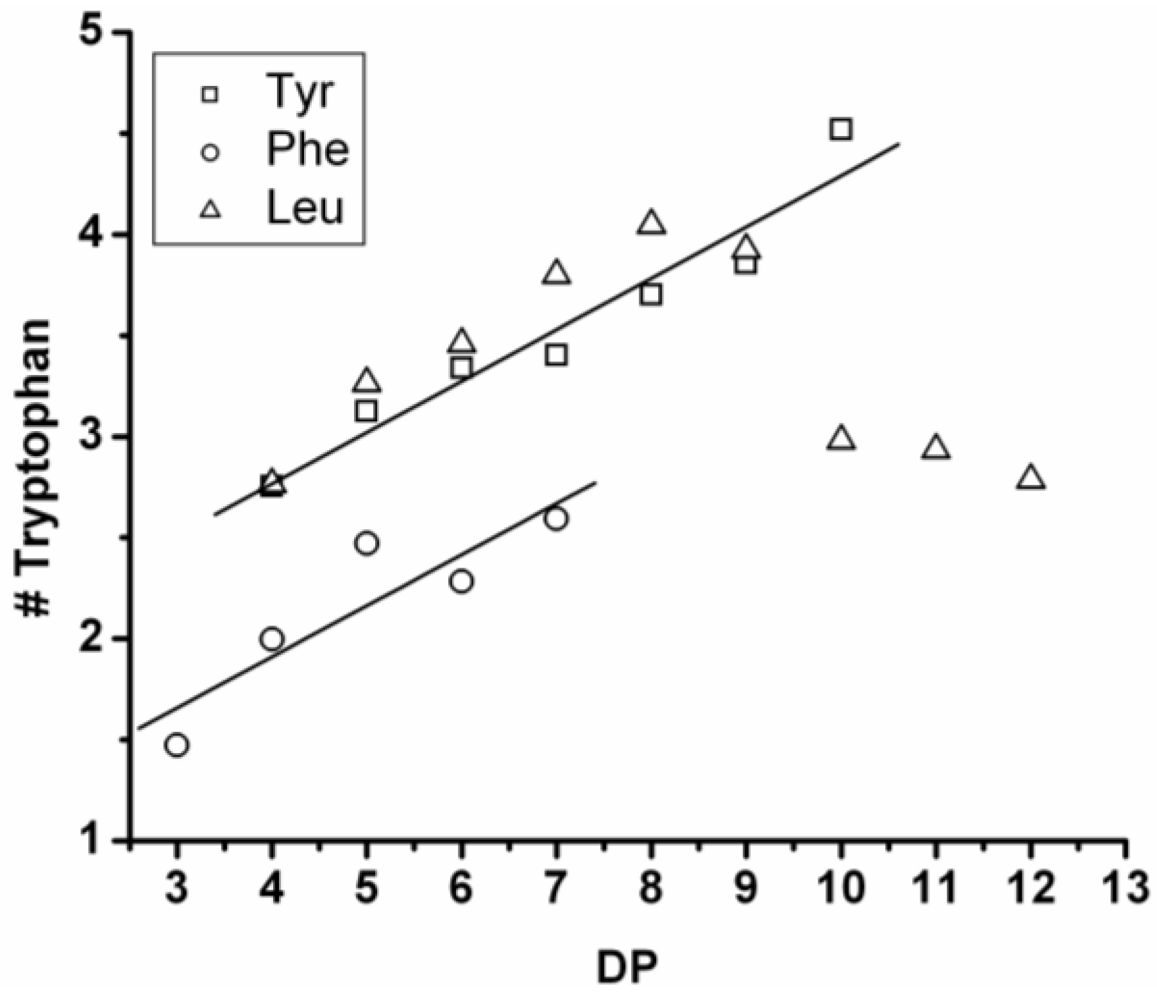
In
Figure 9 the average amount of tyrosine and tryptophan are plotted. The amount of tyrosine residues in the chains rises by 0.75 per DP and the amount of tryptophan in the chains with 0.25 per DP. Tyrosine is more reactive than tryptophan, which was also the amino acid with the smallest yield and shortest chains in the homo-oligomerization of the amino acids.
Figure 8.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of the poly(Tyr-co-Trp).
Figure 8.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of the poly(Tyr-co-Trp).
Figure 9.
Average number of amino acids in poly(Tyr-co-Trp) for every observed DP.
Figure 9.
Average number of amino acids in poly(Tyr-co-Trp) for every observed DP.
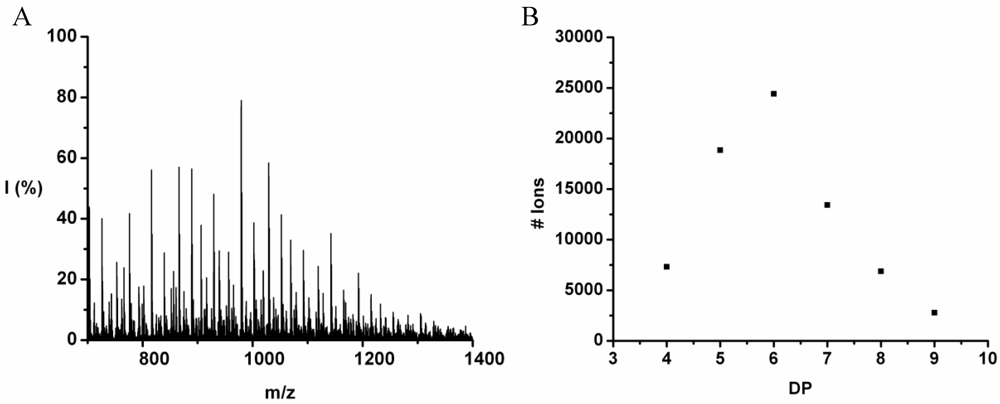
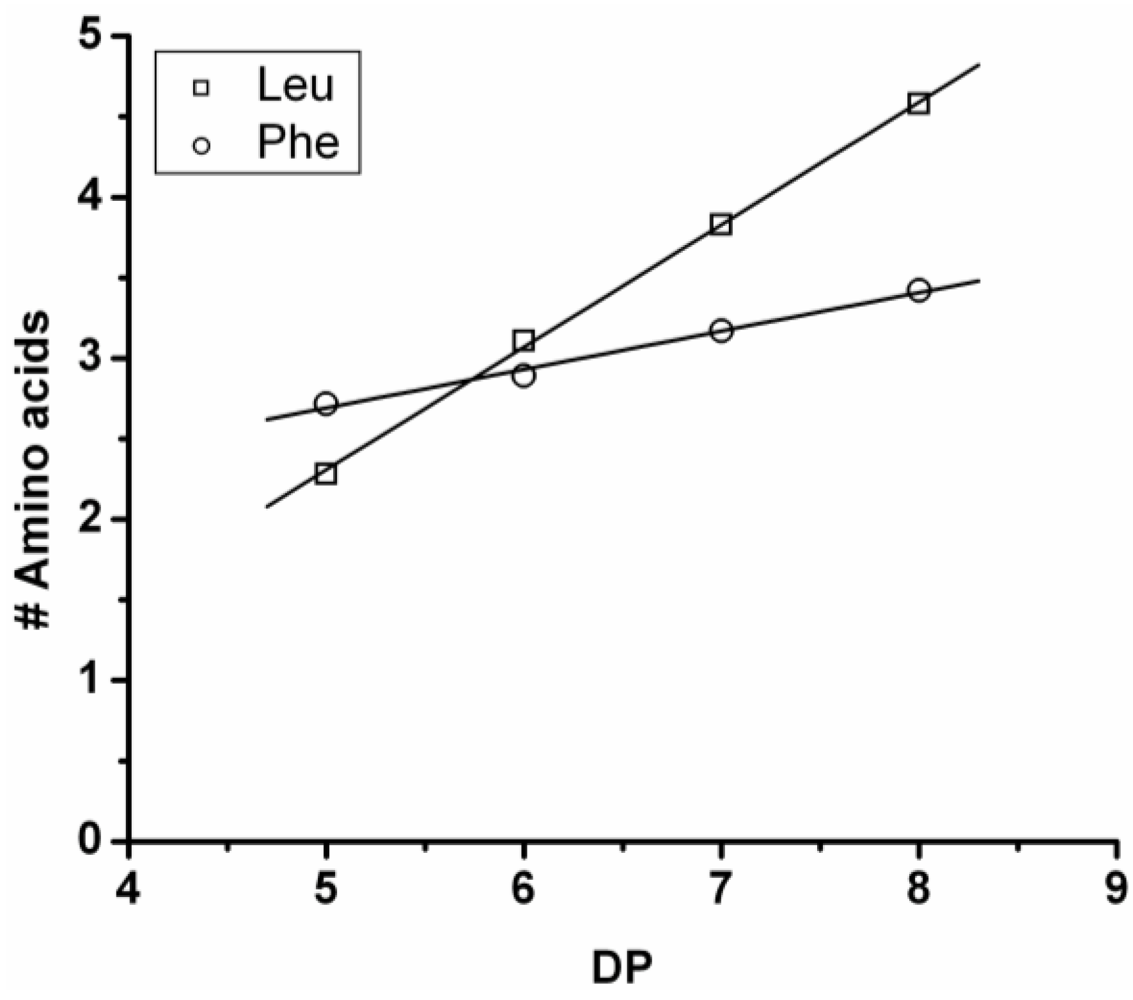
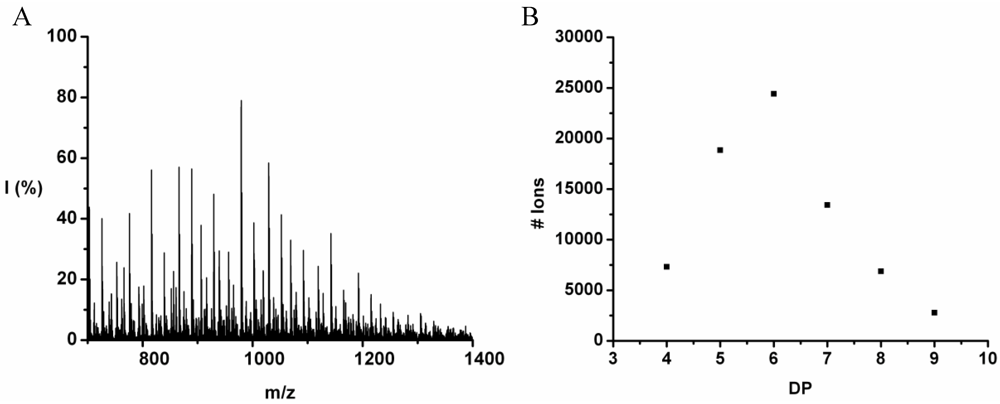
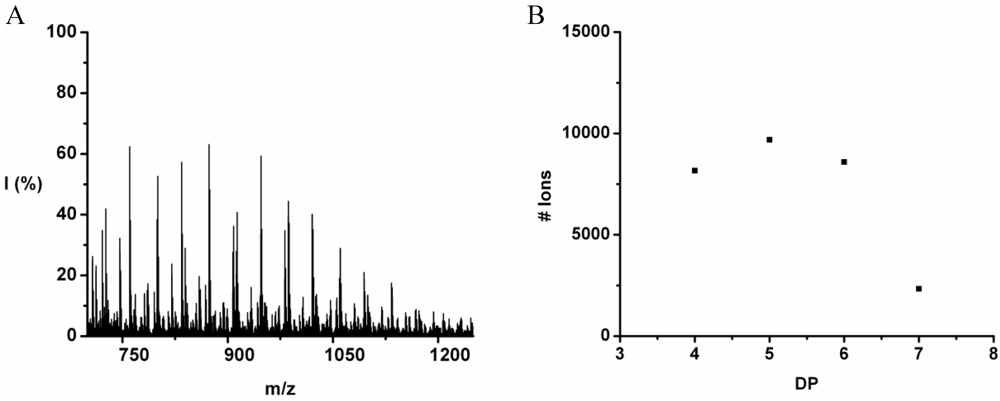
Poly(Leucine-Co-Phenylalanine)
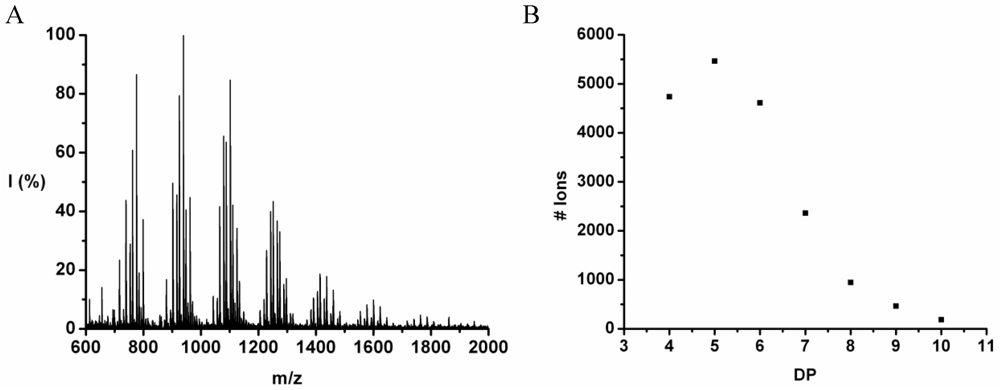
Leucine and phenylalanine were co-oligomerized in 52% yield. The MALDI ToF MS spectrum of this co-oligomer is shown in
Figure 10A. The peaks with m/z values 753–1027 were analyzed and could be assigned to chains with a DP 5–9. The most abundant ion is the 948 m/z, 4_3, [M-Na]
+ ester terminated molecular ion.
An ion of a chain with a DP 9 was found at 1276 m/z belonging to the 3_6 [M-Na]+ ester terminated molecular ion. However, not enough data points of the 9-mer could be found to include these in the discussion.
The chain length distribution is plotted in
Figure 10B and shows a maximum between at DP 7. A DP
avg of 6.4 was calculated from the ion counts as described before.
In
Figure 11 the average amount of each amino acid per DP is plotted. The chains grow by adding more leucine (slope 0.76) than phenylalanine (slope 0.24). Leucine is therefore more reactive than phenylalanine.
Figure 10.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of the poly(Leu-co-Phe).
Figure 10.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of the poly(Leu-co-Phe).
Figure 11.
Average amount of amino acids in poly(Leu-co-Phe) for every observed DP.
Figure 11.
Average amount of amino acids in poly(Leu-co-Phe) for every observed DP.
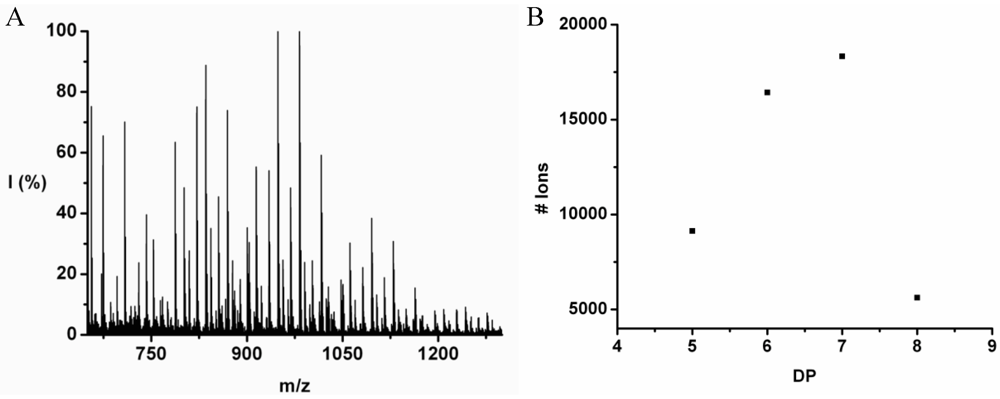
Poly(Tryptophan-Co-Phenylalanine)
Poly(Trp-co-Phe) was obtained in 35% yield. The MALDI-ToF MS spectrum of this co-oligomer is shown in
Figure 12(A). The peaks with m/z values 499–1201 were assigned to chains with DP 4–7. The only chains that were found with DP 7 have a composition 5_2 and 4_3. In
Figure 12(B) the distribution of the chain lengths in the sample is plotted with a maximum DP 4 with a calculated DP
avg 4.4.
The DP rises by adding on average 0.25 tryptophan and 0.90 phenylalanine, see
Figure 13. The slopes add up to more than 1 amino acid residue per DP. This is not possible since chains are elongated with only one amino acid at a time. It is probably the result of a linear fit through all the data points while the values at DP 3 are too low for both amino acids. Phenylalanine is more reactive than tryptophan in this combination.
Figure 12.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of poly(Trp-co-Phe).
Figure 12.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of poly(Trp-co-Phe).
Figure 13.
Average amount of amino acids in poly(Trp-co-Phe) for every observed DP.
Figure 13.
Average amount of amino acids in poly(Trp-co-Phe) for every observed DP.
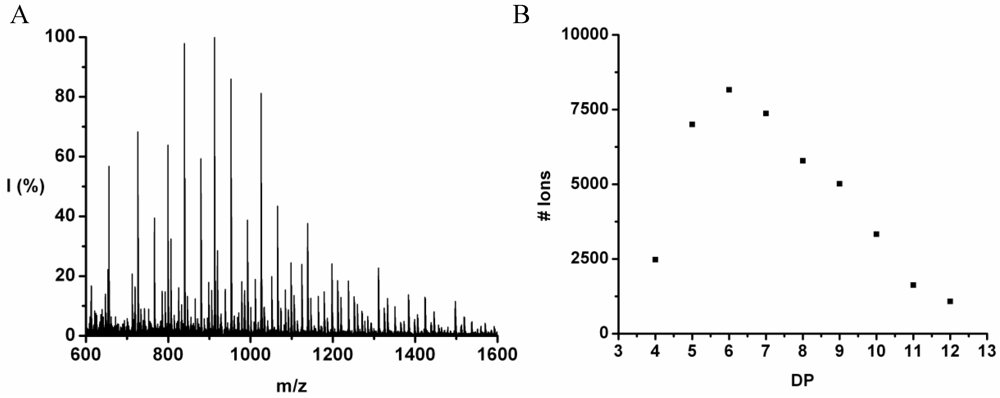
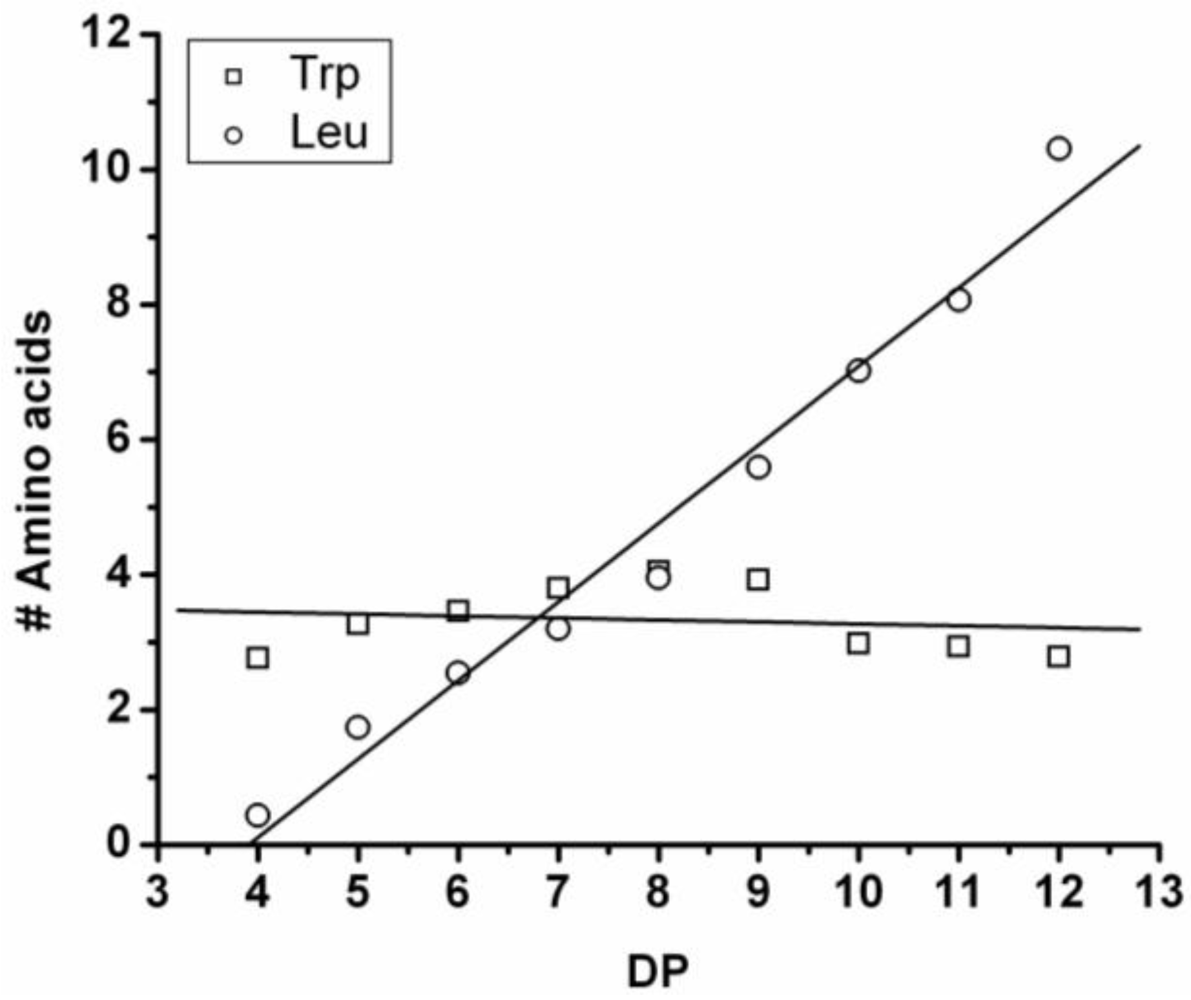
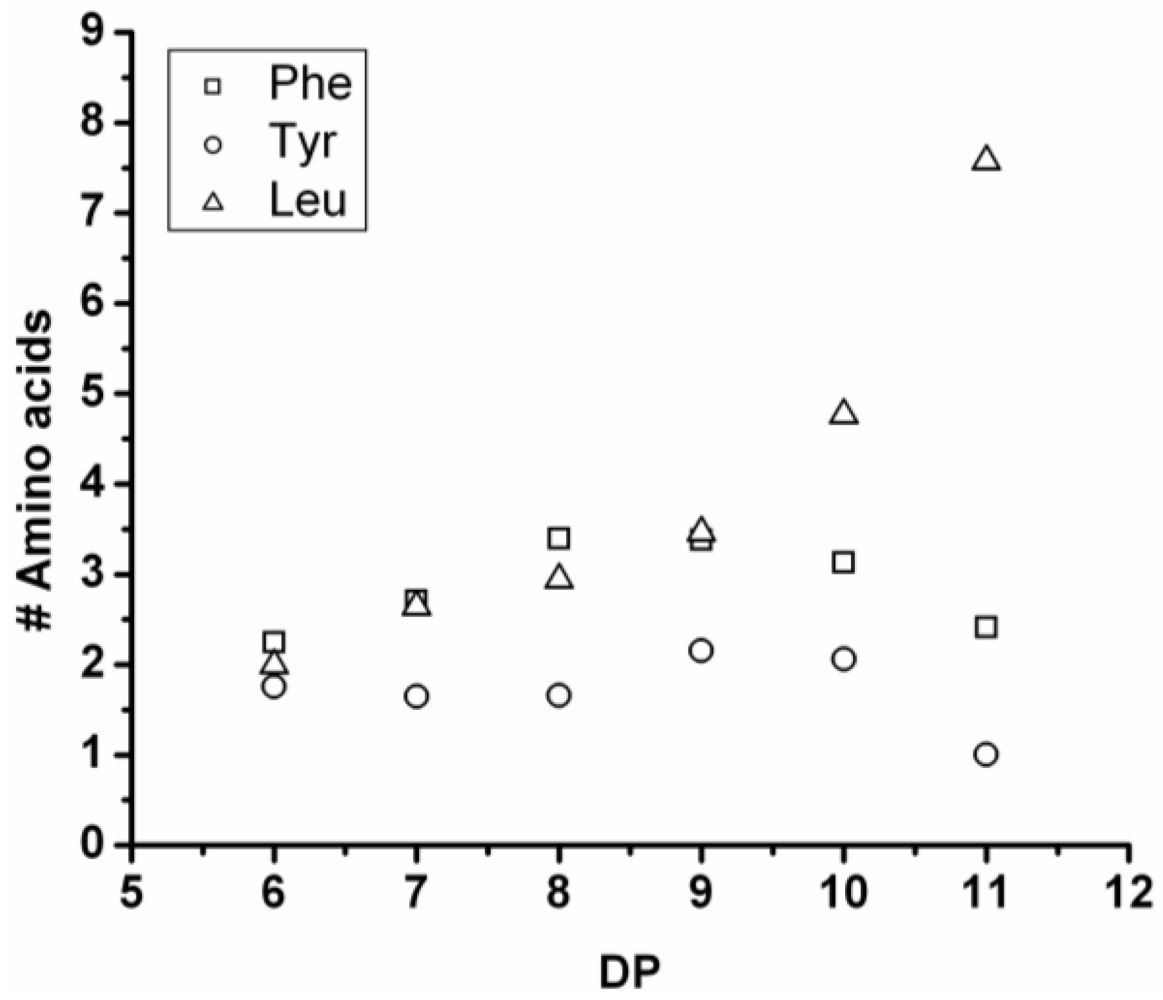
Poly(Tryptophan-Co-Leucine)
Poly(Trp-co-Leu) was obtained in 60% yield. The MALDI-ToF MS spectrum of this co-oligomer is shown in
Figure 14(A). The peaks with m/z values of 704–1745 were assigned to chains with a DP of 4–13. The most abundant ion is the 1025 m/z, 2_4, [M-Na]
+ ester terminated molecular ion. In the high-end tail of the distribution the 1744 m/z, 10_3 [M-Na]
+ ester terminated molecular ion can be found but it is only one of the possible 13-mer ions and therefore not included in the discussion. The chain length distribution (
Figure 14(B)) shows a maximum at DP 7 in good agreement with the calculated the DP
avg of 7.2.
The average amount of each amino acid was calculated and plotted against DP in
Figure 15. The amount of leucine rises with DP and as the chains grow after DP 8 the amount of Trp in the chains drops. One explanation could be that the amount of Trp decreases after some time when the chains redistribute by transamidation or hydrolysis reactions. Leucine has a higher reactivity than tryptophan.
Figure 14.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of the poly(Trp-co-Leu).
Figure 14.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of the poly(Trp-co-Leu).
Figure 15.
Average number of amino acids in poly(Trp-co-Leu) for every observed DP.
Figure 15.
Average number of amino acids in poly(Trp-co-Leu) for every observed DP.
3.3. Reactivities of the Amino Acids in Binary Co-Oligomerization
In the four graphs below, the number of Tyr, Phe, Leu or Trp in combination with the other amino acids per degree of oligomerization are presented. A linear fit through the data points shows how much of each amino acid is added when the chains grow by one amino acid residue. From the slopes a reactivity order is derived that concludes this paragraph.
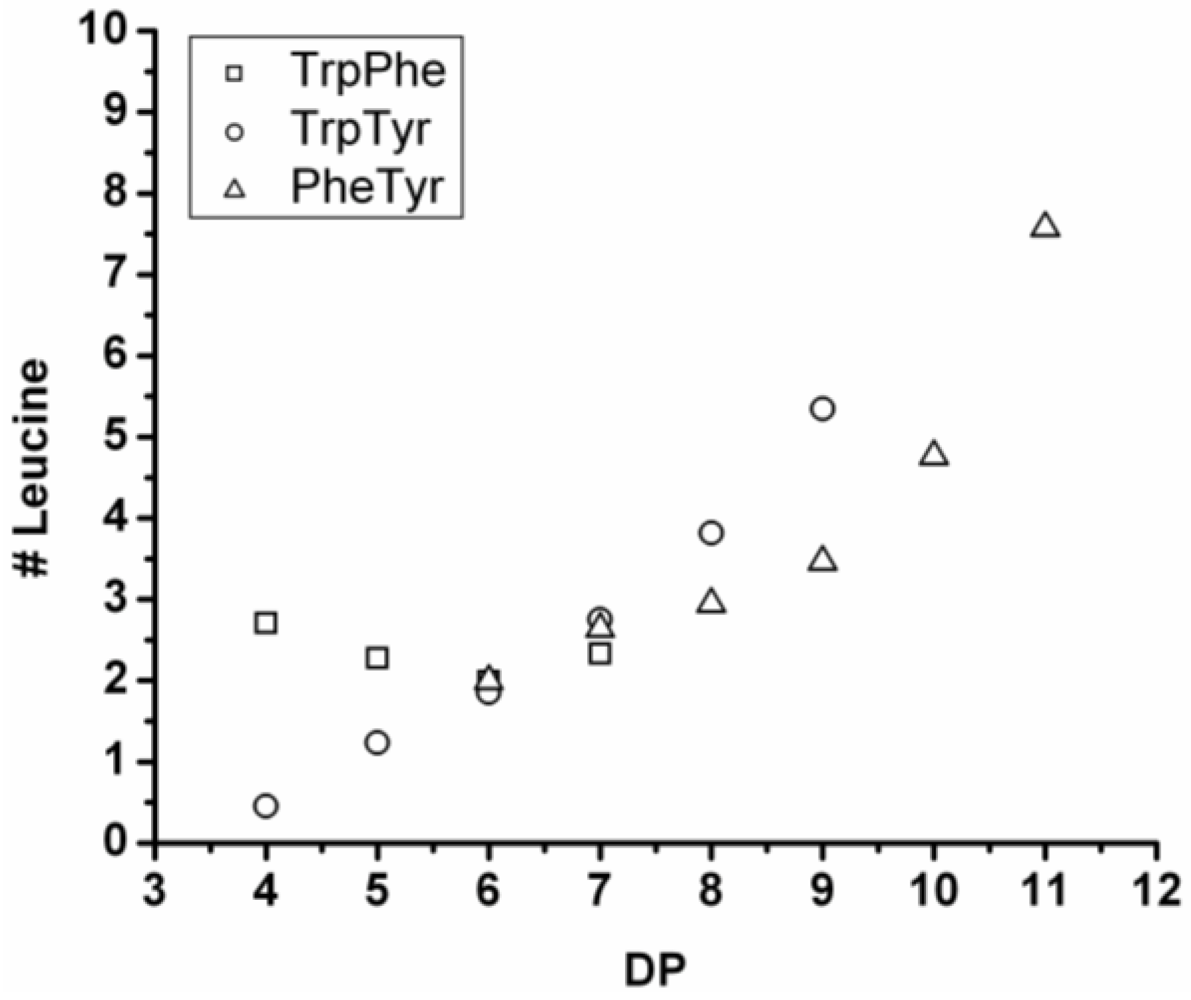
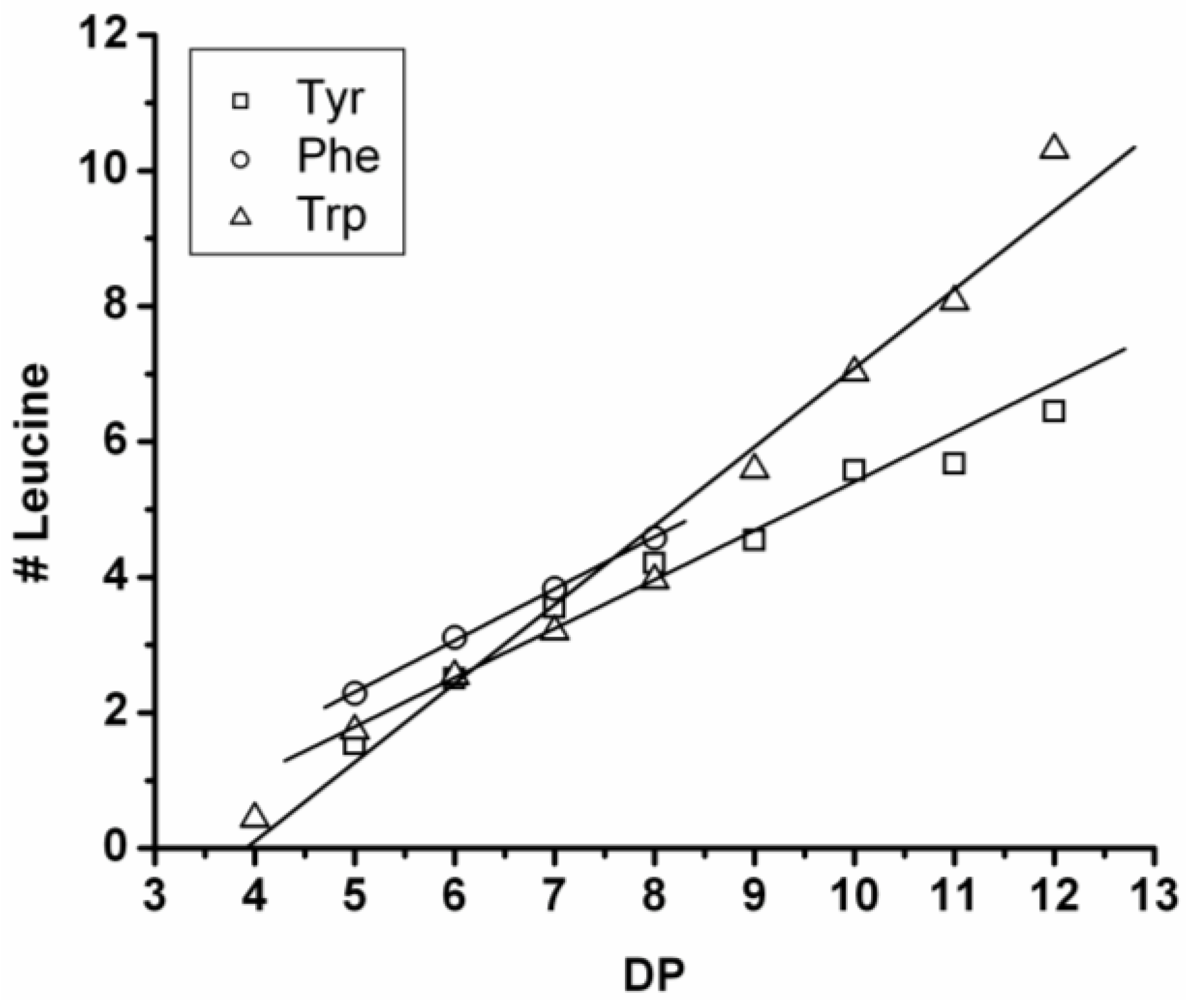
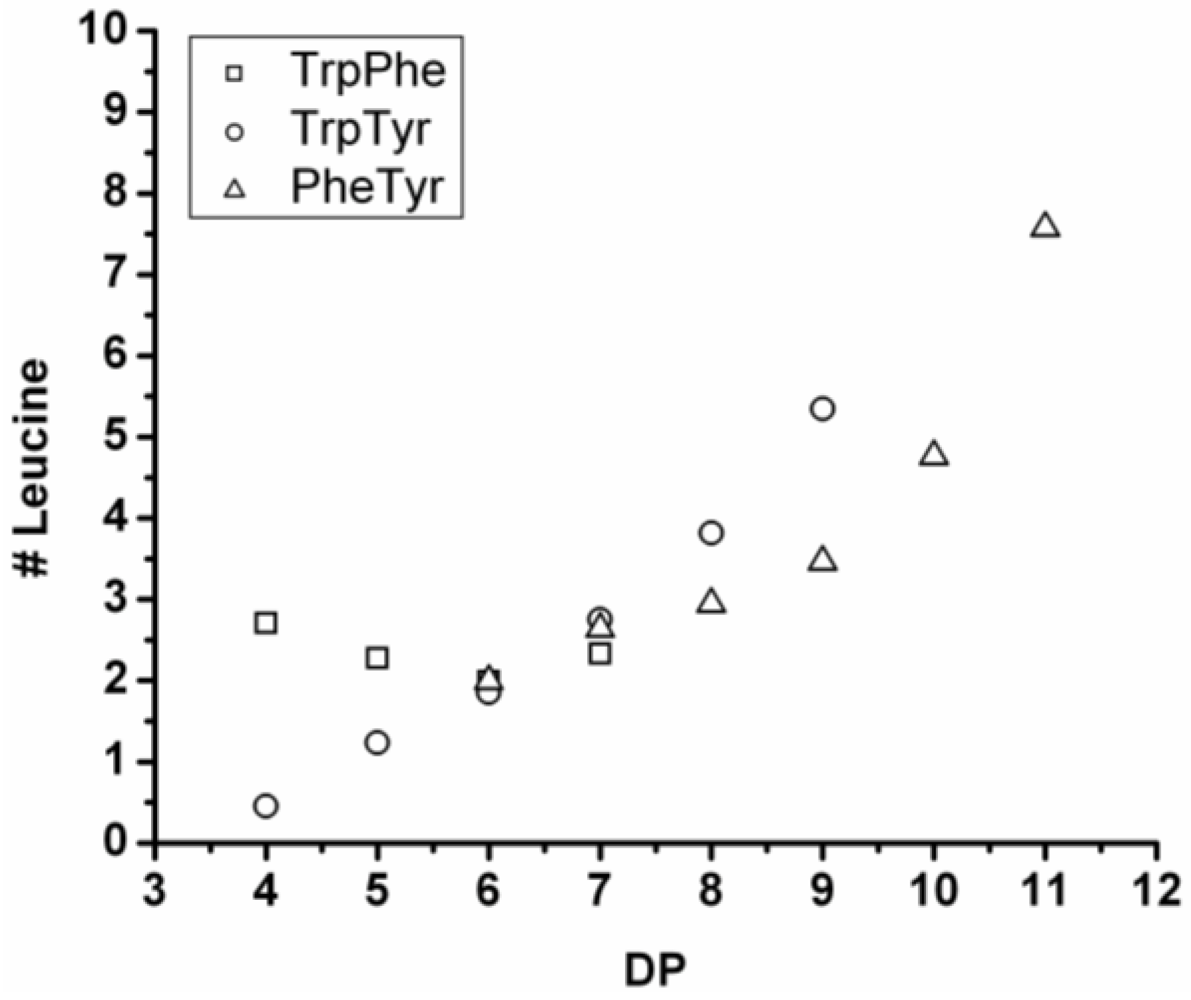
3.3.1. Reactivity of Leucine in Co-Oligomerization with Tyr, Phe or Trp
Figure 16 shows the average amount of leucine in chains obtained when leucine is co-oligomerized with tyrosine, phenylalanine or tryptophan. It can be concluded that leucine is the amino acid that is the most reactive of the four amino acids. This is shown in two ways. First, in all cases the slope of the amount of leucine with increasing DP is higher than 0.5. The slopes are 0.72 in combination with tyrosine and 0.76 in combination with phenylalanine. The slope is 1.16 in combination with tryptophan which can be explained by redistribution reactions and hydrolysis. Second, co-oligomers with leucine always show a higher DP
avg than the homo-oligomers.
Figure 16.
Average number of leucine residues in co-oligomers with Tyr, Phe or Trp.
Figure 16.
Average number of leucine residues in co-oligomers with Tyr, Phe or Trp.
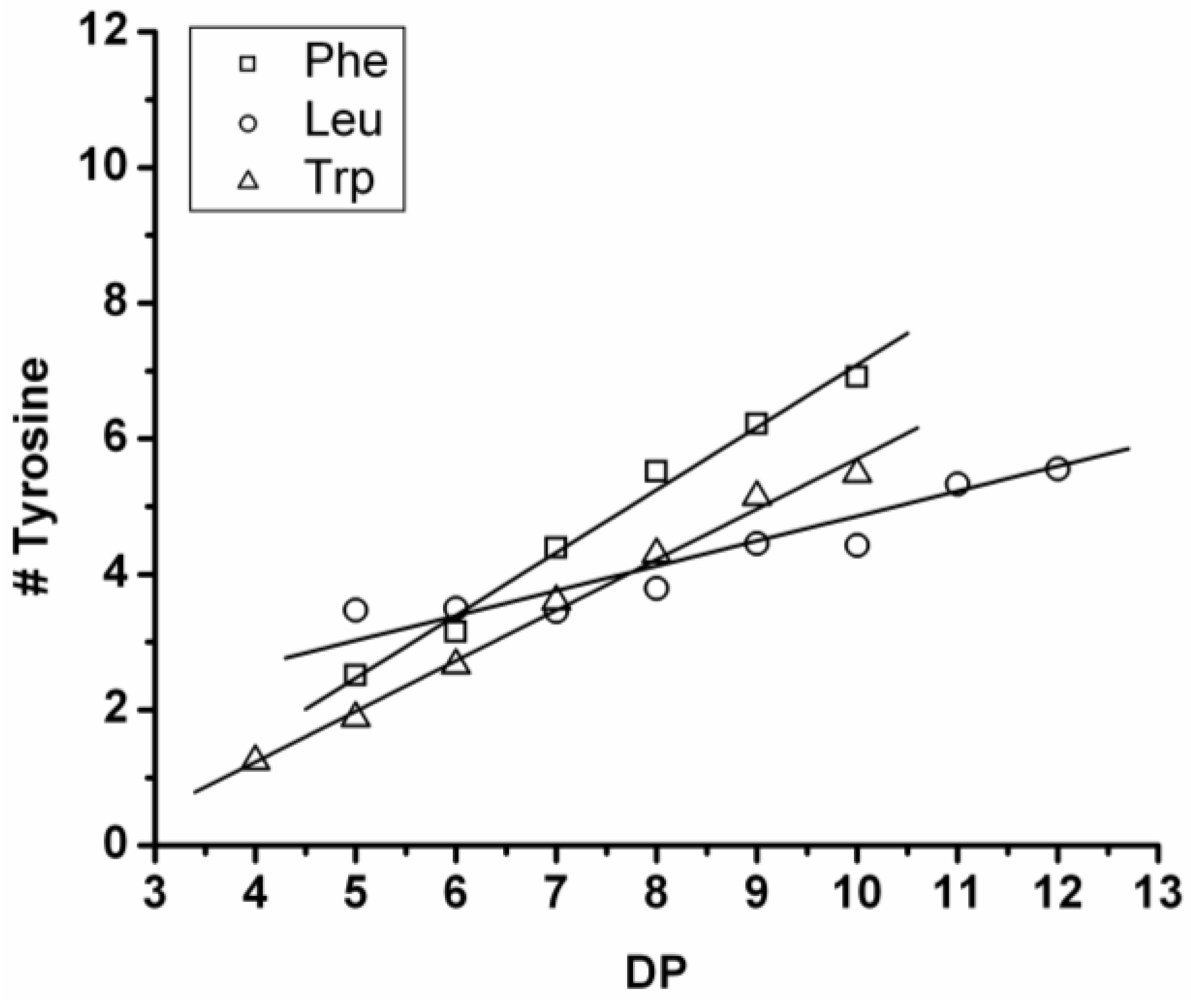
3.3.2. Reactivity of Tyrosine in Co-Oligomerization with Phe, Leu or Trp
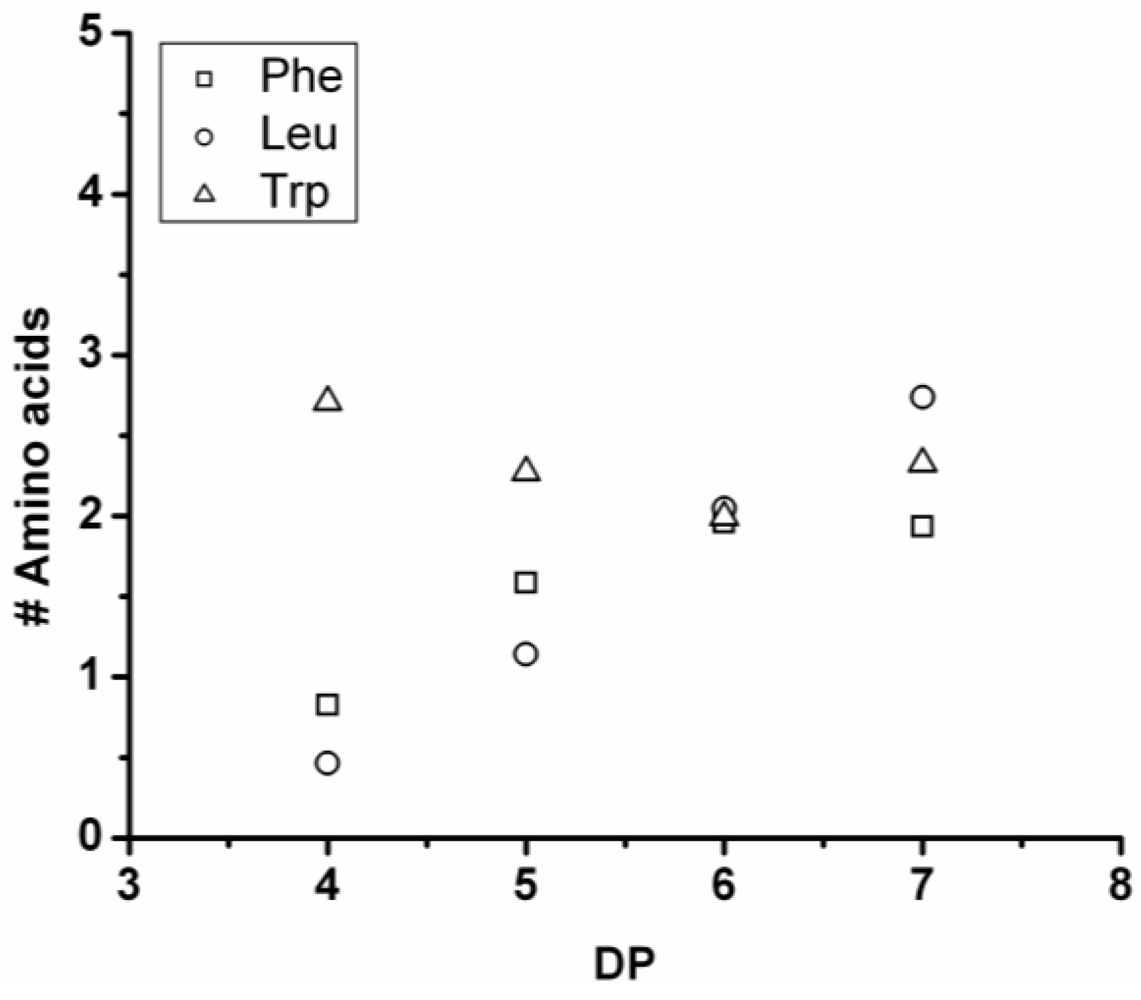
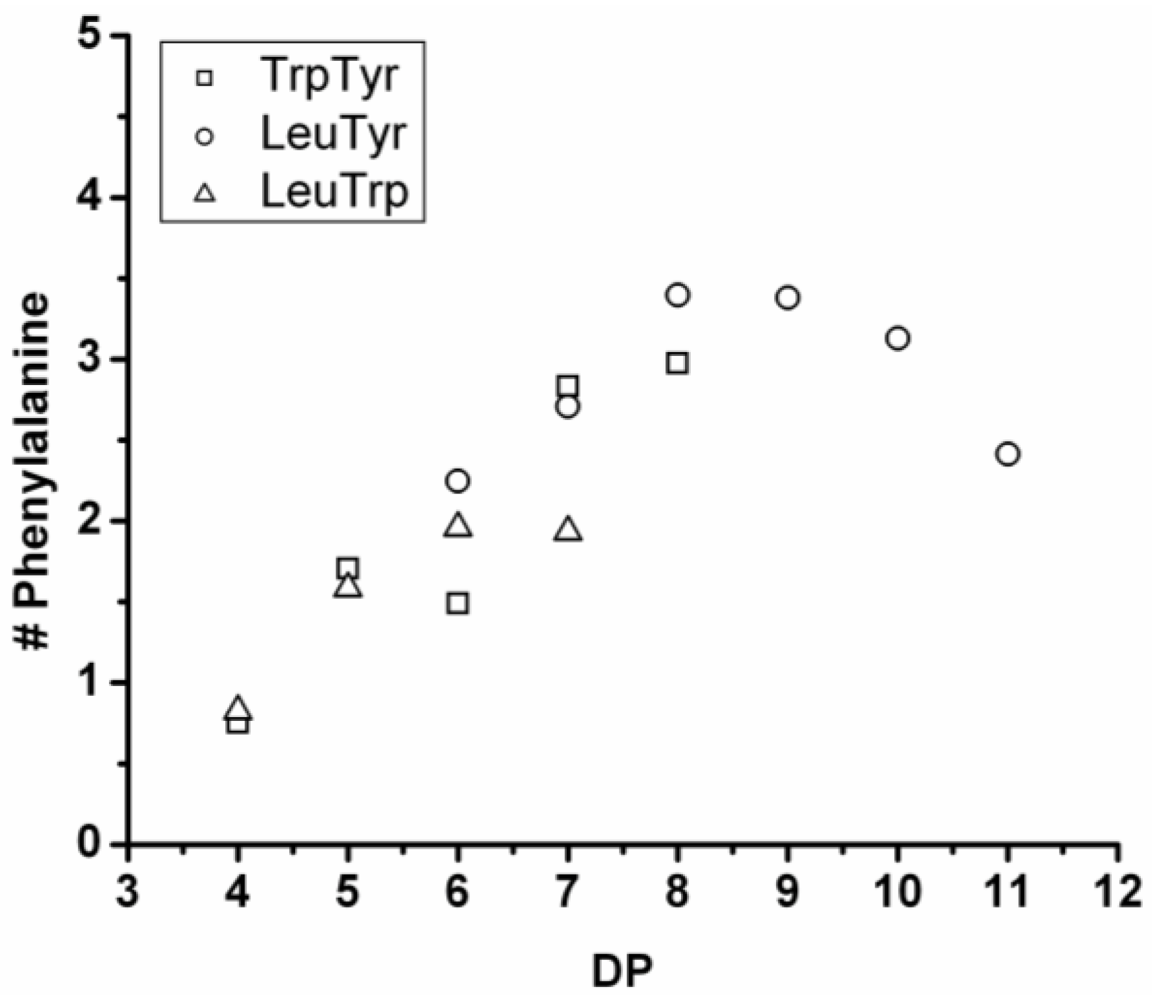
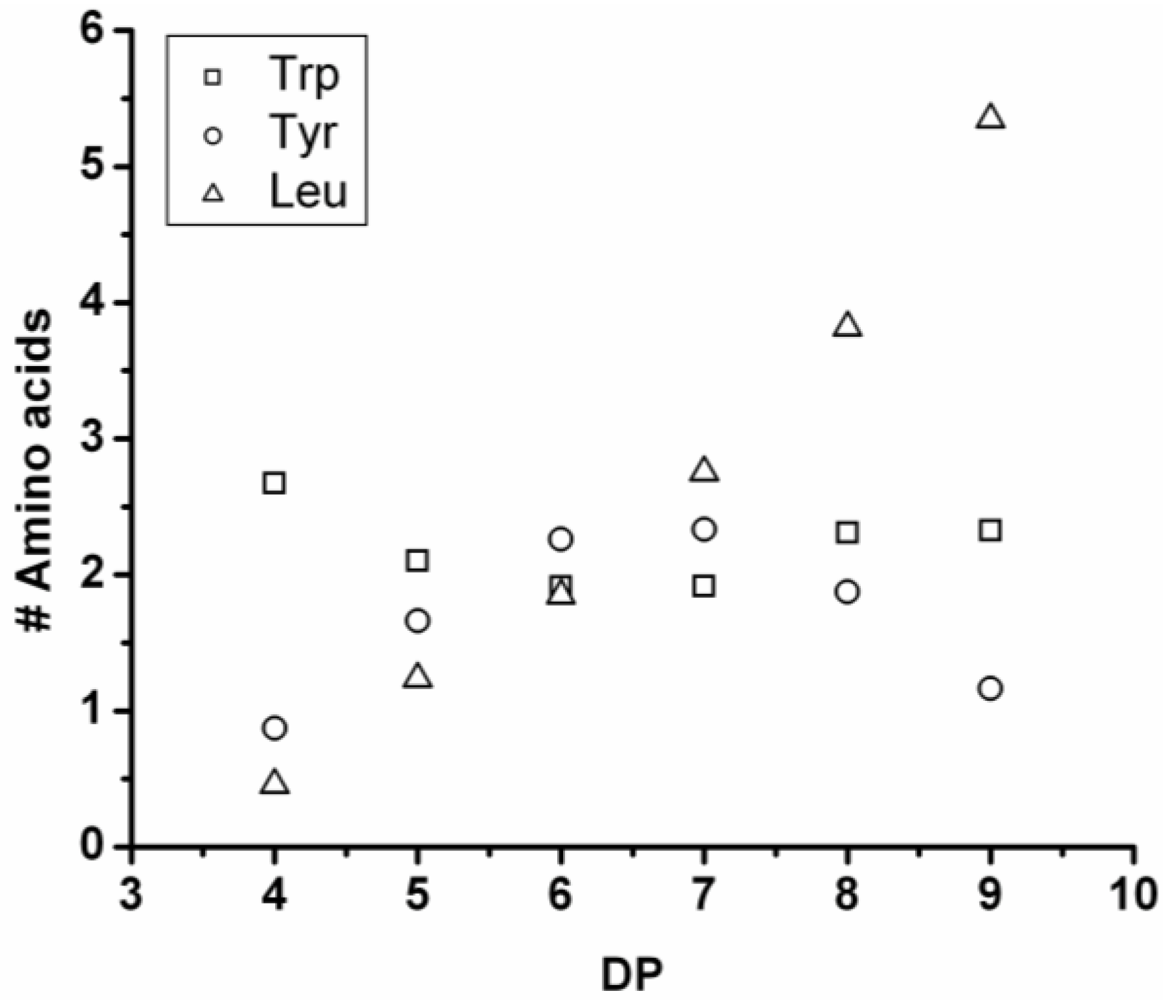
Figure 17 shows the average amount of tyrosine in chains obtained when it is co-oligomerized with Phe, Leu or Tryp. The chains grow by the addition of 0.92 tyrosine residues in combination with Phe, 0.75 with Trp and 0.37 with Leu. From the slopes it can be concluded that Tyr is preferred over Phe and Trp while it is less reactive than Leu.
Figure 17.
Average number of tyrosine residues in co-oligomers with Phe, Leu or Trp.
Figure 17.
Average number of tyrosine residues in co-oligomers with Phe, Leu or Trp.
Compared to the homoligomerizations of Leu, Phe and Trp the binary co-oligomers with tyrosine always have a higher DPavg. However this effect of Leu is stronger. Tyrosine is therefore the second‑ most reactive amino acid.
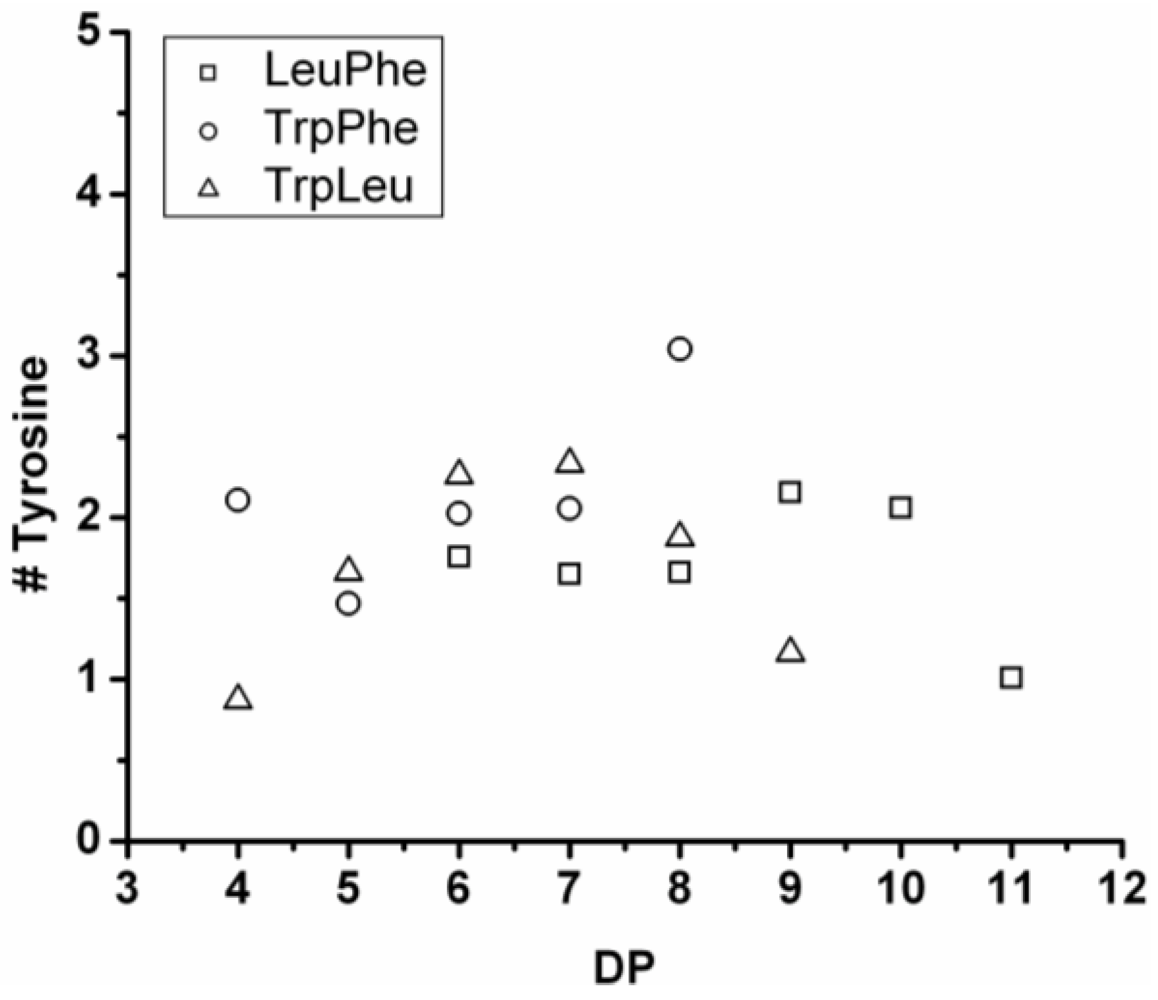
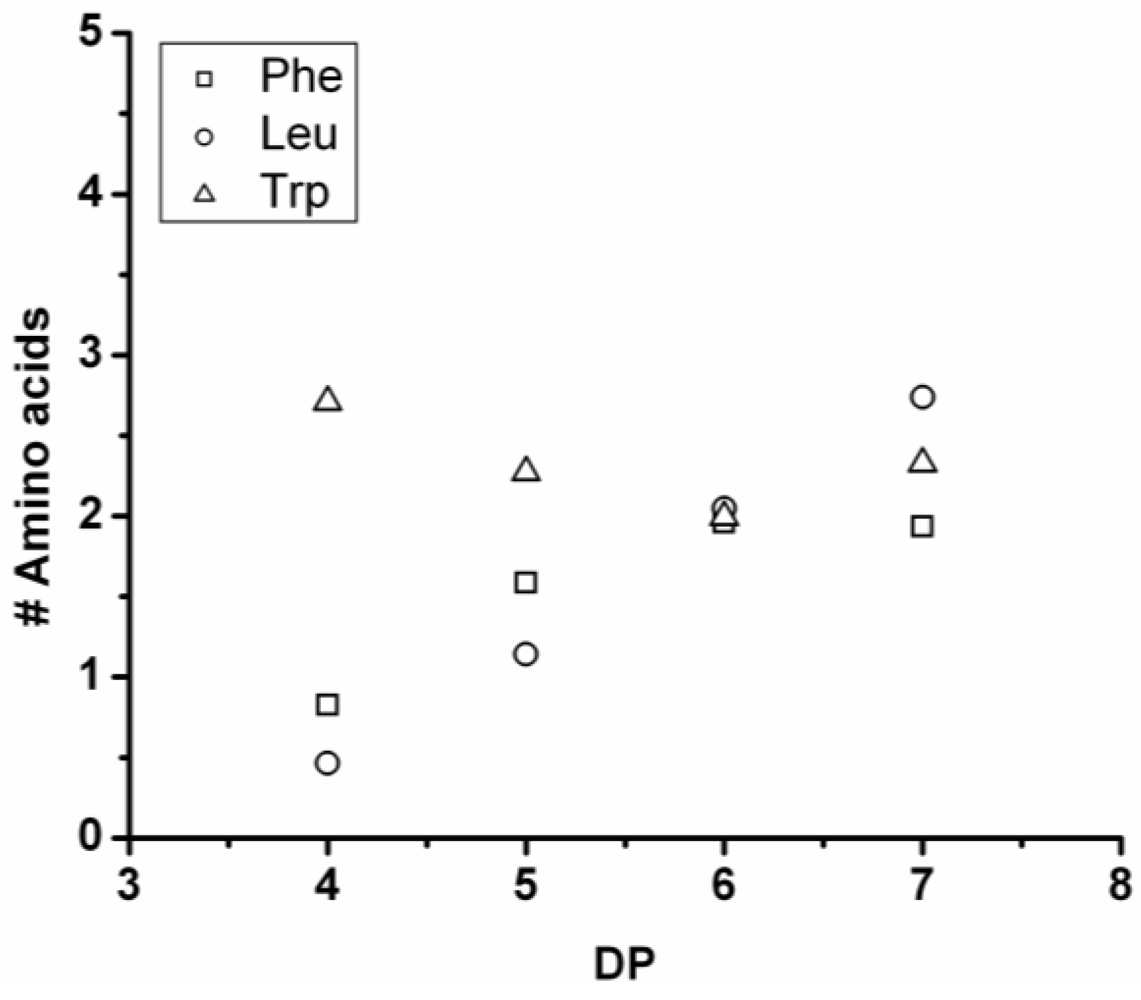
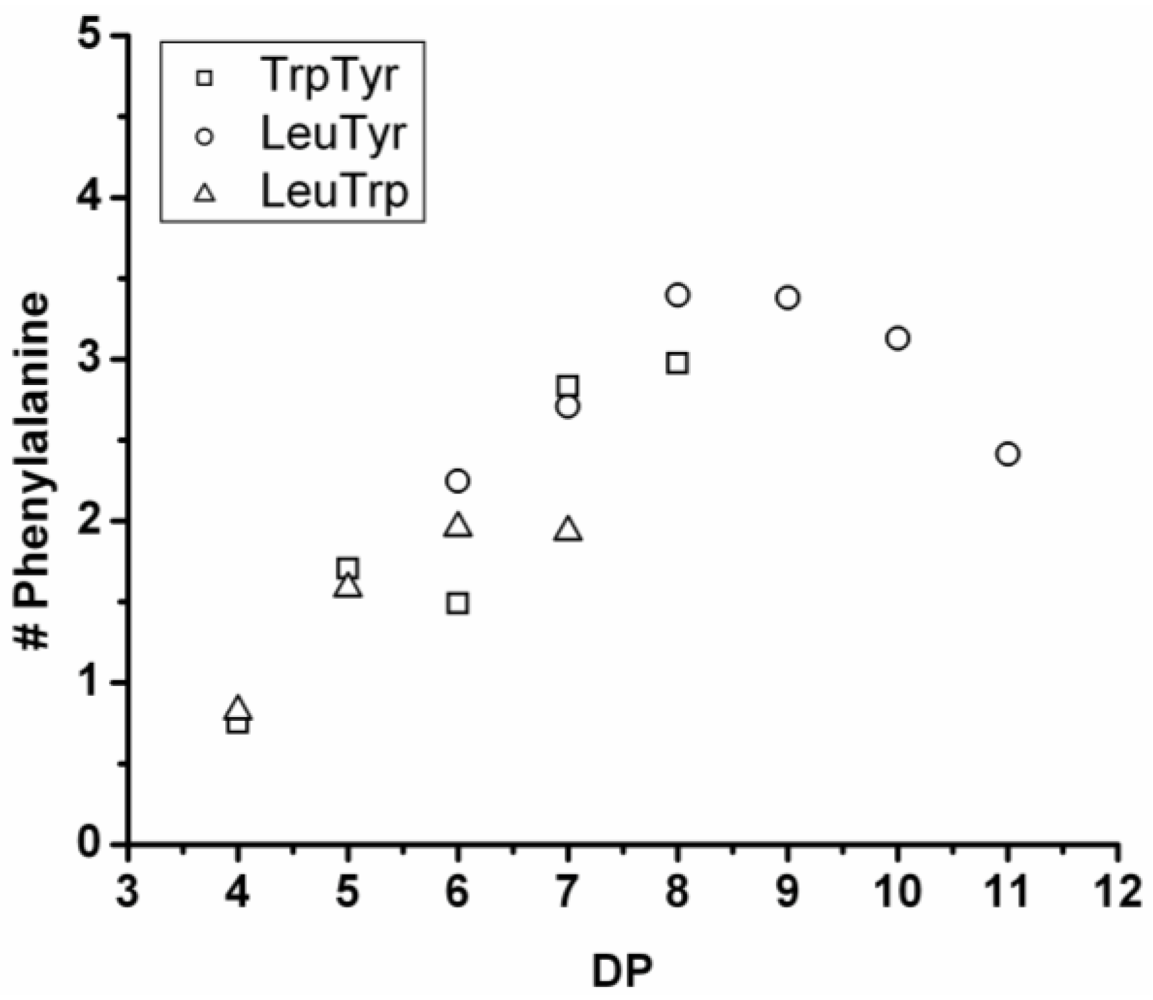
3.3.3. Reactivity of Phenylalanine in Co-Oligomerization with Tyr, Leu or Trp
Figure 18 shows the average amount of phenylalanine in the chains obtained when phenylalanine is co-oligomerized with Tyr, Leu and Trp. The slope of the lines show that the co-oligomers of phenylalanine grow by adding 0.90 units per DP when combined with Trp, 0.24 with Leu and 0.08 with Tyr. Phenylalanine is more reactive than Trp but less than Tyr and Leu. Compared to the homo-oligomers phenylalanine does not really influence the DP
avg. In conclusion, phenylalanine is the third-most reactive amino acid in the synthesis of binary co-oligomers.
Figure 18.
Average number of phenylalanine residues in co-oligomers with Tyr, Leu or Trp.
Figure 18.
Average number of phenylalanine residues in co-oligomers with Tyr, Leu or Trp.
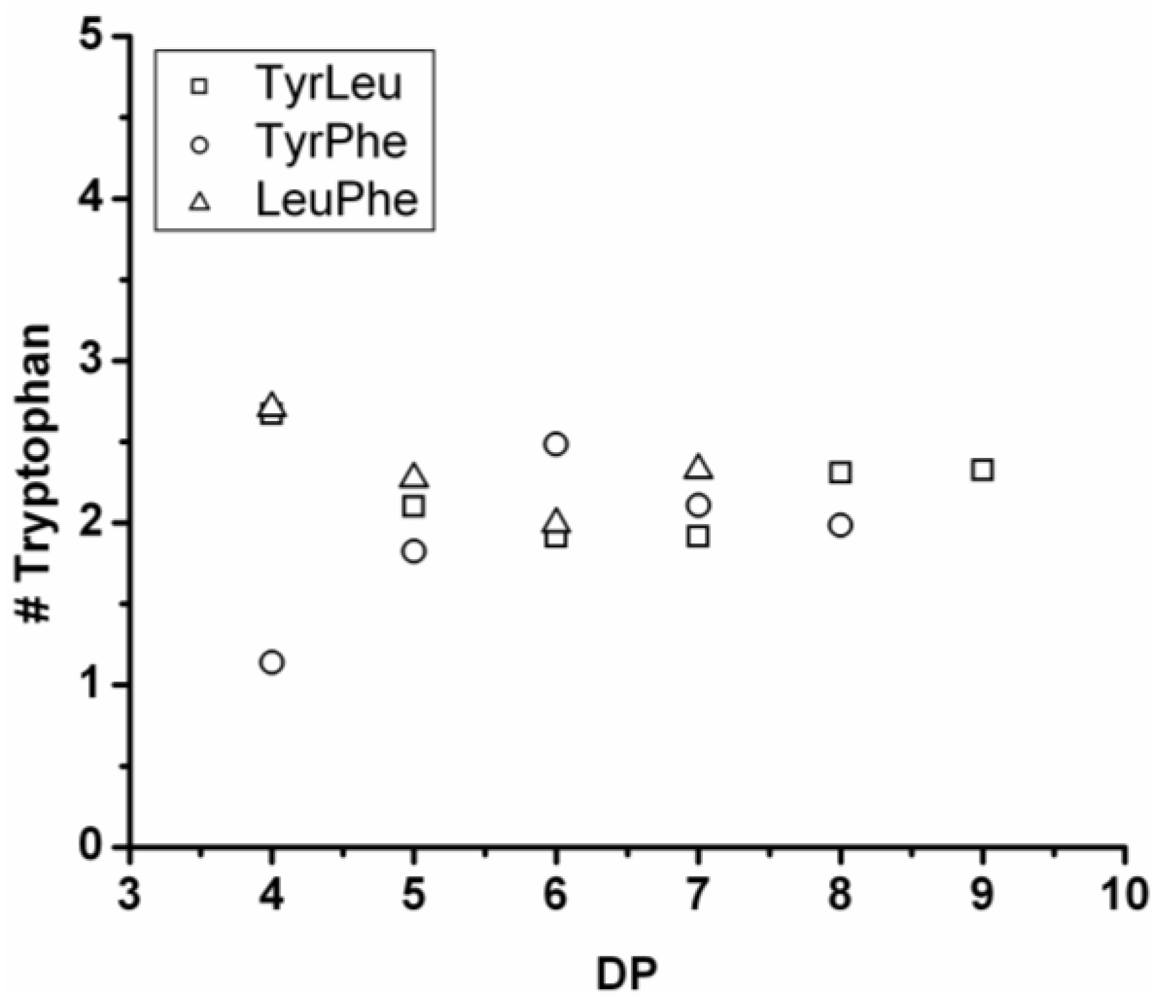
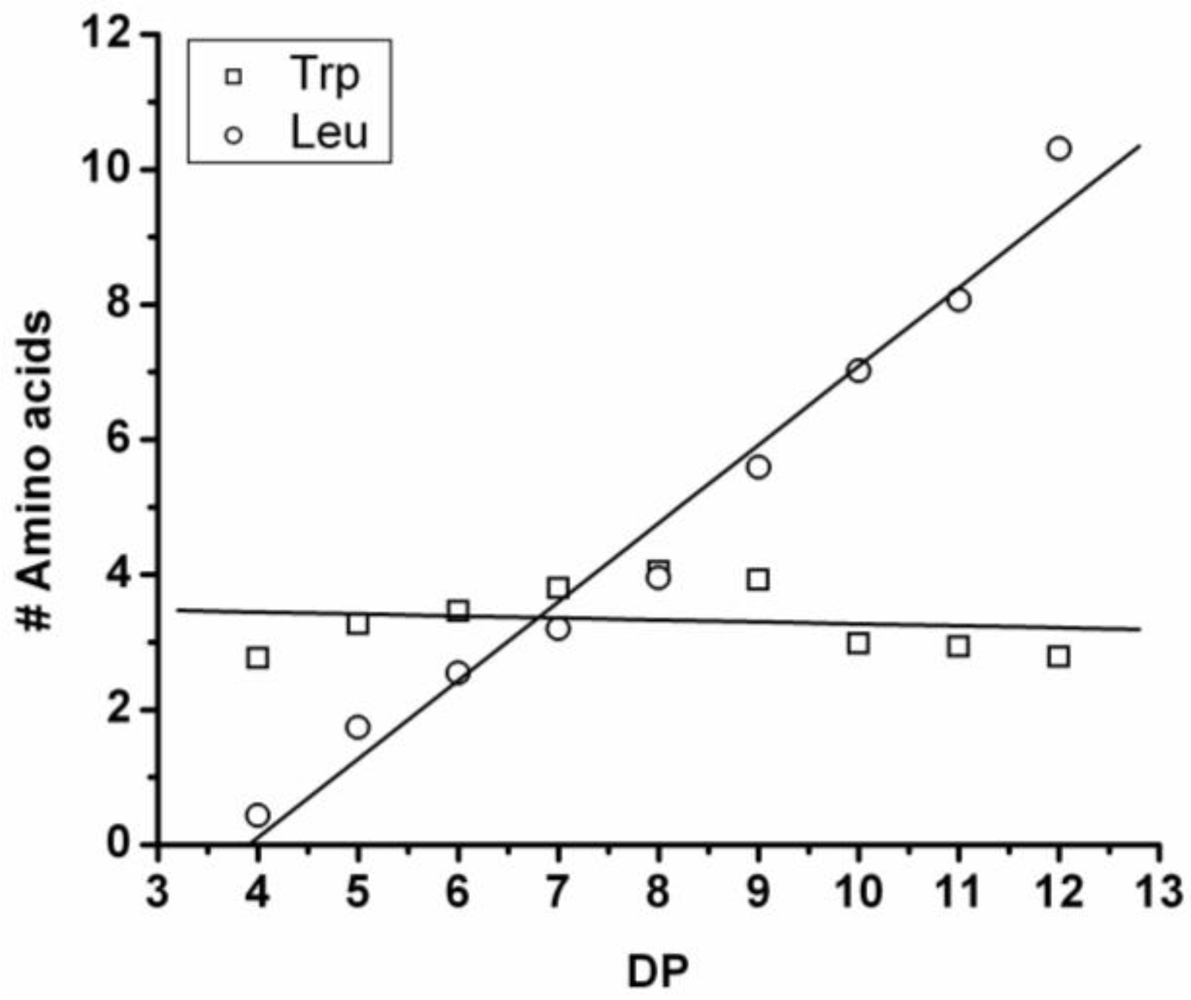
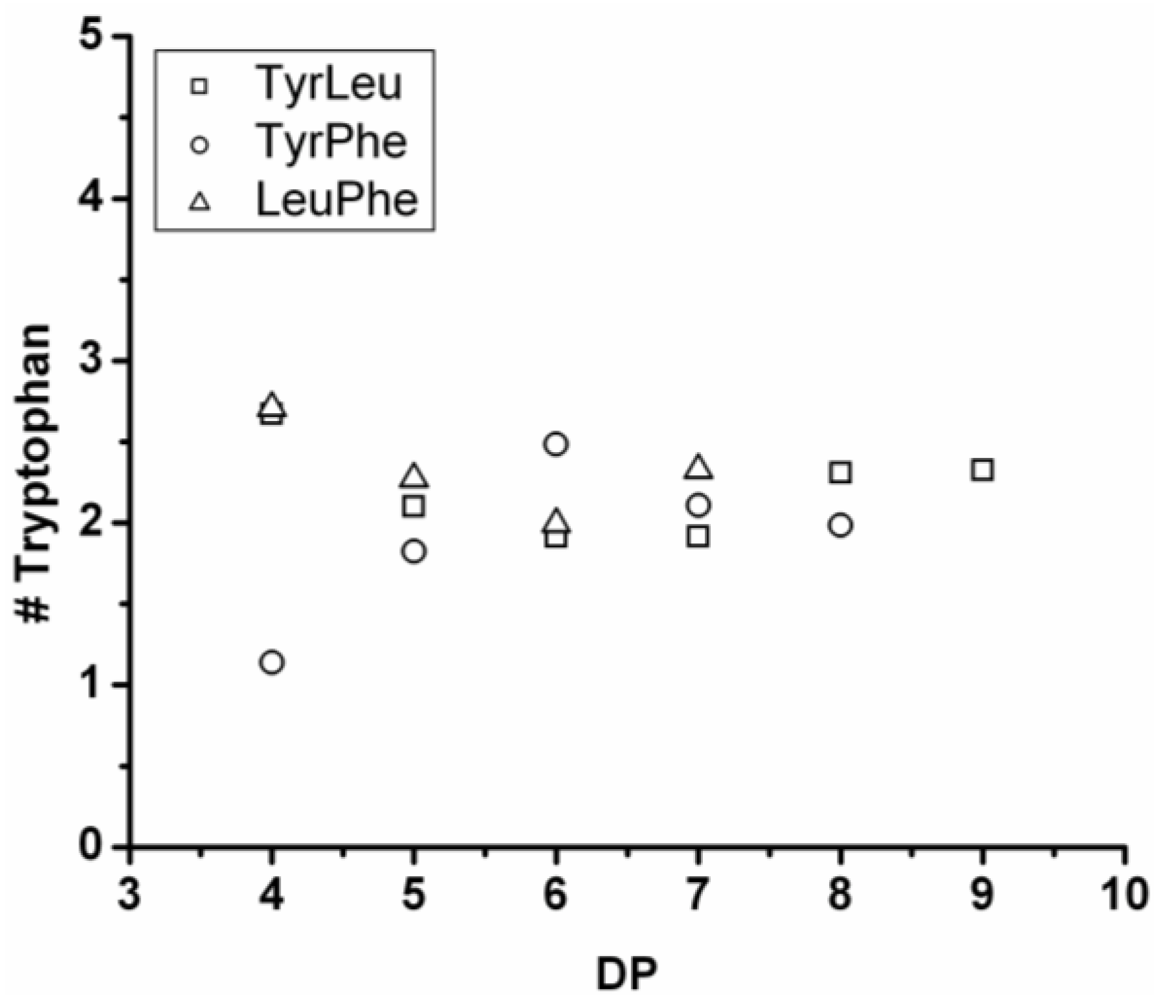
3.3.4. Reactivity of Tryptophan in Co-Oligomerization with Tyr, Phe or Leu
Figure 19 shows the amount of tryptophan found in the chains after co-oligomerization with Tyr, Phe and Leu. In the homo-oligomerization tryptophan was oligomerized in the lowest yield and with the lowest DP
avg. This is also found in combination with other amino acids. The chains grow by adding on average 0.25 Trp units per DP in combination with Tyr and Phe. In combination with leucine the number of tryptophan residues is decreasing for chains with DP 9–12. The decreasing amount of tryptophan can be explained with chain elongation by transamidation or by partial hydrolysis of the chains. From the slopes it is concluded that in all co-oligomerizations the other amino acid is more reactive.
Figure 19.
Average number of tryptophan residues in co-oligomers with Tyr, Phe or Leu.
Figure 19.
Average number of tryptophan residues in co-oligomers with Tyr, Phe or Leu.
The reactivity of the amino acids in the co-oligomerization of two amino acids can be summarized as Leu > Tyr > Phe > Trp. This result is almost in accordance with the reactivity series found for the homo-oligomerizations. Leucine and tyrosine are very close in reactivity because they reach a comparable DPavg in the synthesis of homo-oligomers. The co-oligomers with leucine are probably better soluble and therefore reach a higher DPavg.
3.4. Composition of Ternary Co-Oligo Amino Acids
In
Table 5 the DP
avg of the ternary co-oligo amino acids and the composition at DP
avg are listed. Unlike the co-oligomerization with two amino acids, the chains do not represent the composition of a random co-oligomer. The reactivity differences are more pronounced in these co-oligomers. Below the composition of the different co-oligomers and the reactivity differences are discussed in more detail.
Table 5.
Average composition of co-oligomers containing three amino acids.
Table 5.
Average composition of co-oligomers containing three amino acids.
| entry | % homo | Co-oligomer | DPavg | AA1 | AA2 | AA3 |
|---|
| 7 | 9.5 | Phe-Tyr-Trp | 6.0 | 2.13 | 1.88 | 1.99 |
| 8 | 3.5 | Phe-Tyr-Leu | 8.3 | 3.08 | 1.84 | 3.37 |
| 9 | 1.3 | Trp-Tyr-Leu | 6.0 | 2.21 | 1.92 | 1.90 |
| 10 | 5.8 | Phe-Leu-Trp | 5.2 | 1.48 | 1.26 | 2.44 |
3.4.1. Oligo(Phenylalanine-Co-Tyrosine-Co-Tryptophan)
In phosphate buffer this co-oligomer was obtained in 41% yield. The MALDI-ToF MS spectrum of this co-oligomer is shown in
Figure 20(A). Peaks with m/z values of 656–1445 were assigned to the chains with a DP 4–8 see
Figure 20(B). From the chain length distribution DP
avg is calculated to be 6.0.
Figure 20.
(A) MALDI-ToF mass spectrum of oligo(Phe-co-Tyr-co-Trp) and (B) chain length distribution of oligo(Phe-co-Tyr-co-Trp).
Figure 20.
(A) MALDI-ToF mass spectrum of oligo(Phe-co-Tyr-co-Trp) and (B) chain length distribution of oligo(Phe-co-Tyr-co-Trp).
In
Figure 21 the average amount of the amino acids in this co-oligomer is plotted for each DP. The chains contain progressively more Phe and Tyr. As more redistribution reactions take place the amount of tryptophan drops and again after DP 6. The same reactions cause an increase in the amount of Tyr and Phe. Trp is the least reactive amino acid in the formation of binary co-oligomers and that trend is reflected here as well.
Figure 21.
Average number of amino acids in oligo(Phe-co-Tyr-co-Trp) for every observed DP.
Figure 21.
Average number of amino acids in oligo(Phe-co-Tyr-co-Trp) for every observed DP.
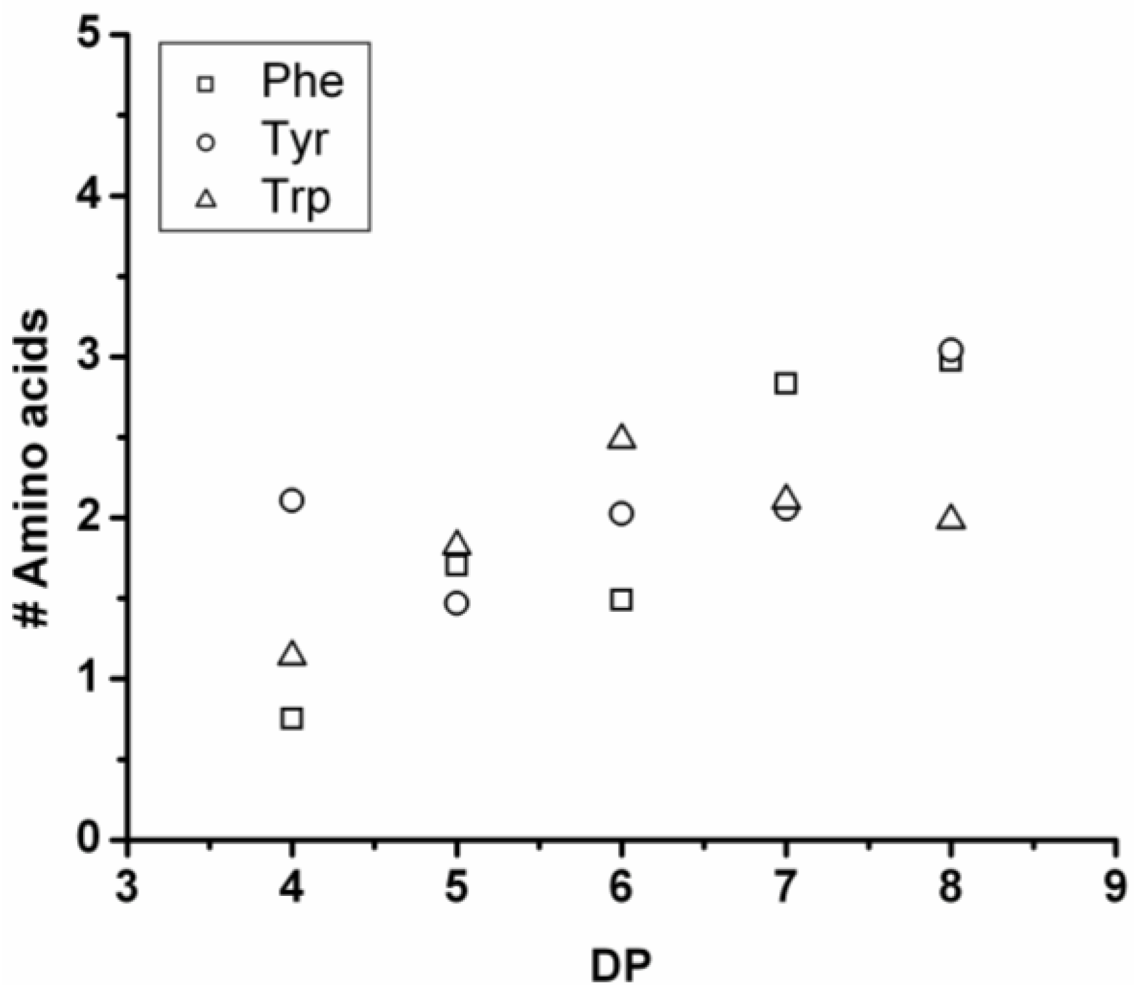
3.4.2. Oligo(Phenylalanine-Co-Tyrosine-Co-Leucine)
The co-oligomer of phenylalanine, tyrosine and leucine was synthesized in 28% yield. The MALDI-ToF MS spectrum of this co-oligomer is shown in
Figure 22(A). The peaks with an m/z value 753–1523 were assigned to chains with a DP 6–11. The chain length distribution of the oligomer was determined and plotted in
Figure 22(B) it shows a maximum ion counts at DP 8. The DP
avg of the co-oligomer was calculated as 8.3.
Figure 22.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of Oligo(Phe-co-Tyr-co-Leu).
Figure 22.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of Oligo(Phe-co-Tyr-co-Leu).
In
Figure 23 the average number of each amino acid in the co-oligomer is plotted for every DP. Leu is the most reactive amino acid in this product, as was also found for the co-oligomers derived from two amino acids. This is because, after DP 9, the amount of Phe and Tyr drop and these amino acids are replaced by leucine, showing an increase in the oligomers with DP 9–12.
Figure 23.
Average number of amino acids in Oligo(Phe-co-Tyr-co-Leu) for every observed DP.
Figure 23.
Average number of amino acids in Oligo(Phe-co-Tyr-co-Leu) for every observed DP.
3.4.3. Oligo(Tryptophan-Co-Tyrosine-Co-Leucine)
The co-oligomer of tyrosine, leucine and tryptophan was obtained in 36% yield. The MALDI-ToF MS spectrum of this co-oligomer is shown in
Figure 24(A). The peaks with m/z values 703–1329 were assigned to chains with DP 4–9 with a distribution as plotted in
Figure 24(B) with a DP
avg 6.0.
Figure 24.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of oligo(Trp-co-Tyr-co-Leu).
Figure 24.
(A) MALDI-ToF mass spectrum and (B) chain length distribution of oligo(Trp-co-Tyr-co-Leu).
For every DP the average number of every containing amino acid was calculated and depicted in
Figure 25. The average amount of leucine rises throughout the distribution with 0.95 residues per DP. Tyrosine shows a maximum number of residues at DP 7 and a decrease for DP 8 and 9. Tryptophan seems to arrive at around 2.3 residues on average per chain. Decreasing amounts of any amino acid can be the result of redistribution reactions like hydrolysis or transamidation.
Figure 25.
Average number of amino acids in oligo(Trp-co-Tyr-co-Leu).
Figure 25.
Average number of amino acids in oligo(Trp-co-Tyr-co-Leu).
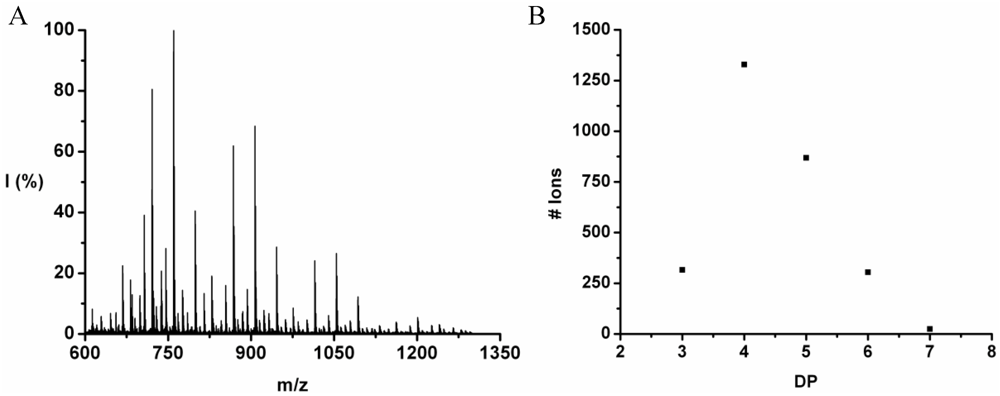
3.4.4. Oligo(Phenylalanine-Co-Leucine-Co-Tryptophan)
Phenylalanine, leucine and tryptophan were ized in 39% yield. The MALDI-ToF MS spectrum of this co-oligomer is shown in
Figure 26(A). Peaks with an m/z value 673–1123 were assigned to chains with DP 4–7 with its distribution depicted in
Figure 26(B). The DP
avg was calculated as 5.2.
Figure 26.
(A) MALDI-ToF mass spectrum of oligo(Phe-co-Leu-co-Trp) and (B) chain length distribution of oligo(Phe-co-Leu-co-Trp).
Figure 26.
(A) MALDI-ToF mass spectrum of oligo(Phe-co-Leu-co-Trp) and (B) chain length distribution of oligo(Phe-co-Leu-co-Trp).
For every DP the average amount of the amino acids is plotted in
Figure 27. The amount of Leu rises in the oligomer with every elongation. The amount of Trp and Phe in the chain arrives at a constant value.
Figure 27.
Average amount of amino acids in oligo(Phe-co-Leu-co-Trp).
Figure 27.
Average amount of amino acids in oligo(Phe-co-Leu-co-Trp).
3.5. Reactivities of the Amino Acids in Ternary Co-Oligomerization
In the four graphs below the number of Leu, Tyr, Phe or Trp in combination with the other amino acids per degree of oligomerization is presented, as already shown for the binary co-oligomers. The plots show how much of each amino acid is added when the chains grow by one amino acid residue (Dp+1). From the average amount of every amino acid and the effect of adding an amino acid to the binary co-oligomer on the DPavg, an order of reactivity was determined.
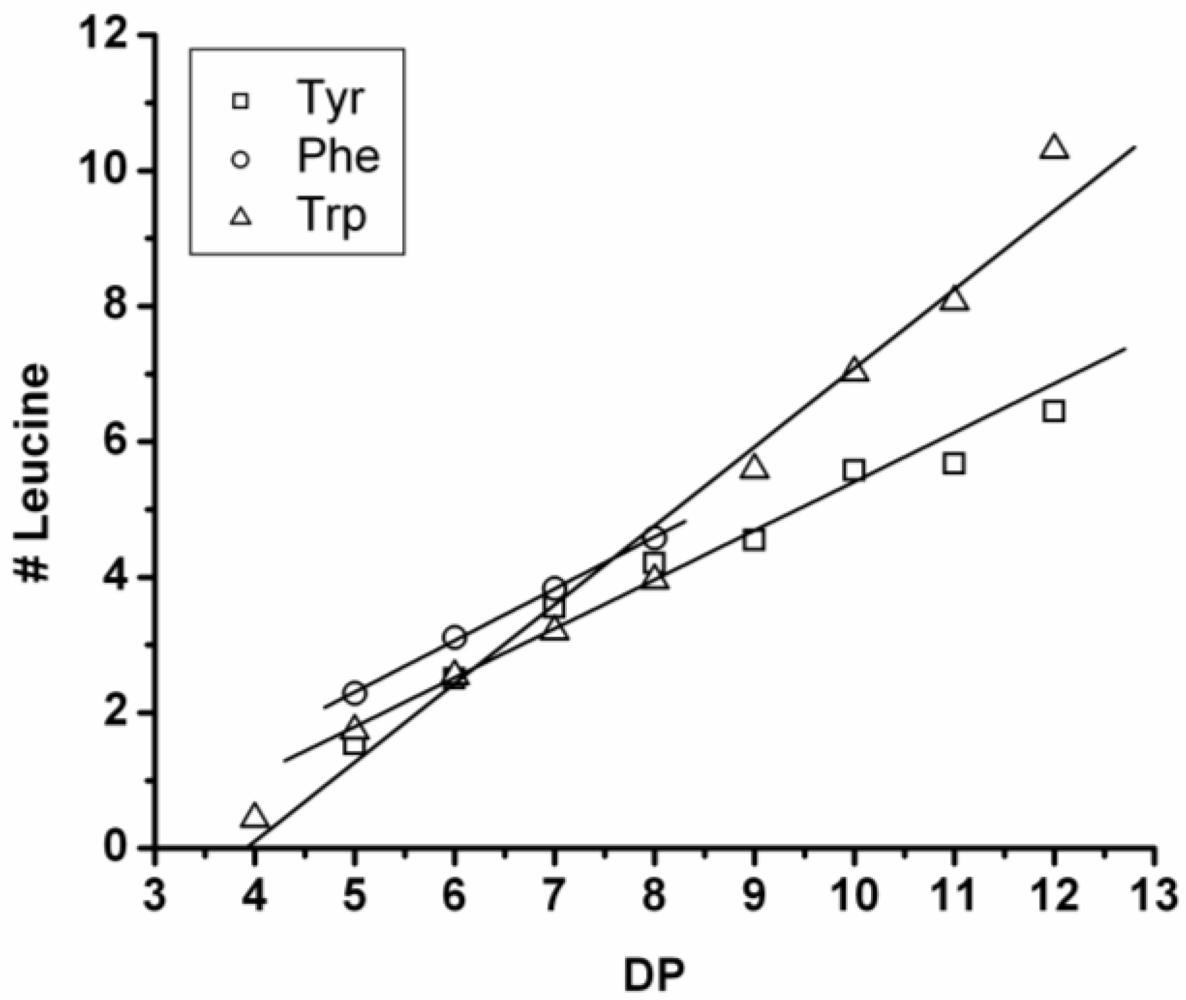
3.5.1. Leucine Containing Ternary Co-Oligomers
All of the co-oligomers containing leucine have more leucine residues incorporated as the chains grow longer (see
Figure 28). Elongation with leucine is often combined with a reducing amount of one of the other components, like tyrosine, when combined with Leu-Trp and Phe in combination with Leu-Trp and Leu-Tyr. This was also observed for Trp in combination with Leu in the series of binary co-oligomers.
Figure 28.
Average amount of leucine residues in ternary co-oligo amino acids.
Figure 28.
Average amount of leucine residues in ternary co-oligo amino acids.
When compared with the binary co-oligomers, the influence of leucine on the DPavg is very clear. Adding leucine produces ternary co-oligomers that have a higher DPavg than the corresponding binary co-oligomers. In accordance with the results from the binary co-oligomers, leucine has the highest reactivity in the ternary co-oligomerizations.
3.5.2. Tyrosine Containing Ternary Co-Oligomers
Tyrosine is present in the chains varying from 1.0 to 3.0 residues (see
Figure 29). However, most chains contain 1.7–2.2 tyrosine residues, and this seems to be rather constant for all the co-oligomers throughout the distribution.
Figure 29.
Average amount of tyrosine residues in ternary co-oligo amino acids.
Figure 29.
Average amount of tyrosine residues in ternary co-oligo amino acids.
When only combined with Trp-Leu (triangles in
Figure 29), the amount of tyrosine rises with DP and drops again after DP 7. Again this is explained by redistribution reactions taking place.
When the DPavg of the co-oligomers is compared to the DPavg of the binary co-oligomers, the addition of tyrosine leads to a lower DPavg in combination with Trp-Leu and an increasing DPavg for the combinations with Phe-Leu and Phe-Trp. From this it is concluded that Tyr is the second-most reactive amino acid in the ternary co-oligomerizations.
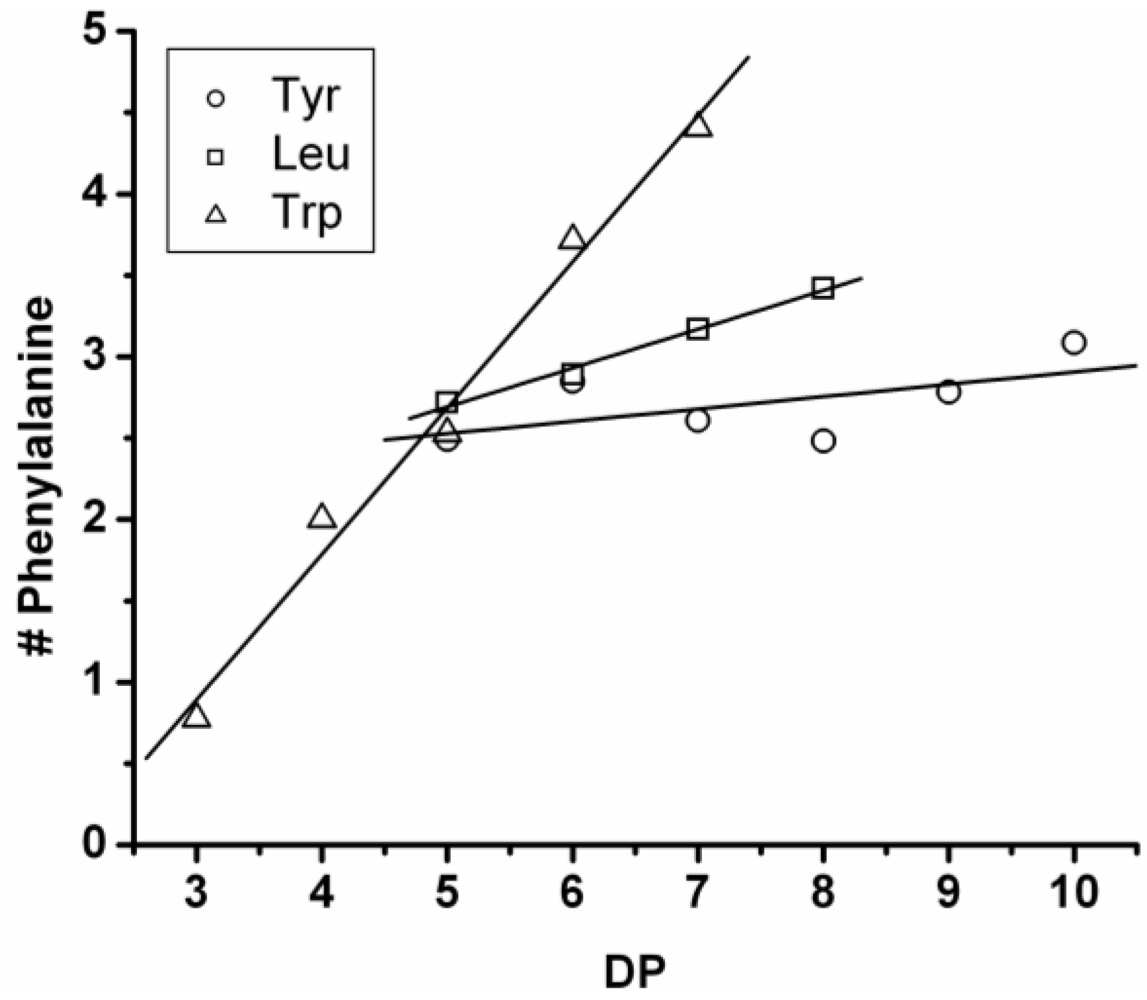
3.5.3. Phenylalanine Containing Ternary Co-Oligomers
The amount of phenylalanine rises with DP and starts to go down again after DP 8 for the combination with Leu-Tyr (see
Figure 30). Again, the explanation for this is a hydrolysis or transamidation reaction leading to a redistribution of the amino acids. In combination with Trp-Tyr and Leu-Trp, the amount of phenylalanine seems to reach a maximum, but not enough data points are available (due to precipitation) to conclude if it is there.
Comparing the DPavg of the ternary co-oligomers with the binary co-oligomers shows that phenylalanine had no influence on the DPavg when combined with Tyr-Trp. It increased the DPavg when combined with Tyr-Leu and decreased DPavg in combination with Leu-Trp. In conclusion, phenylalanine is the third-most reactive amino acid, as was also seen for the binary co-oligomers.
Figure 30.
Average amount of phenylalanine in ternary co-oligo amino acids.
Figure 30.
Average amount of phenylalanine in ternary co-oligo amino acids.
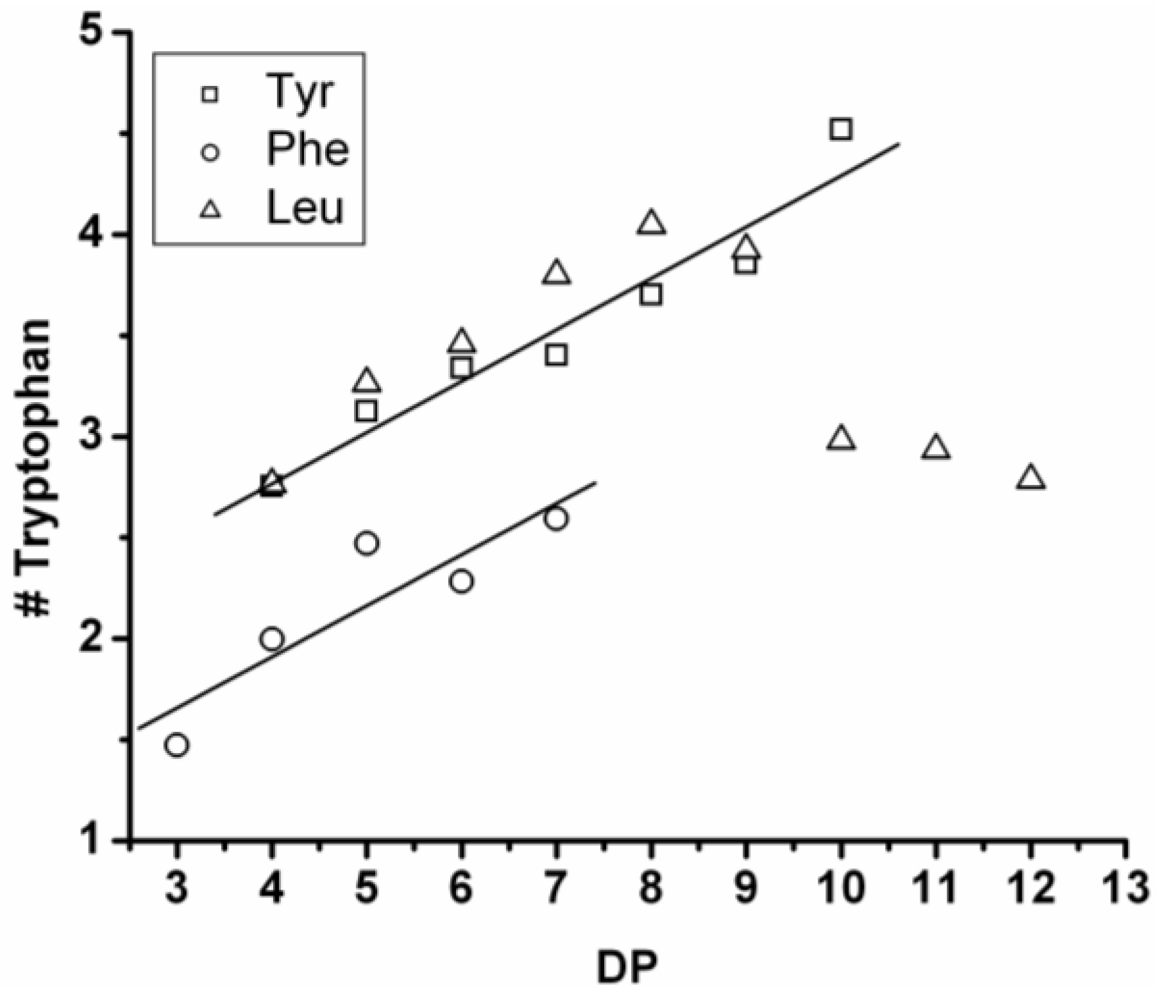
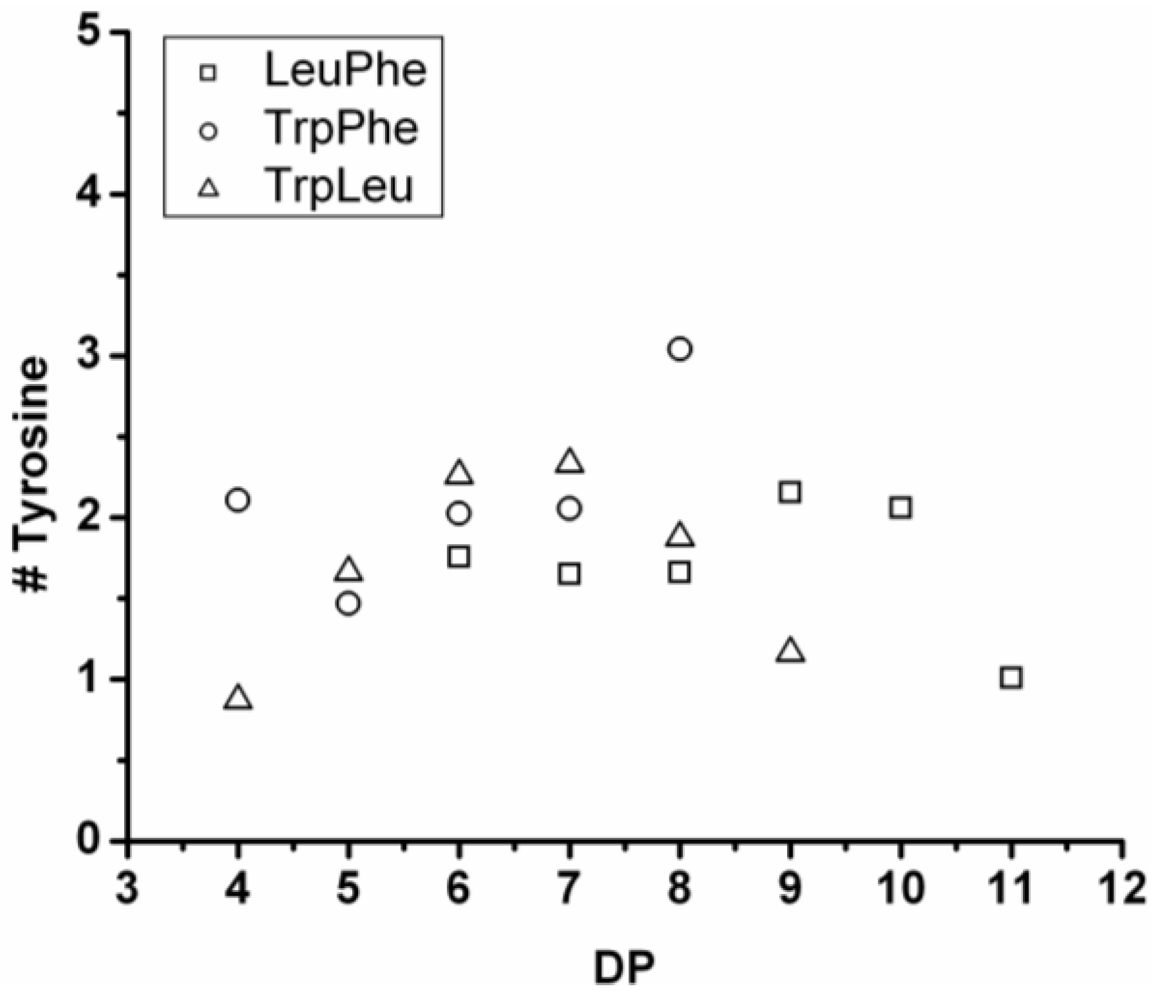
3.5.4. Tryptophan Containing Ternary Co-Oligomers
The number of tryptophan residues in the ternary co-oligomers lies between 1.8 and 2.4 (see
Figure 31). In the binary co-oligomer of Trp-Leu a redistribution was seen. The redistribution reactions are not obvious in the ternary co-oligomers. When the DP
avg of the ternary co-oligomers is compared to the DP
avg of the corresponding binary co-oligomers Trp lowers the DP
avg for all chains. Tryptophan is again found to be the least-reactive amino acid.
Based on the above discussed reactivities of the amino acids in the ternary co-oligomerizations, it is concluded that the reactivity order of the amino acids is Leu > Tyr > Phe > Trp for these oligomerizations.
Figure 31.
Average amount of tryptophan in co-oligomers based on three amino acids.
Figure 31.
Average amount of tryptophan in co-oligomers based on three amino acids.
3.6. Thermogravimetric Analysis of Peptides
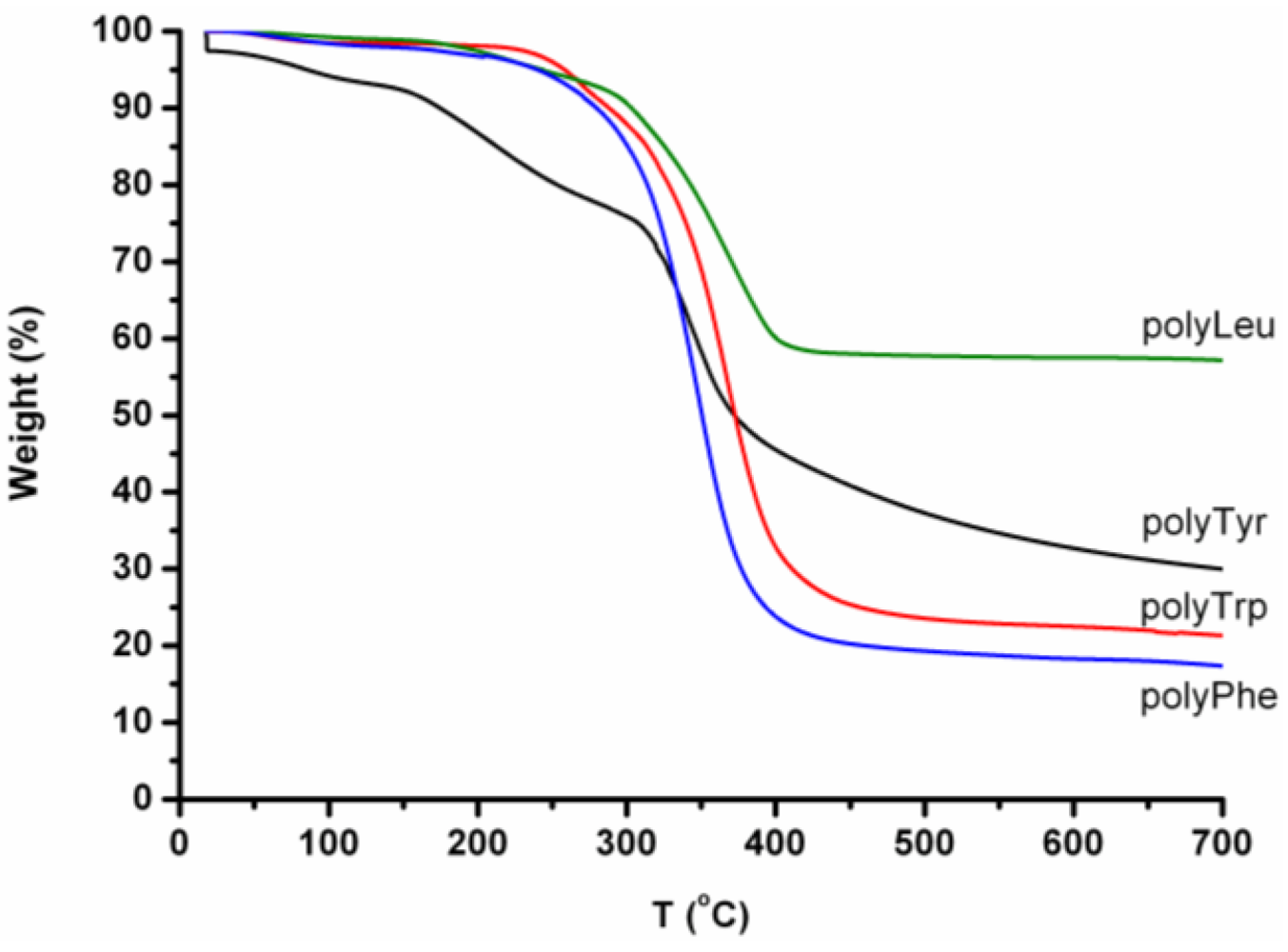
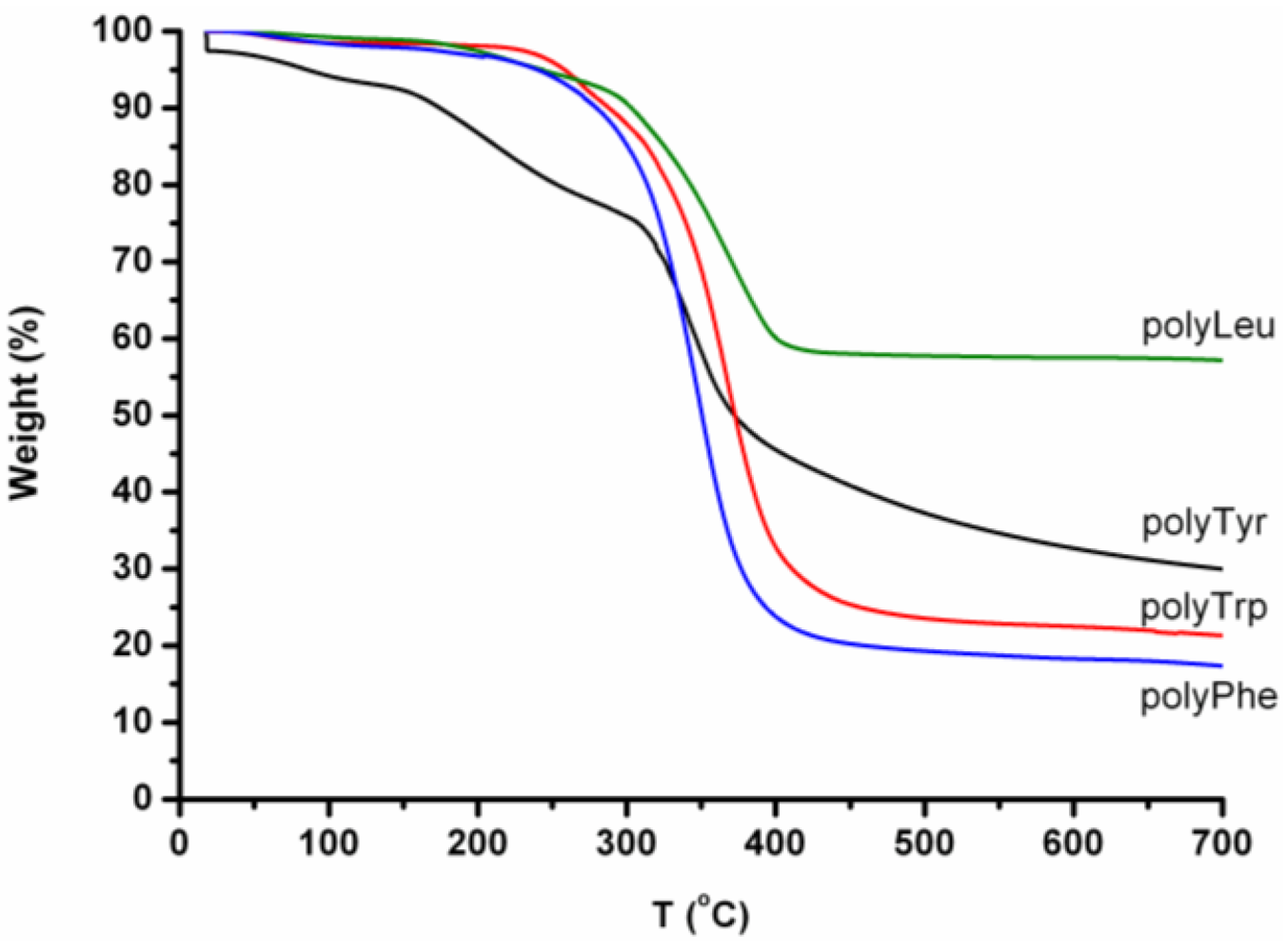
The oligo amino acids oligoleucine, oligotyrosine, oligophenylalanine and oligotryptophan were pyrolysed by heating the samples from 20 to 800 °C. In
Table 6 the TGA data are presented. The oligo amino acids have a main degradation step between 300–320 °C. The oligoleucine and the oligotryptophan and oligophenylalanine have an extra degradation step between 160 °C and 239 °C.
Table 6.
Oligomer degradation temperatures of oligo amino acids.
Table 6.
Oligomer degradation temperatures of oligo amino acids.
| Oligomer | T1 | T2 | wt% char |
|---|
| Leu | 178 | 306 | 57 |
| Tyr | 158 | 317 | 40 |
| Phe | 239 | 331 | 24 |
| Trp | | 302 | 18 |
In
Figure 32, the TGA-curves of the homo-oligomers are presented. All the oligomers except oligoTrp show a weight loss step before the main degradation step (300–340 °C). After the thermal treatment a significant amount of char is left. Oligo(leucine) in particular degrades only half, since 57% by weight is left as a char.
Figure 32.
TGA curves of oligoLeu, oligoTyr, oligoTrp and oligoPhe.
Figure 32.
TGA curves of oligoLeu, oligoTyr, oligoTrp and oligoPhe.
The weight loss before the main degradation is caused by the loss of water, while cyclic peptides are formed [
24]—the so-called diketopiperazides. During the main degradation step these cyclic structures fall apart and a char is left. The main degradation step at around 300–350 °C can be explained by two processes. The pyrolysis of oligotyrosine [
34] and oligoleucine [
23] proceeds through the degradation of cyclic structures under the release of ammonia and water. Polyphenylalanine [
24,
31] degrades by falling apart in nitrile and olefin fragments that further degrade by the loss of CN, HCN and CH
2-R (R = amino acid side group) [
34].
Char left after the thermal treatment can be stabilized by phenolic residues in the case of polytyrosine [
34,
35]. The other oligo amino acids also leave a char behind.
3.6.1. Pyrolysis of Co-Oligo amino Acids
The co-oligo amino acids were also subjected to thermal degradation. The results of this degradation are summarized in
Table 7. All the co-oligomers show the main degradation between 300 and 325 °C. After heating the samples to 800 °C, at least 10% char was still left for all the oligomers. A combination of phenylalanine and tryptophan left 29 wt% char and a co-oligomer of tyrosine, phenylalanine and tryptophan left 26 wt% char. The other co-oligomers left <20 wt% char. Degradation already starts at temperatures between 150–200 °C.
Table 7.
Degradation temperatures of the co-oligomers.
Table 7.
Degradation temperatures of the co-oligomers.
| Entry | Co-oligomer | PDT a (°C) | loss (wt%) | Char (wt%) |
|---|
| 1 | Tyr-Phe | 320 | 88 | 12 |
| 2 | Tyr-Leu | 312 | 80 | 10 |
| 3 | Tyr-Trp | 308 | 81 | 18 |
| 4 | Phe-Leu | 301 | 88 | 10 |
| 5 | Phe-Trp | 314 | 69 | 29 |
| 6 | Leu-Trp | 313 | 90 | 10 |
| 7 | Tyr-Phe-Trp | 322 | 63 | 26 |
| 8 | Tyr-Phe-Leu | 316 | 86 | 13 |
| 9 | Tyr-Leu-Trp | 311 | 80 | 16 |
| 10 | Phe-Trp-Leu | 307 | 85 | 11 |
| 11 | Phe-Leu-Trp-Tyr | 302 | 83 | 13 |
The degradation of peptides proceeds through the formation of cyclic peptides and the formation of diketopiperazines. The formation of the diketopiperazines is accompanied by the release of water, in the TGA diagrams this first step was visible as a small decrease of ~2 wt% before the start of the main degradation step.
3.7. Discussion
The oligomerizations described in the former paragraphs all yield oligomers as an insoluble product after 24 h of reaction. Since all homo-oligomers are obtained as polydisperse solid precipitates, it is assumed that the precipitation starts and the chains present at that time are dragged out of the solution. This seems a reasonable assumption for the co-oligomers as well. According to literature, the precipitates of polypeptides contain a distribution of chains as shown by Uyama and Li for the enzymatic (co)oligomerization of glutamic acid derivatives [
22,
23] and by Fukuoka for the enzymatic oligomerization of tyrosine derivatives [
41].
Chains of amino acids precipitate as a solid powder or in crystal structures. The crystal structure peptides and oligo amino acids usually formed are the α-helix and the β-sheet. An α-helix is only stabilized when at least two turns can be formed, each containing 3.6 amino acids. Chains with a DP 5–10 usually form β-sheets. The oligopeptides in this research all have a DPavg of 8 or lower and therefore precipitate predominantly in β-sheets.
Not all the amino acids used like to form β-sheets. The propensities of amino acids to form β-sheets was discussed in a review article by Nesloney and Kelly [
42]. From a series of amino acids the four amino acids used in this chapter can be ranked, as shown in
Table 8, according to four different studies. Each group used different computations to find energy minima for the β-sheet formation. These studies agree that polyleucine has the least propensity to form β-sheets. For the other amino acids, it is not clear which is the right order when looking at the β-sheet propensities.
Table 8.
β-sheet propensities of Tyr, Trp ,Phe and Leu ranked from high to low propensity.
Table 8.
β-sheet propensities of Tyr, Trp ,Phe and Leu ranked from high to low propensity.
| Chou and Fasman [43,44] | Kim and Berg [45] | Smith and Regan [46] | Minor and Kim [47,48] |
|---|
| Tyr | Phe | Tyr | Tyr |
| Trp | Tyr | Phe | Phe |
| Phe | Trp | Trp | Trp |
| Leu | Leu | Leu | Leu |
For α-helix formation similar studies were reported in literature summarized in
Table 9. It can be seen that leucine has the highest propensity to form α-helical structures. The order of the other three amino acids is not that clear. As well in α-helix as in β-sheet propensities, leucine stands out and the others have a similar preference for β-sheets. Chains with a DP >10 are found for a few (co)oligomers, these chains either precipitate as an α-helix or, they are included in the β-sheets.
Table 9.
α-helix propensities of Tyr, Trp, Phe and Leu.
Table 9.
α-helix propensities of Tyr, Trp, Phe and Leu.
| Pace and Scholtz [49] | Rohl et al. [450] | Munoz and Serrano [51] | Williams et al. [52] | Chou and Fasman [43,44] | Luque et al. [53] |
|---|
| Leu | Leu | Leu | Leu | Leu | Leu |
| Trp | Tyr | Trp | Phe | Trp | Trp |
| Tyr | Trp | Tyr | Trp | Phe | Phe |
| Phe | Phe | Phe | Tyr | Tyr | Tyr |
A polydispersity is also found in the precipitate obtained in the homo-oligomerization of Leu, Phe, Tyr and Trp. Once β-sheet formation is started oligomers of different DP are included in the precipitate.
Co-oligomers will precipitate in β-sheets for two reasons. First, amino acids show similar propensities towards β-sheet formation. Second, the DPavg found for the co-oligomers is 8.3 or less and this implies β-sheet formation for all the co-oligomers.
In order to obtain longer oligomers in buffer systems, monomers can be included that are more hydrophilic. Another approach is changing to non-aqueous media. This also needs enhancement of papain for the use in these media. From literature, it is known that this can be achieved by immobilization [
54,
55,
56], modification with polyethylene glycol [
57,
58,
59] or site directed mutagenesis [
60].
3.7.1. Selectivity by Papain
Although the solubility of the oligomers limits the oligomerization for all co-oligomers, different reactivities of the amino acids are found. This can be the result of the inherent selectivity of papain. It is known that papain has a preference for aromatic hydrophobic residues in the S2 position [
36,
37], with a preference for tyrosine over phenylalanine [
38]. In particular, cysteine proteases and papain are reported to be tolerant with respect to binding amino acids in other subsites [
39]. In the homo-oligomerizations this preference for tyrosine is found. In the co-oligomerizations, leucine is found in a higher ratio than the other amino acids with tyrosine as the second-most reactive amino acid.
3.7.2. Reactivity of the Amino Acids
The nucleophilicity of the amino acids does not vary that much judged by their pK values listed in
Table 10. Based on this data it is not expected that there is a big difference in reactivity of the amino acids. And indeed this is not found in the oligomerizations.
Table 10.
pK1 and pK2 of Leu, Trp, Phe and Ty [
40].
Table 10.
pK1 and pK2 of Leu, Trp, Phe and Ty [40].
| | pK1 (α-COOH) | pK2 (α-NH3+) |
|---|
| Leu | 2.33 | 9.74 |
| Trp | 2.46 | 9.41 |
| Phe | 2.20 | 9.31 |
| Tyr | 2.20 | 9.21 |
4. Conclusions
Papain catalyzed the oligomerization of the amino acids leucine, tyrosine, phenylalanine and tryptophan. Binary and ternary co-oligomerization of the above-mentioned amino acids were also catalyzed by the enzyme. The observed reactivity of the amino acids is the result of an interplay between the selectivity of the enzyme, reactivity of the amino acids and the solubility of the oligomers. Further research needs to be done to clarify the contribution of each of these factors.
The homo-oligomerization of the amino acids revealed that papain catalyzes the oligomerization with a reactivity order of Tyr > Leu > Phe > Trp. Although the DPavg of oligoTyr (6.98) and oligoLeu (6.85) is comparable the yields of both oligomerizations (55% and 80% respectively) determined the order presented here.
Binary co-oligomerization led to oligomers obtained in yields of 35–60% and DPavg varying between 4.3 and 7.7. The variations in chain length and in composition of the chains were determined and evaluated.
The reactivity of the amino acids was determined, based on the changing amount of each amino acid with DP. The reactivity of the amino acids was Leu > Tyr > Phe > Trp. This order of reactivity was also found in relation to the corresponding homo-oligomers.
Ternary co-oligomers were synthesized by papain; the composition of the chains was calculated. Leucine turned out to be most present at high DP. The amount of leucine shows an increase per DP for all of the oligomers. Tryptophan was found to be present in constant, but low amounts. Phenylalanine and tyrosine were less pronounced. However, when the effect of the amino acids on the DP of the parent 2-amino acid co-oligomer was evaluated it showed that the reactivity order was the same as for the 2-amino acid co-oligomers Leu > Tyr > Phe > Trp.
The degradation of the homo-oligomers proceeded in one or two steps. The main degradation step is similar for the homo-oligomers and the co-oligomers, ranging from 300–325 °C. All the oligomers left char behind after heating to 800 °C. Homo-oligomers left 18–57% char while the co-oligomers left only 10–29% char. Char formation is explained by cross-linking reactions by the phenolic residues of tyrosine. Co-oligomerization enabled more complete degradation of the products.
In the future, research should be directed to improve the solubility of reaction products in aqueous media by incorporating hydrophilic amino acids or by changing to non-aqueous media with challenges for the activity of papain. Experiments towards the consumption of monomers during the reaction can reveal a selectivity by the papain in this reaction.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}