
Stabilization of Keratinocyte Monolayer Integrity in the Presence of Anti-Desmoglein-3 Antibodies through FcRn Blockade with Efgartigimod: Novel Treatment Paradigm for Pemphigus?

 ,
,
Abstract
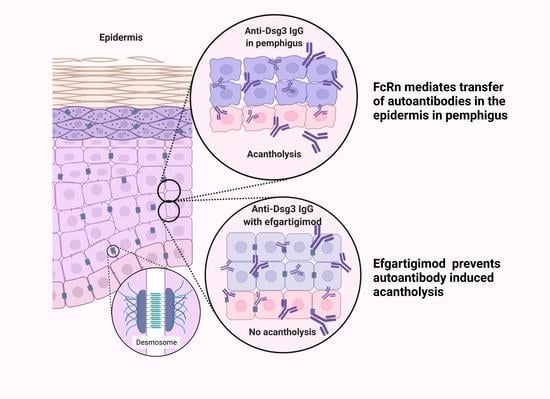
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibody Production and Purification
2.3. Dispase-Based Monolayer Dissociation Assay
2.4. Cell Lysis, Gel Electrophoresis and Western Blot
2.5. Sequential Detergent Extraction
2.6. Immunofluorescence
2.7. Anti-Dsg3 ELISA
2.8. AK23 Competition ELISA and Apparent Affinity Determination
2.9. RNA Isolation and cDNA Synthesis
2.10. Quantitative Real-Time PCR
2.11. Transcytosis Assay
2.12. Quantification of Transcytosed Fc Fragment and IgG Variants by ELISA
2.13. PV Patient and IgG Isolation
2.14. Statistical Analysis
3. Results
3.1. Reformatted Antibodies hAK23 and 4B3 Recognize Dsg3 in Indirect Immunofluorescnce of Human Keratinocytes
3.2. Recombinant Anti-Dsg3 Antibodies hAK23 and 4B3 Induce Acantholysis, Reduce Monolayer Integrity and Alter Dsg3 Localization in Human Keratinocytes
3.3. Treatment with hAK23 and 4B3 Results in Reduced Dsg3 Amount, and 4B3 Induces FcRn Depletion
3.4. Efgartigimod Treatment Prevents the Loss of Monolayer Integrity Induced by hAK23 and 4B3 Monoclonal Antibodies and by PV IgG
3.5. Efgartigimod Treatment Does Not Result in Degradation of hAK23 and 4B3 Antibodies
3.6. Efgartigimod Does Not Rescue Dsg3 Mislocalization Induced by Treatment with hAK23 or 4B3
3.7. Efgartigimod Treatment Does Not Prevent Dsg3 Degradation Induced by 4B3
3.8. Efgartigimod Treatment Prevents 4B3-Induced FcRn Degradation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hegazy, M.; Perl, A.L.; Svoboda, S.A.; Green, K.J. Desmosomal Cadherins in Health and Disease. Annu. Rev. Pathol. 2021, 17, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Spindler, V.; Waschke, J. Pemphigus-A Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front. Immunol. 2018, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.; Amber, K.T.; Agnoletti, A.F.; Wang, C.; Grando, S.A. Synergy among non-desmoglein antibodies contributes to the immunopathology of desmoglein antibody-negative pemphigus vulgaris. J. Biol. Chem. 2019, 294, 4520–4528. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.; Patel, K.G.; Grando, S.A. Mechanisms of synergy of autoantibodies to M3 muscarinic acetylcholine receptor and secretory pathway Ca(2+)/Mn(2+)-ATPase isoform 1 in patients with non-desmoglein pemphigus vulgaris. Int. Immunopharmacol. 2020, 80, 106149. [Google Scholar] [CrossRef] [PubMed]
- Amber, K.T.; Valdebran, M.; Grando, S.A. Non-Desmoglein Antibodies in Patients With Pemphigus Vulgaris. Front. Immunol. 2018, 9, 1190. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Arredondo, J.; Kitajima, Y.; Sato-Nagai, M.; Grando, S.A. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: Characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J. Biol. Chem. 2007, 282, 13804–13812. [Google Scholar] [CrossRef] [PubMed]
- Spindler, V.; Waschke, J. Desmosomal cadherins and signaling: Lessons from autoimmune disease. Cell Commun. Adhes 2014, 21, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.; Waschke, J. Autoantibody-Specific Signalling in Pemphigus. Front. Med. 2021, 8, 701809. [Google Scholar] [CrossRef] [PubMed]
- Spindler, V.; Eming, R.; Schmidt, E.; Amagai, M.; Grando, S.; Jonkman, M.F.; Kowalczyk, A.P.; Muller, E.J.; Payne, A.S.; Pincelli, C.; et al. Mechanisms Causing Loss of Keratinocyte Cohesion in Pemphigus. J. Investig. Dermatol. 2018, 138, 32–37. [Google Scholar] [CrossRef]
- Tsunoda, K.; Ota, T.; Aoki, M.; Yamada, T.; Nagai, T.; Nakagawa, T.; Koyasu, S.; Nishikawa, T.; Amagai, M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J. Immunol. 2003, 170, 2170–2178. [Google Scholar] [CrossRef]
- Payne, A.S.; Ishii, K.; Kacir, S.; Lin, C.; Li, H.; Hanakawa, Y.; Tsunoda, K.; Amagai, M.; Stanley, J.R.; Siegel, D.L. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J. Clin. Investig. 2005, 115, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Ober, R.J.; Radu, C.G.; Ghetie, V.; Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: Implications for therapeutic antibodies. Int. Immunol. 2001, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Daba, M.B.; Berntzen, G.; Michaelsen, T.E.; Sandlie, I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J. Biol. Chem. 2010, 285, 4826–4836. [Google Scholar] [CrossRef] [PubMed]
- Gornowicz-Porowska, J.; Kowalczyk, M.J.; Seraszek-Jaros, A.; Bowszyc-Dmochowska, M.; Kaczmarek, E.; Zaba, R.; Dmochowski, M. A Comparative Analysis of CD32A and CD16A Polymorphisms in Relation to Autoimmune Responses in Pemphigus Diseases and Subepithelial Autoimmune Blistering Disorders. Genes 2020, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Recke, A.; Konitzer, S.; Lemcke, S.; Freitag, M.; Sommer, N.M.; Abdelhady, M.; Amoli, M.M.; Benoit, S.; El-Chennawy, F.; Eldarouti, M.; et al. The p.Arg435His Variation of IgG3 With High Affinity to FcRn Is Associated With Susceptibility for Pemphigus Vulgaris-Analysis of Four Different Ethnic Cohorts. Front. Immunol. 2018, 9, 1788. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.; Bata-Csorgo, Z.; De Simone, C.; Didona, B.; Remenyik, E.; Reznichenko, N.; Stoevesandt, J.; Ward, E.S.; Parys, W.; de Haard, H.; et al. Treatment of pemphigus vulgaris and foliaceus with efgartigimod, a neonatal Fc receptor inhibitor: A phase 2 multicentre, open-label feasibility trial. Br. J. Dermatol. 2022, 186, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Werth, V.P.; Culton, D.A.; Concha, J.S.S.; Graydon, J.S.; Blumberg, L.J.; Okawa, J.; Pyzik, M.; Blumberg, R.S.; Hall, R.P., 3rd. Safety, Tolerability, and Activity of ALXN1830 Targeting the Neonatal Fc Receptor in Chronic Pemphigus. J. Investig. Dermatol. 2021, 141, 2858–2865.e4. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Christianson, G.J.; Sproule, T.J.; Brown, A.C.; Akilesh, S.; Jung, N.; Petkova, S.; Avanessian, L.; Choi, E.Y.; Shaffer, D.J.; et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J. Immunol. 2003, 170, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, C.; Mehnaz, S.; Robinson, J.M.; Hayton, W.L.; Pearl, D.K.; Roopenian, D.C.; Anderson, C.L. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 2003, 197, 315–322. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Huber, A.H.; Bjorkman, P.J. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994, 372, 379–383. [Google Scholar] [CrossRef] [PubMed]
- West, A.P., Jr.; Bjorkman, P.J. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,). Biochemistry 2000, 39, 9698–9708. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin binding to FcRn: Distinct from the FcRn-IgG interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Abdiche, Y.N.; Yeung, Y.A.; Chaparro-Riggers, J.; Barman, I.; Strop, P.; Chin, S.M.; Pham, A.; Bolton, G.; McDonough, D.; Lindquist, K.; et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. MAbs 2015, 7, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Dee Qian, J.; Sandlie, I. The conserved histidine 166 residue of the human neonatal Fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur. J. Immunol. 2006, 36, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Sesarman, A.; Vidarsson, G.; Sitaru, C. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol. Life Sci. 2010, 67, 2533–2550. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Ober, R.J. Chapter 4: Multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv. Immunol. 2009, 103, 77–115. [Google Scholar] [CrossRef]
- Ward, E.S.; Ober, R.J. Targeting FcRn to Generate Antibody-Based Therapeutics. Trends Pharmacol. Sci. 2018, 39, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Grevys, A.; Nilsen, J.; Sand, K.M.K.; Daba, M.B.; Oynebraten, I.; Bern, M.; McAdam, M.B.; Foss, S.; Schlothauer, T.; Michaelsen, T.E.; et al. A human endothelial cell-based recycling assay for screening of FcRn targeted molecules. Nat. Commun. 2018, 9, 621. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, C.; Zhou, J.; Ober, R.J.; Ward, E.S. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat. Biotechnol. 2005, 23, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, G.M.; Finn, P.W.; Ward, E.S.; Dumont, J.; Dickinson, B.L.; Blumberg, R.S.; Lencer, W.I. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J. Exp. Med. 2002, 196, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.L.; Badizadegan, K.; Wu, Z.; Ahouse, J.C.; Zhu, X.; Simister, N.E.; Blumberg, R.S.; Lencer, W.I. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J. Clin. Investig. 1999, 104, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Bern, M.; Nilsen, J.; Ferrarese, M.; Sand, K.M.K.; Gjolberg, T.T.; Lode, H.E.; Davidson, R.J.; Camire, R.M.; Baekkevold, E.S.; Foss, S.; et al. An engineered human albumin enhances half-life and transmucosal delivery when fused to protein-based biologics. Sci. Transl. Med. 2020, 12, eabb0580. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Qiao, S.W.; Kuo, T.T.; Aveson, V.G.; Platzer, B.; Andersen, J.T.; Sandlie, I.; Chen, Z.; de Haar, C.; Lencer, W.I.; et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9927–9932. [Google Scholar] [CrossRef]
- Baker, K.; Rath, T.; Pyzik, M.; Blumberg, R.S. The Role of FcRn in Antigen Presentation. Front. Immunol. 2014, 5, 408. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.W.; Kobayashi, K.; Johansen, F.E.; Sollid, L.M.; Andersen, J.T.; Milford, E.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Dependence of antibody-mediated presentation of antigen on FcRn. Proc. Natl. Acad. Sci. USA 2008, 105, 9337–9342. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Cao, Y. In Translation: FcRn across the Therapeutic Spectrum. Int. J. Mol. Sci. 2021, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Cauza, K.; Hinterhuber, G.; Dingelmaier-Hovorka, R.; Brugger, K.; Klosner, G.; Horvat, R.; Wolff, K.; Foedinger, D. Expression of FcRn, the MHC class I-related receptor for IgG, in human keratinocytes. J. Investig. Dermatol. 2005, 124, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.F., Jr.; Bril, V.; Burns, T.M.; Mantegazza, R.; Bilinska, M.; Szczudlik, A.; Beydoun, S.; Garrido, F.; Piehl, F.; Rottoli, M.; et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 2019, 92, e2661–e2673. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.F., Jr.; Bril, V.; Vu, T.; Karam, C.; Peric, S.; Margania, T.; Murai, H.; Bilinska, M.; Shakarishvili, R.; Smilowski, M.; et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): A multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021, 20, 526–536. [Google Scholar] [CrossRef]
- Newland, A.C.; Sanchez-Gonzalez, B.; Rejto, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Beckert, B.; Panico, F.; Pollmann, R.; Eming, R.; Banning, A.; Tikkanen, R. Immortalized Human hTert/KER-CT Keratinocytes a Model System for Research on Desmosomal Adhesion and Pathogenesis of Pemphigus Vulgaris. Int. J. Mol. Sci. 2019, 20, 3113. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, K.; Ota, T.; Saito, M.; Hata, T.; Shimizu, A.; Ishiko, A.; Yamada, T.; Nakagawa, T.; Kowalczyk, A.P.; Amagai, M. Pathogenic relevance of IgG and IgM antibodies against desmoglein 3 in blister formation in pemphigus vulgaris. Am. J. Pathol. 2011, 179, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.E.; Chen, J.; Sexton, D.J.; Muruganandam, A.; Bitonti, A.J.; Dumont, J.; Viswanathan, M.; Martik, D.; Wassaf, D.; Mezo, A.; et al. Fully human monoclonal antibody inhibitors of the neonatal fc receptor reduce circulating IgG in non-human primates. Front. Immunol. 2015, 6, 176. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://imagej.net/software/fiji/ (accessed on 30 September 2021).
- Stahley, S.N.; Saito, M.; Faundez, V.; Koval, M.; Mattheyses, A.L.; Kowalczyk, A.P. Desmosome Assembly and Disassembly Are Membrane Raft-Dependent. PLoS ONE 2014, 9, e87809. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Amagai, M.; Hall, R.P.; Hashimoto, T.; Takayanagi, A.; Gamou, S.; Shimizu, N.; Nishikawa, T. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J. Immunol. 1997, 159, 2010–2017. [Google Scholar]
- Ishii, K.; Harada, R.; Matsuo, I.; Shirakata, Y.; Hashimoto, K.; Amagai, M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J. Inv. Dermat. 2005, 124, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Abasq, C.; Mouquet, H.; Gilbert, D.; Tron, F.; Grassi, V.; Musette, P.; Joly, P. ELISA testing of anti-desmoglein 1 and 3 antibodies in the management of pemphigus. Arch. Dermatol. 2009, 145, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.; Vielmuth, F.; Rotkopf, L.; Sardy, M.; Horvath, O.N.; Goebeler, M.; Schmidt, E.; Eming, R.; Hertl, M.; Spindler, V.; et al. Different signaling patterns contribute to loss of keratinocyte cohesion dependent on autoantibody profile in pemphigus. Sci. Rep. 2017, 7, 3579. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.S.; Siegel, D.L.; Stanley, J.R. Targeting pemphigus autoantibodies through their heavy-chain variable region genes. J. Investig. Dermatol. 2007, 127, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Vollner, F.; Ali, J.; Kurrle, N.; Exner, Y.; Eming, R.; Hertl, M.; Banning, A.; Tikkanen, R. Loss of flotillin expression results in weakened desmosomal adhesion and Pemphigus vulgaris-like localisation of desmoglein-3 in human keratinocytes. Sci. Rep. 2016, 6, 28820. [Google Scholar] [CrossRef] [PubMed]
- Delva, E.; Jennings, J.M.; Calkins, C.C.; Kottke, M.D.; Faundez, V.; Kowalczyk, A.P. Pemphigus vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism. J. Biol. Chem. 2008, 283, 18303–18313. [Google Scholar] [CrossRef] [PubMed]
- Calkins, C.C.; Setzer, S.V.; Jennings, J.M.; Summers, S.; Tsunoda, K.; Amagai, M.; Kowalczyk, A.P. Desmoglein Endocytosis and Desmosome Disassembly Are Coordinated Responses to Pemphigus Autoantibodies. J. Biol. Chem. 2006, 281, 7623–7634. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.M.; Tucker, D.K.; Kottke, M.D.; Saito, M.; Delva, E.; Hanakawa, Y.; Amagai, M.; Kowalczyk, A.P. Desmosome Disassembly in Response to Pemphigus Vulgaris IgG Occurs in Distinct Phases and Can Be Reversed by Expression of Exogenous Dsg3. J. Inv. Dermat. 2011, 131, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, S.E.; Kowalczyk, A.P. The desmosome as a model for lipid raft driven membrane domain organization. Biochim. Biophys Acta. Biomembr. 2020, 1862, 183329. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chernyavsky, A.; Webber, R.J.; Grando, S.A.; Wang, P.H. Critical Role of the Neonatal Fc Receptor (FcRn) in the Pathogenic Action of Antimitochondrial Autoantibodies Synergizing with Anti-desmoglein Autoantibodies in Pemphigus Vulgaris. J. Biol. Chem. 2015, 290, 23826–23837. [Google Scholar] [CrossRef] [PubMed]
- Ben Suleiman, Y.; Yoshida, M.; Nishiumi, S.; Tanaka, H.; Mimura, T.; Nobutani, K.; Yamamoto, K.; Takenaka, M.; Aoganghua, A.; Miki, I.; et al. Neonatal Fc receptor for IgG (FcRn) expressed in the gastric epithelium regulates bacterial infection in mice. Mucosal. Immunol. 2012, 5, 87–98. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gao, Y.; Zhao, L.; Li, Y.; Zhang, Y.; Wang, S.; Zhang, H.; Lu, G.; Guo, X. The expression and function of the neonatal Fc receptor in thyrocytes of Hashimoto’s thyroiditis. Int. Immunopharmacol. 2017, 44, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Lencer, W.I.; Blumberg, R.S. A passionate kiss, then run: Exocytosis and recycling of IgG by FcRn. Trends Cell Biol. 2005, 15, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Weflen, A.W.; Baier, N.; Tang, Q.J.; Van den Hof, M.; Blumberg, R.S.; Lencer, W.I.; Massol, R.H. Multivalent immune complexes divert FcRn to lysosomes by exclusion from recycling sorting tubules. Mol. Biol. Cell 2013, 24, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, L.; Bai, Y.; Mojidi, H.; Simister, N.E.; Zhu, X. Activation of the JAK/STAT-1 signaling pathway by IFN-gamma can down-regulate functional expression of the MHC class I-related neonatal Fc receptor for IgG. J. Immunol. 2008, 181, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, E.H.; Kneibner, D.; Kridin, K.; Amber, K.T. Serum and blister fluid levels of cytokines and chemokines in pemphigus and bullous pemphigoid. Autoimmun. Rev. 2019, 18, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, W.; Fauty, S.; Fang, Y.; Hamuro, L.; Hussain, A.; Prueksaritanont, T. The effect of the neonatal Fc receptor on human IgG biodistribution in mice. MAbs 2014, 6, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Balthasar, J.P. Investigation of the influence of FcRn on the distribution of IgG to the brain. AAPS J. 2009, 11, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, M.; Hilario-Vargas, J.; Prisayanh, P.; Warren, S.; Diaz, L.A.; Roopenian, D.C.; Liu, Z. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J. Clin. Investig. 2005, 115, 3440–3450. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakrzewicz, A.; Würth, C.; Beckert, B.; Feldhoff, S.; Vanderheyden, K.; Foss, S.; Andersen, J.T.; Haard, H.d.; Verheesen, P.; Bobkov, V.; et al. Stabilization of Keratinocyte Monolayer Integrity in the Presence of Anti-Desmoglein-3 Antibodies through FcRn Blockade with Efgartigimod: Novel Treatment Paradigm for Pemphigus? Cells 2022, 11, 942. https://doi.org/10.3390/cells11060942
Zakrzewicz A, Würth C, Beckert B, Feldhoff S, Vanderheyden K, Foss S, Andersen JT, Haard Hd, Verheesen P, Bobkov V, et al. Stabilization of Keratinocyte Monolayer Integrity in the Presence of Anti-Desmoglein-3 Antibodies through FcRn Blockade with Efgartigimod: Novel Treatment Paradigm for Pemphigus? Cells. 2022; 11(6):942. https://doi.org/10.3390/cells11060942
Chicago/Turabian StyleZakrzewicz, Anna, Celina Würth, Benedikt Beckert, Simon Feldhoff, Katrien Vanderheyden, Stian Foss, Jan Terje Andersen, Hans de Haard, Peter Verheesen, Vladimir Bobkov, and et al. 2022. "Stabilization of Keratinocyte Monolayer Integrity in the Presence of Anti-Desmoglein-3 Antibodies through FcRn Blockade with Efgartigimod: Novel Treatment Paradigm for Pemphigus?" Cells 11, no. 6: 942. https://doi.org/10.3390/cells11060942