
Pediatric Beta Blocker Therapy: A Comprehensive Review of Development and Genetic Variation to Guide Precision-Based Therapy in Children, Adolescents, and Young Adults


Abstract
:1. Introduction
2. Dose–Exposure–Response Paradigm and Pediatric Trials
3. Evolution of Beta Blocker Therapy in Children
4. Contributions of Ontogeny and Genetic Variation in Beta Blocker Disposition and Response
5. Fundamental Issues for Evaluating Variability in Drug Disposition in Pediatrics
6. Absorption
7. Distribution
8. Metabolism
9. Excretion
10. Response
11. Genetic Determinants of Beta Blocker Disposition
12. Genetic Determinants of Beta Blocker Response
13. Developmental Differences in Beta Blocker Disposition
14. Developmental Differences in ADRB1
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Wong, K.; Potts, J.; Etheridge, S.; Sanatani, S. Medications used to manage supraventricular tachycardia in the infant a North American survey. Pediatr. Cardiol. 2006, 27, 199–203. [Google Scholar] [CrossRef]
- Kikano, S.; Kannankeril, P.J. Precision Medicine in Pediatric Cardiology. Pediatr. Ann. 2022, 51, e390–e395. [Google Scholar] [CrossRef]
- Rodriguez, W.; Selen, A.; Avant, D.; Chaurasia, C.; Crescenzi, T.; Gieser, G.; Di Giacinto, J.; Huang, S.-M.; Lee, P.; Mathis, L.; et al. Improving pediatric dosing through pediatric initiatives: What we have learned. Pediatrics 2008, 121, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Rosi, J.; Bertho, G.; Mansuy, D. Cytochromes P450 catalyze both steps of the major pathway of clopidogrel bioactivation, whereas paraoxonase catalyzes the formation of a minor thiol metabolite isomer. Chem. Res. Toxicol. 2012, 25, 348–356. [Google Scholar] [CrossRef]
- Kazui, M.; Nishiya, Y.; Ishizuka, T.; Hagihara, K.; Farid, N.A.; Okazaki, O.; Ikeda, T.; Kurihara, A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2010, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.T.; Close, S.L.; Iturria, S.J.; Payne, C.D.; Farid, N.A.; Ernest, C.S.; Lachno, D.R.; Salazar, D.; Winters, K.J. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J. Thromb. Haemost. 2007, 5, 2429–2436. [Google Scholar] [CrossRef]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; et al. Cytochrome P-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Simon, T.; Collet, J.-P.; Anderson, J.L.; Antman, E.M.; Bliden, K.; Cannon, C.P.; Danchin, N.; Giusti, B.; Gurbel, P.; et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: A meta-analysis. JAMA 2010, 304, 1821–1830. [Google Scholar] [CrossRef]
- Rudez, G.; Bouman, H.J.; van Werkum, J.W.; Leebeek, F.W.; Kruit, A.; Ruven, H.J.; Berg, J.M.T.; de Maat, M.P.; Hackeng, C.M. Common variation in the platelet receptor P2RY12 gene is associated with residual on-clopidogrel platelet reactivity in patients undergoing elective percutaneous coronary interventions. Circ. Cardiovasc. Genet. 2009, 2, 515–521. [Google Scholar] [CrossRef]
- Wagner, J.; Abdel-Rahman, S.M. Pediatric pharmacokinetics. Pediatr. Rev. 2013, 34, 258–269. [Google Scholar] [CrossRef]
- Koukouritaki, S.B.; Manro, J.R.; Marsh, S.A.; Stevens, J.C.; Rettie, A.E.; McCarver, D.G.; Hines, R.N. Developmental expression of human hepatic CYP2C9 and CYP2C19. J. Pharmacol. Exp. Ther. 2004, 308, 965–974. [Google Scholar] [CrossRef]
- Kaley, V.R.; Aregullin, E.O.; Samuel, B.P.; Vettukattil, J.J. Trends in the off-label use of β-blockers in pediatric patients. Pediatr. Int. 2019, 61, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, R.P.; Lynch, G.S.; Ryall, J.G.; Saifeddine, M.; Seymour, M.L.; Xiao, Y.-P.; Compton, S.J.; Houle, S.; Ramachandran, R.; MacNaughton, W.K.; et al. A study of the adrenotropic receptors. Am. J. Physiol. 1948, 153, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Black, J.; Crowther, A.; Shanks, R.; Smith, L.; Dornhorst, A. A New Adrenergic: Betareceptor Antagonist. Lancet 1964, 283, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Minobe, W.; Cubicciotti, R.S.; Sageman, W.S.; Lurie, K.; Billingham, M.E.; Harrison, D.C.; Stinson, E.B. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982, 307, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Staley, N.A.; Noren, G.R.; Einzig, S.; Rublein, T.G. Effect of early propranolol treatment in an animal model of congestive cardiomyopathy. II. beta-adrenergic receptors and left ventricular function. Cardiovasc. Res. 1984, 18, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A. Improving clinical outcomes with drug treatment in heart failure: What have trials taught? Am. J. Cardiol. 2003, 91, 9D–14D. [Google Scholar] [CrossRef]
- Wilkinson, J.D.; Landy, D.C.; Colan, S.D.; Towbin, J.A.; Sleeper, L.A.; Orav, E.J.; Cox, G.F.; Canter, C.E.; Hsu, D.T.; Webber, S.A.; et al. The pediatric cardiomyopathy registry and heart failure: Key results from the first 15 years. Heart Fail. Clin. 2010, 6, 401–413. [Google Scholar] [CrossRef]
- Kantor, P.F.; Abraham, J.R.; Dipchand, A.I.; Benson, L.N.; Redington, A.N. The impact of changing medical therapy on transplantation-free survival in pediatric dilated cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 1377–1384. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Shaddy, R.E.; Boucek, M.M.; Hsu, D.T.; Boucek, R.J.; Canter, C.E.; Mahony, L.; Ross, R.D.; Pahl, E.; Blume, E.D.; Dodd, D.A.; et al. Carvedilol for children and adolescents with heart failure: A randomized controlled trial. JAMA 2007, 298, 1171–1179. [Google Scholar] [CrossRef]
- Alabed, S.; Sabouni, A.; Al Dakhoul, S.; Bdaiwi, Y. Beta-blockers for congestive heart failure in children. Cochrane Database Syst. Rev. 2020, 7, CD007037. [Google Scholar] [CrossRef] [PubMed]
- Garson, A.; Gillette, P.C.; McNamara, D.G. Supraventricular tachycardia in children: Clinical features, response to treatment, and long-term follow-up in 217 patients. J. Pediatr. 1981, 98, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Sanatani, S.; Potts, J.E.; Reed, J.H.; Saul, J.P.; Stephenson, E.A.; Gibbs, K.A.; Anderson, C.C.; Mackie, A.S.; Ro, P.S.; Tisma-Dupanovic, S.; et al. The study of antiarrhythmic medications in infancy (SAMIS): A multicenter, randomized controlled trial comparing the efficacy and safety of digoxin versus propranolol for prophylaxis of supraventricular tachycardia in infants. Circ. Arrhythmia Electrophysiol. 2012, 5, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Bruder, D.; Weber, R.; Gass, M.; Balmer, C.; Cavigelli-Brunner, A. Antiarrhythmic Medication in Neonates and Infants with Supraventricular Tachycardia. Pediatr. Cardiol. 2022, 43, 1311–1318. [Google Scholar] [CrossRef]
- Lim, Y.T.; Kim, Y.H.; Kwon, J.E. Effective Control of Supraventricular Tachycardia in Neonates May Requires Combination Pharmacologic Therapy. J. Clin. Med. 2022, 11, 3279. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Marelli, A.; Burchill, L.J.; Chubb, H.; Roche, S.L.; Cedars, A.M.; Khairy, P.; Zaidi, A.N.; Janousek, J.; Crossland, D.S.; et al. Management of Heart Failure with Arrhythmia in Adults with Congenital Heart Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 80, 2224–2238. [Google Scholar] [CrossRef] [PubMed]
- Zarem, H.A.M.; Edgerton, M.T.M. Induced resolution of cavernous hemangiomas following prednisolone therapy. Plast. Reconstr. Surg. 1967, 39, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Léauté-Labrèze, C.; de la Roque, E.D.; Hubiche, T.; Boralevi, F.; Thambo, J.-B.; Taïeb, A. Propranolol for severe hemangiomas of infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef]
- Mabeta, P.; Pepper, M.S. Hemangiomas—Current therapeutic strategies. Int. J. Dev. Biol. 2011, 55, 431–437. [Google Scholar] [CrossRef]
- Léauté-Labrèze, C.; Hoeger, P.; Mazereeuw-Hautier, J.; Guibaud, L.; Baselga, E.; Posiunas, G.; Phillips, R.J.; Caceres, H.; Gutierrez, J.C.L.; Ballona, R.; et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N. Engl. J. Med. 2015, 372, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Ågesen, F.N.; Weeke, P.E.; Tfelt-Hansen, P.; Tfelt-Hansen, J.; Escape-Net, F. Pharmacokinetic variability of beta-adrenergic blocking agents used in cardiology. Pharmacol. Res. Perspect. 2019, 7, e00496. [Google Scholar] [CrossRef] [PubMed]
- Pope, E.; Lara-Corrales, I.; Sibbald, C.; Liy-Wong, C.; Kanigsberg, N.; Drolet, B.; Ma, J. Noninferiority and Safety of Nadolol vs Propranolol in Infants with Infantile Hemangioma: A Randomized Clinical Trial. JAMA Pediatr. 2022, 176, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Tierney, E.S.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Shores, J.; Berger, K.R.; Murphy, E.A.; Pyeritz, R.E. Progression of aortic dilatation and the benefit of long-term β-adrenergic blockade in Marfan’s syndrome. N. Engl. J. Med. 1994, 330, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Sleeper, L.A.; Gelb, B.D.; Morris, S.A.; Dietz, H.C.; Forbus, G.A.; Goldmuntz, E.; Hoskoppal, A.; James, J.; Lee, T.M.; et al. Variants in ADRB1 and CYP2C9: Association with Response to Atenolol and Losartan in Marfan Syndrome. J. Pediatr. 2020, 222, 213–220.e5. [Google Scholar] [CrossRef] [PubMed]
- Leeder, J.S.; Kearns, G.L.; Spielberg, S.P.; Anker, J.v.D.; Kearns, G.L.; Anker, J.v.D. Understanding the relative roles of pharmacogenetics and ontogeny in pediatric drug development and regulatory science. J. Clin. Pharmacol. 2010, 50, 1377–1387. [Google Scholar] [CrossRef]
- Wagner, J.; Leeder, J.S. Pediatric pharmacogenomics: A systematic assessment of ontogeny and genetic variation to guide the design of statin studies in children. Pediatr. Clin. N. Am. 2012, 59, 1017–1037. [Google Scholar] [CrossRef]
- Cojocariu, S.A.; Maștaleru, A.; Sascău, R.A.; Stătescu, C.; Mitu, F.; Leon-Constantin, M.M. Neuropsychiatric Consequences of Lipophilic β-Blockers. Medicina 2021, 57, 155. [Google Scholar] [CrossRef]
- Kirch, W.; Görg, K.G. Clinical pharmacokinetics of atenolol—A review. Eur. J. Drug Metab. Pharmacokinet. 1982, 7, 81–91. [Google Scholar] [CrossRef]
- Alam, M.; Awal, M.; Suban, N.; Mostofa, M. In-vitro relationship between protein-binding and free drug concentrations of a water-soluble selective β-adrenoreceptor antagonist (atenolol) and its interaction with arsenic. J. Health Popul. Nutr. 2009, 27, 20–30. [Google Scholar] [CrossRef]
- Carlson, W.; Oberg, K. Clinical Pharmacology of Carvedilol. J. Cardiovasc. Pharmacol. Ther. 1999, 4, 205–218. [Google Scholar] [CrossRef]
- Morgan, T. Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin. Pharmacokinet. 1994, 26, 335–346. [Google Scholar] [CrossRef]
- Sager, G.; Nilsen, O.G.; Jacobsen, S. Variable binding of propranolol in human serum. Biochem. Pharmacol. 1979, 28, 905–911. [Google Scholar] [CrossRef]
- Regardh, C.-G.; Johnsson, G. Clinical pharmacokinetics of metoprolol. Clin. Pharmacokinet. 1980, 5, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.H.; Hutchinson, V.E.; Pippenger, C.E.; Blumenfeld, T.A.; Feldman, B.R.; Davis, W.J. Effect of dietary protein and carbohydrate on theophylline metabolism in children. Pediatrics 1980, 66, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Kanakoudi, F.; Drossou, V.; Tzimouli, V.; Diamanti, E.; Konstantinidis, T.; Germenis, A.; Kremenopoulos, G. Serum concentrations of 10 acute-phase proteins in healthy term and preterm infants from birth to age 6 months. Clin. Chem. 1995, 41, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Walle, U.K.; Walle, T.; A Bai, S.; Olanoff, L.S. Stereoselective binding of propranolol to human plasma, α1-acid glycoprotein, and albumin. Clin. Pharmacol. Ther. 1983, 34, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, W.; Sponer, G.; Strein, K.; Kling, L.; Martin, U.; Borbe, H.O.; Müller-Beckmann, B.; Böhm, E. Pharmacological characteristics of the stereoisomers of carvedilol. Eur. J. Clin. Pharmacol. 1990, 38 (Suppl. S2), S104–S107. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Kawanabe, Y.; Kato, A.; Tanaka, T. Interaction of propranolol with S100 proteins of the cardiac muscle. Eur. J. Pharmacol. 1996, 315, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.G. Fruit juice inhibition of uptake transport: A new type of food–drug interaction. Br. J. Clin. Pharmacol. 2010, 70, 645–655. [Google Scholar] [CrossRef]
- Jeon, H.; Jang, I.; Lee, S.; Ohashi, K.; Kotegawa, T.; Ieiri, I.; Cho, J.; Yoon, S.H.; Shin, S.; Yu, K.; et al. Apple juice greatly reduces systemic exposure to atenolol. Br. J. Clin. Pharmacol. 2013, 75, 172–179. [Google Scholar] [CrossRef]
- Mimura, Y.; Yasujima, T.; Ohta, K.; Inoue, K.; Yuasa, H. Functional identification of organic cation transporter 1 as an atenolol transporter sensitive to flavonoids. Biochem. Biophys. Rep. 2015, 2, 166–171. [Google Scholar] [CrossRef]
- Lilja, J.J.; Raaska, K.; Neuvonen, P.J. Effects of orange juice on the pharmacokinetics of atenolol. Eur. J. Clin. Pharmacol. 2005, 61, 337–340. [Google Scholar] [CrossRef]
- Fitzgerald, J.D.; Ruffin, R.; Smedstad, K.G.; Roberts, R.; McAinsh, J. Studies on the pharmacokinetics and pharmacodynamics of atenolol in man. Eur. J. Clin. Pharmacol. 1978, 13, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.R.; McAinsh, J.; McIntosh, D.A.D.; Winrow, M.J. Metabolism of atenolol in man. Xenobiotica 1978, 8, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Koda, R.; Maronde, R. Pharmacokinetics, pharmacology of atenolol and effect of renal disease. Br. J. Clin. Pharmacol. 1979, 7, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Saaby, L.; Helms, H.C.C.; Brodin, B. IPEC-J2 MDR1, a Novel High-Resistance Cell Line with Functional Expression of Human P-glycoprotein (ABCB1) for Drug Screening Studies. Mol. Pharm. 2016, 13, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.L.; Clissold, S.P. Controlled release metoprolol formulations. A review of their pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension and ischaemic heart disease. Drugs 1992, 43, 382–414. [Google Scholar] [CrossRef]
- Routledge, P.A.; Shand, D.G. Clinical pharmacokinetics of propranolol. Clin. Pharmacokinet. 1979, 4, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-K.; Song, I.-S. Organic cation transporters and their pharmacokinetic and pharmacodynamic consequences. Drug Metab. Pharmacokinet. 2008, 23, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zeng, S.; Shu, Y. Drug-Drug Interactions at Organic Cation Transporter 1. Front. Pharmacol. 2021, 12, 628705. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Duan, H.; Shirasaka, Y.; Prasad, B.; Wang, J. Atenolol Renal Secretion Is Mediated by Human Organic Cation Transporter 2 and Multidrug and Toxin Extrusion Proteins. Drug Metab. Dispos. 2015, 43, 1872–1881. [Google Scholar] [CrossRef]
- Alayan, J.; Vaquette, C.; Farah, C.; Ivanovski, S. A histomorphometric assessment of collagen-stabilized anorganic bovine bone mineral in maxillary sinus augmentation—A prospective clinical trial. Clin. Oral Implant. Res. 2016, 27, 850–858. [Google Scholar] [CrossRef]
- Iwaki, M.; Niwa, T.; Bandoh, S.; Itoh, M.; Hirose, H.; Kawase, A.; Komura, H. Application of substrate depletion assay to evaluation of CYP isoforms responsible for stereoselective metabolism of carvedilol. Drug Metab. Pharmacokinet. 2016, 31, 425–432. [Google Scholar] [CrossRef]
- Lennard, M.S.; Silas, J.H.; Freestone, S.; Ramsay, L.E.; Tucker, G.T.; Woods, H.F. Oxidation phenotype—A major determinant of metoprolol metabolism and response. N. Engl. J. Med. 1982, 307, 1558–1560. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Hosokawa, S.; Horie, T.; Suzuki, T.; Ohmori, S.; Kitada, M.; Narimatsu, S. Cytochrome P450 isozymes involved in propranolol metabolism in human liver microsomes. The role of CYP2D6 as ring-hydroxylase and CYP1A2 as N-desisopropylase. Drug Metab. Dispos. 1994, 22, 909–915. [Google Scholar]
- McGourty, J.; Silas, J.; Lennard, M.S.; Tucker, G.; Woods, H. Metoprolol metabolism and debrisoquine oxidation polymorphism-population and family studies. Br. J. Clin. Pharmacol. 1985, 20, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Oldham, H.; E Clarke, S. In vitro identification of the human cytochrome P450 enzymes involved in the metabolism of R(+)- and S(-)-carvedilol. Drug Metab. Dispos. 1997, 25, 970–977. [Google Scholar] [PubMed]
- Brown, H.C.; Carruthers, S.G.; Johnston, G.D.; Kelly, J.G.; McAinsh, J.; McDevitt, D.G.; Shanks, R.G. Clinical pharmacologic observations on atenolol, a β-adrenoceptor blocker. Clin. Pharmacol. Ther. 1976, 20, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.D.; Winer, N.; Kochak, G.; Cohen, I.; Bell, R. Kinetics and absolute bioavailability of atenolol. Clin. Pharmacol. Ther. 1979, 25, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Burlew, B.S. Metoprolol metabolism via cytochrome P4502D6 in ethnic populations. Drug Metab. Dispos. 1996, 24, 350–355. [Google Scholar]
- Borg, K.O.; Carlsson, E.; Hoffmann, K.; Jönsson, T.; Thorin, H.; Wallin, B. Metabolism of metoprolol-(3H) in man, the dog and the rat. Acta Pharmacol. Toxicol. 1975, 36 (Suppl. S5), 125–135. [Google Scholar] [CrossRef]
- Murthy, S.S.; Shetty, H.; Nelson, W.L.; Jackson, P.R.; Lennard, M.S. Enantioselective and diastereoselective aspects of the oxidative metabolism of metoprolol. Biochem. Pharmacol. 1990, 40, 1637–1644. [Google Scholar] [CrossRef]
- Antunes, N.d.J.; Cavalli, R.C.; Marques, M.P.; Moisés, E.C.D.; Lanchote, V.L. Influence of gestational diabetes on the stereoselective pharmacokinetics and placental distribution of metoprolol and its metabolites in parturients. Br. J. Clin. Pharmacol. 2015, 79, 605–616. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; McCrink, K.A.; Brill, A. Impact of CYP2D6 Genetic Variation on the Response of the Cardiovascular Patient to Carvedilol and Metoprolol. Curr. Drug Metab. 2015, 17, 30–36. [Google Scholar] [CrossRef]
- Neugebauer, G.; Neubert, P. Metabolism of carvedilol in man. Eur. J. Drug Metab. Pharmacokinet. 1991, 16, 257–260. [Google Scholar] [CrossRef]
- Abrudan, M.B.; Popa, D.S.; Muntean, D.M.; Gheldiu, A.M.; Vlase, L. Pharmacokinetic interactions study between carvedilol and some antidepressants in rat liver microsoms—A comparative study. Med. Pharm. Rep. 2019, 92, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Graff, D.W.; Williamson, K.M.; Pieper, J.A.; Carson, S.W.; Adams, K.F.; Cascio, W.E.; Patterson, J.H. Effect of fluoxetine on carvedilol pharmacokinetics, CYP2D6 activity, and autonomic balance in heart failure patients. J. Clin. Pharmacol. 2001, 41, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Gerards, L.J.; Vermeulen-Meiners, C.; Bruinse, H.W. Neonatal thyrotoxicosis treated with exchange transfusion and Lugol’s iodine. Eur. J. Pediatr. 1985, 143, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Walle, U.K.; Cowart, T.D.; Conradi, E.C. Pathway-selective sex differences in the metabolic clearance of propranolol in human subjects. Clin. Pharmacol. Ther. 1989, 46, 257–263. [Google Scholar] [CrossRef]
- Ou-Yang, D.-S.; Huang, S.-L.; Wang, W.; Xie, H.-G.; Xu, Z.-H.; Shu, Y.; Zhou, H.-H. Phenotypic polymorphism and gender-related differences of CYP1A2 activity in a Chinese population. Br. J. Clin. Pharmacol. 2000, 49, 145–151. [Google Scholar] [CrossRef]
- Olanoff, L.S.; Walle, T.; Walle, U.K.; Cowart, T.D.; E Gaffney, T. Stereoselective clearance and distribution of intravenous propranolol. Clin. Pharmacol. Ther. 1984, 35, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Walle, U.; Wilson, M.; Fagan, T.; Gaffney, T. Stereoselective ring oxidation of propranolol in man. Br. J. Clin. Pharmacol. 1984, 18, 741–748. [Google Scholar] [CrossRef]
- Hanioka, N.; Hayashi, K.; Shimizudani, T.; Nagaoka, K.; Koeda, A.; Naito, S.; Narimatsu, S. Stereoselective glucuronidation of propranolol in human and cynomolgus monkey liver microsomes: Role of human hepatic UDP-glucuronosyltransferase isoforms, UGT1A9, UGT2B4 and UGT2B7. Pharmacology 2008, 82, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Hanioka, N.; Tanaka, S.; Moriguchi, Y.; Narimatsu, S. Stereoselective glucuronidation of carvedilol in human liver and intestinal microsomes. Pharmacology 2012, 90, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ohno, A.; Saito, Y.; Hanioka, N.; Jinno, H.; Saeki, M.; Ando, M.; Ozawa, S.; Sawada, J.-I. Involvement of human hepatic UGT1A1, UGT2B4, and UGT2B7 in the glucuronidation of carvedilol. Drug Metab. Dispos. 2004, 32, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Sten, T.; Qvisen, S.; Uutela, P.; Luukkanen, L.; Kostiainen, R.; Finel, M. Prominent but reverse stereoselectivity in propranolol glucuronidation by human UDP-glucuronosyltransferases 1A9 and 1A10. Drug Metab. Dispos. 2006, 34, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Miyano, J.; Yamamoto, S.; Hanioka, N.; Narimatsu, S.; Ishikawa, T.; Ogura, K.; Watabe, T.; Nishimura, M.; Ueda, N.; Naito, S. Involvement of SULT1A3 in elevated sulfation of 4-hydroxypropranolol in Hep G2 cells pretreated with beta-naphthoflavone. Biochem. Pharmacol. 2005, 69, 941–950. [Google Scholar] [CrossRef] [PubMed]
- The International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Giessmann, T.; Modess, C.; Hecker, U.; Zschiesche, M.; Dazert, P.; Kunert-Keil, C.; Warzok, R.; Engel, G.; Weitschies, W.; Cascorbi, I.; et al. CYP2D6 genotype and induction of intestinal drug transporters by rifampin predict presystemic clearance of carvedilol in healthy subjects. Clin. Pharmacol. Ther. 2004, 75, 213–222. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd. Adrenergic signaling polymorphisms and their impact on cardiovascular disease. Physiol. Rev. 2010, 90, 1013–1062. [Google Scholar] [CrossRef]
- Bristow, M.R.; Ginsburg, R.; Umans, V.; Fowler, M.; Minobe, W.; Rasmussen, R.; Zera, P.; Menlove, R.; Shah, P.; Jamieson, S. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ. Res. 1986, 59, 297–309. [Google Scholar] [CrossRef]
- Cooper-DeHoff, R.M.; Niemi, M.; Ramsey, L.B.; Luzum, J.A.; Tarkiainen, E.K.; Straka, R.J.; Gong, L.; Tuteja, S.; Wilke, R.A.; Wadelius, M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and Statin-Associated Musculoskeletal Symptoms. Clin. Pharmacol. Ther. 2022, 111, 1007–1021. [Google Scholar] [CrossRef]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Gong, L.; Lee, S.; Wagner, J.B.; Zhou, X.; Sangkuhl, K.; Adams, S.M.; Straka, R.J.; Empey, P.E.; Boone, E.C.; et al. PharmVar GeneFocus: SLCO1B1. Clin. Pharmacol. Ther. 2023, 113, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Nakajima, M.; Tamai, I.; Noda, K.; Nezu, J.-I.; Sai, Y.; Tsuji, A.; Yokoi, T. Genetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): Allele frequencies in the Japanese population and functional analysis. J. Pharmacol. Exp. Ther. 2002, 302, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Imai, K.; Nezu, J.-I.; Tsuji, A.; Tamai, I. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J. Pharmacol. Exp. Ther. 2004, 308, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Imanaga, J.; Kotegawa, T.; Imai, H.; Tsutsumi, K.; Yoshizato, T.; Ohyama, T.; Shirasaka, Y.; Tamai, I.; Tateishi, T.; Ohashi, K. The effects of the SLCO2B1 c.1457C>T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet. Genom. 2011, 21, 84–93. [Google Scholar] [CrossRef]
- Shu, Y.; Brown, C.; Castro, R.; Shi, R.; Lin, E.; Owen, R.; Sheardown, S.; Yue, L.; Burchard, E.; Brett, C.; et al. effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 2008, 83, 273–280. [Google Scholar] [CrossRef]
- Nofziger, C.; Turner, A.J.; Sangkuhl, K.; Whirl-Carrillo, M.; Agúndez, J.A.G.; Black, J.L.; Dunnenberger, H.M.; Ruano, G.; Kennedy, M.A.; Phillips, M.S.; et al. PharmVar GeneFocus: CYP2D6. Clin. Pharmacol. Ther. 2020, 107, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef]
- Sehrt, D.; Meineke, I.; Tzvetkov, M.; Gültepe, S.; Brockmöller, J. Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics 2011, 12, 783–795. [Google Scholar] [CrossRef]
- Honda, M.; Nozawa, T.; Igarashi, N.; Inoue, H.; Arakawa, R.; Ogura, Y.; Okabe, H.; Taguchi, M.; Hashimoto, Y. Effect of CYP2D6*10 on the pharmacokinetics of R- and S-carvedilol in healthy Japanese volunteers. Biol. Pharm. Bull. 2005, 28, 1476–1479. [Google Scholar] [CrossRef] [PubMed]
- Takekuma, Y.; Takenaka, T.; Kiyokawa, M.; Yamazaki, K.; Okamoto, H.; Kitabatake, A.; Tsutsui, H.; Sugawara, M. Contribution of polymorphisms in UDP-glucuronosyltransferase and CYP2D6 to the individual variation in disposition of carvedilol. J. Pharm. Pharm Sci. 2006, 9, 101–112. [Google Scholar] [PubMed]
- Zhou, H.-H.; Wood, A.J.J. Stereoselective disposition of carvedilol is determined by CYP2D6. Clin. Pharmacol. Ther. 1995, 57, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chuang, S.; Cheng, C.; Lai, M. Pharmacokinetics of metoprolol enantiomers in Chinese subjects of major CYP2D6 genotypes. Clin. Pharmacol. Ther. 1999, 65, 402–407. [Google Scholar] [CrossRef]
- Blake, C.M.; Kharasch, E.D.; Schwab, M.; Nagele, P. A Meta-analysis of CYP2D6 metabolizer phenotype and metoprolol pharmacokinetics. Clin. Pharmacol. Ther. 2013, 94, 394–399. [Google Scholar] [CrossRef]
- Bijl, M.; Visser, L.; van Schaik, R.; Kors, J.; Witteman, J.; Hofman, A.; Vulto, A.; van Gelder, T.; Stricker, B. Genetic variation in the CYP2D6 gene is associated with a lower heart rate and blood pressure in beta-blocker users. Clin. Pharmacol. Ther. 2009, 85, 45–50. [Google Scholar] [CrossRef]
- Collett, S.; Massmann, A.; Petry, N.J.; Van Heukelom, J.; Schultz, A.; Hellwig, T.; Baye, J.F. Metoprolol and CYP2D6: A Retrospective Cohort Study Evaluating Genotype-Based Outcomes. J. Pers. Med. 2023, 13, 416. [Google Scholar] [CrossRef]
- Lai, M.L.; Wang, S.L.; Lai, M.D.; Lin, E.T.; Tse, M.; Huang, J.D. Propranolol disposition in Chinese subjects of different CYP2D6 genotypes. Clin. Pharmacol. Ther. 1995, 58, 264–268. [Google Scholar] [CrossRef]
- Huang, C.-W.; Lai, M.-L.; Lin, M.-S.; Lee, H.-L.; Huang, J.-D. Dose-response relationships of propranolol in Chinese subjects with different CYP2D6 genotypes. J. Chin. Med. Assoc. 2003, 66, 57–62. [Google Scholar]
- Lennard, M.S.; Jackson, P.; Freestone, S.; Tucker, G.; Ramsay, L.; Woods, H. The relationship between debrisoquine oxidation phenotype and the pharmacokinetics and pharmacodynamics of propranolol. Br. J. Clin. Pharmacol. 1984, 17, 679–685. [Google Scholar] [CrossRef]
- Raghuram, T.C.; Koshakji, R.P.; Wilkinson, G.R.; Wood, A.J.J. Polymorphic ability to metabolize propranolol alters 4-hydroxypropranolol levels but not beta blockade. Clin. Pharmacol. Ther. 1984, 36, 51–56. [Google Scholar] [CrossRef]
- Shaheen, O.; Biollaz, J.; Koshakji, R.P.; Wilkinson, G.R.; Wood, A.J.J. Influence of debrisoquin phenotype on the inducibility of propranolol metabolism. Clin. Pharmacol. Ther. 1989, 45, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, K.M.; Burlew, B.S. Impact of CYP2D6 poor metabolizer phenotype on propranolol pharmacokinetics and response. Pharmacotherapy 1997, 17, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.C.; Ikediobi, O.N. The clinical application of UGT1A1 pharmacogenetic testing: Gene-environment interactions. Hum. Genom. 2010, 4, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Baudhuin, L.M.; Miller, W.L.; Train, L.; Bryant, S.; Hartman, K.A.; Phelps, M.; LaRock, M.; Jaffe, A.S. Relation of ADRB1, CYP2D6, and UGT1A1 polymorphisms with dose of, and response to, carvedilol or metoprolol therapy in patients with chronic heart failure. Am. J. Cardiol. 2010, 106, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.; Moran, M.; McCormick, F.; Pawson, T. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature 1990, 343, 377–381. [Google Scholar] [CrossRef]
- Hillgren, K.M.; Keppler, D.; A Zur, A.; Giacomini, K.M.; Stieger, B.; E Cass, C.; Zhang, L. Emerging transporters of clinical importance: An update from the International Transporter Consortium. Clin. Pharmacol. Ther. 2013, 94, 52–63. [Google Scholar] [CrossRef]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Sangha, V.; Shen, H.; Feng, B.; Wittwer, M.B.; Varma, M.V.S.; Liang, X.; Sugiyama, Y.; Zhang, L.; Bendayan, R.; et al. Transporters in Drug Development: International Transporter Consortium Update on Emerging Transporters of Clinical Importance. Clin. Pharmacol. Ther. 2022, 112, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Terra, S.G. β-adrenergic receptor polymorphisms: Cardiovascular disease associations and pharmacogenetics. Pharm. Res. 2002, 19, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Zineh, I.; Puckett, B.J.; McGorray, S.P.; Yarandi, H.N.; Pauly, D.F. β1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin. Pharmacol. Ther. 2003, 74, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Small, K.M.; McGraw, D.W.; Liggett, S.B. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 381–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Meyers, D.; Javorsky, G.; Burstow, D.; Lolekha, P.; Lucas, M.; Semmler, A.B.; Savarimuthu, S.M.; Fong, K.M.; Yang, I.A.; et al. Arg389Gly-beta1-adrenergic receptors determine improvement in left ventricular systolic function in nonischemic cardiomyopathy patients with heart failure after chronic treatment with carvedilol. Pharmacogenet. Genom. 2007, 17, 941–949. [Google Scholar] [CrossRef]
- Huntgeburth, M.; La Rosée, K.; ten Freyhaus, H.; Böhm, M.; Schnabel, P.; Hellmich, M.; Rosenkranz, S. The Arg389Gly β1-adrenoceptor gene polymorphism influences the acute effects of β-adrenoceptor blockade on contractility in the human heart. Clin. Res. Cardiol. 2011, 100, 641–647. [Google Scholar] [CrossRef]
- Kurnik, D.; Muszkat, M.; Li, C.; Sofowora, G.G.; Friedman, E.A.; Scheinin, M.; Wood, A.J.; Stein, C.M. Genetic variations in the α2A-adrenoreceptor are associated with blood pressure response to the agonist dexmedetomidine. Circ. Cardiovasc. Genet. 2011, 4, 179–187. [Google Scholar] [CrossRef]
- Liggett, S.B.; Mialet-Perez, J.; Thaneemit-Chen, S.; Weber, S.A.; Greene, S.M.; Hodne, D.; Nelson, B.; Morrison, J.; Domanski, M.J.; Wagoner, L.E.; et al. A polymorphism within a conserved β1-adrenergic receptor motif alters cardiac function and β-blocker response in human heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 11288–11293. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Yu, B.; Xu, F.; Mo, W.; Zhou, G.; Liu, Y.; Li, Q.; Zhou, H. β1-Adrenergic receptor polymorphisms influence the response to metoprolol monotherapy in patients with essential hypertension. Clin. Pharmacol. Ther. 2006, 80, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.M.; A Rathz, D.; Petrashevskaya, N.N.; Hahn, H.S.; E Wagoner, L.; Schwartz, A.; Dorn, G.W.; Liggett, S.B. β1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat. Med. 2003, 9, 1300–1305. [Google Scholar] [CrossRef]
- Parvez, B.; Chopra, N.; Rowan, S.; Vaglio, J.C.; Muhammad, R.; Roden, D.M.; Darbar, D. A common β1-adrenergic receptor polymorphism predicts favorable response to rate-control therapy in atrial fibrillation. J. Am. Coll. Cardiol. 2012, 59, 49–56. [Google Scholar] [CrossRef]
- Rau, T.; Düngen, H.-D.; Edelmann, F.; Waagstein, F.; Lainščak, M.; Dimković, S.; Apostolović, S.; Nešković, A.N.; Haverkamp, W.; Gelbrich, G.; et al. Impact of the β1-adrenoceptor Arg389Gly polymorphism on heart-rate responses to bisoprolol and carvedilol in heart-failure patients. Clin. Pharmacol. Ther. 2012, 92, 21–28. [Google Scholar] [CrossRef]
- Sofowora, G.G.; Dishy, V.; Muszkat, M.; Xie, H.G.; Kim, R.B.; Harris, P.A.; Prasad, H.C.; Byrne, D.W.; Nair, U.B.; Wood, A.J.J.; et al. A common β1-adrenergic receptor polymorphism (Arg389Gly) affects blood pressure response to β-blockade. Clin. Pharmacol. Ther. 2003, 73, 366–371. [Google Scholar] [CrossRef]
- Terra, S.G.; Pauly, D.F.; Lee, C.R.; Patterson, J.H.; Adams, K.F.; Schofield, R.S.; Belgado, B.S.; Hamilton, K.K.; Aranda, J.M.; Hill, J.A.; et al. β-adrenergic receptor polymorphisms and responses during titration of metoprolol controlled release/extended release in heart failure. Clin. Pharmacol. Ther. 2005, 77, 127–137. [Google Scholar] [CrossRef]
- Mason, D.A.; Moore, J.D.; Green, S.A.; Liggett, S.B. A Gain-of-function polymorphism in a G-protein coupling domain of the human β1-adrenergic receptor. J. Biol. Chem. 1999, 274, 12670–12674. [Google Scholar] [CrossRef] [PubMed]
- Lobmeyer, M.T.; Gong, Y.; Terra, S.G.; Beitelshees, A.L.; Langaee, T.Y.; Pauly, D.F.; Schofield, R.S.; Hamilton, K.K.; Patterson, J.H.; Adams, K.F.; et al. Synergistic polymorphisms of β1 and α2C-adrenergic receptors and the influence on left ventricular ejection fraction response to β-blocker therapy in heart failure. Pharmacogenet. Genom. 2007, 17, 277–282. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Fiuzat, M.; Carson, P.E.; Anand, I.S.; Plehn, J.F.; Gottlieb, S.S.; Silver, M.A.; Lindenfeld, J.; Miller, A.B.; White, M.; et al. Combinatorial pharmacogenetic interactions of bucindolol and β1, α2C adrenergic receptor polymorphisms. PLoS ONE 2012, 7, e44324. [Google Scholar] [CrossRef] [PubMed]
- de Groote, P.; Helbecque, N.; Lamblin, N.; Hermant, X.; Mc Fadden, E.; Foucher-Hossein, C.; Amouyel, P.; Dallongeville, J.; Bauters, C. Association between β-1 and β-2 adrenergic receptor gene polymorphisms and the response to β-blockade in patients with stable congestive heart failure. Pharmacogenet. Genom. 2005, 15, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Filigheddu, F.; Argiolas, G.; Degortes, S.; Zaninello, R.; Frau, F.; Pitzoi, S.; Bulla, E.; Bulla, P.; Troffa, C.; Glorioso, N. Haplotypes of the adrenergic system predict the blood pressure response to β-blockers in women with essential hypertension. Pharmacogenomics 2010, 11, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Metra, M.; Covolo, L.; Pezzali, N.; Zacà, V.; Bugatti, S.; Lombardi, C.; Bettari, L.; Romeo, A.; Gelatti, U.; Giubbini, R.; et al. Role of β-adrenergic receptor gene polymorphisms in the long-term effects of β-blockade with carvedilol in patients with chronic heart failure. Cardiovasc. Drugs Ther. 2010, 24, 49–60. [Google Scholar] [CrossRef]
- On behalf of the MERIT-HF Study Group; White, H.L.; de Boer, R.A.; Maqbool, A.; Greenwood, D.; van Veldhuisen, D.J.; Cuthbert, R.; Ball, S.G.; Hall, A.S.; Balmforth, A.J. An evaluation of the beta-1 adrenergic receptor Arg389Gly polymorphism in individuals with heart failure: A MERIT-HF sub-study. Eur. J. Heart Fail. 2003, 5, 463–468. [Google Scholar] [CrossRef]
- Levin, M.C.; Marullo, S.; Muntaner, O.; Andersson, B.; Magnusson, Y. The myocardium-protective Gly-49 variant of the β1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J. Biol. Chem. 2002, 277, 30429–30435. [Google Scholar] [CrossRef]
- Rathz, D.A.; Brown, K.M.; Kramer, L.A.; Liggett, S.B. Amino acid 49 polymorphisms of the human β1-adrenergic receptor affect agonist-promoted trafficking. J. Cardiovasc. Pharmacol. 2002, 39, 155–160. [Google Scholar] [CrossRef]
- Funk, R.S.; Brown, J.T.; Abdel-Rahman, S.M. Pediatric pharmacokinetics: Human development and drug disposition. Pediatr. Clin. N. Am. 2012, 59, 1001–1016. [Google Scholar] [CrossRef]
- Friis-Hansen, B. Water distribution in the foetus and newborn infant. Acta Paediatr. Scand. Suppl. 1983, 72, 7–11. [Google Scholar] [CrossRef]
- Kyler, K.E.; Wagner, J.; Hosey-Cojocari, C.; Watt, K.; Shakhnovich, V. Drug Dose Selection in Pediatric Obesity: Available Information for the Most Commonly Prescribed Drugs to Children. Pediatr. Drugs 2019, 21, 357–369. [Google Scholar] [CrossRef]
- Aranda, J.V.; Sitar, D.S.; Parsons, W.D.; Loughnan, P.M.; Neims, A.H. Pharmacokinetic aspects of theophylline in premature newborns. N. Engl. J. Med. 1976, 295, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Kingston, H.G.G.; Kendrick, A.; Sommer, K.M.; Olsen, G.D.; Downes, H. Binding of thiopental in neonatal serum. Anesthesiology 1990, 72, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Meistelman, C.; Benhamou, D.; Barre, J.; Levron, J.-C.; Mahe, V.; Mazoit, X.; Ecoffey, C. Effects of age on plasma protein binding of sufentanil. Anesthesiology 1990, 72, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.N. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 2008, 118, 250–267. [Google Scholar] [CrossRef]
- Blake, M.J.; Gaedigk, A.; E Pearce, R.; Bomgaars, L.R.; Christensen, M.L.; Stowe, C.; James, L.P.; Wilson, J.T.; Kearns, G.L.; Leeder, J.S. Ontogeny of dextromethorphan O- and N-demethylation in the first year of life. Clin. Pharmacol. Ther. 2007, 81, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.C.; Marsh, S.A.; Zaya, M.J.; Regina, K.J.; Divakaran, K.; Le, M.; Hines, R.N. Developmental changes in human liver CYP2D6 expression. Drug Metab. Dispos. 2008, 36, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Treluyer, J.; Jacqz-Aigrain, E.; Alvarez, F.; Cresteil, T. Expression of CYP2D6 in developing human liver. Eur. J. Biochem. 1991, 202, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Cazeneuve, C.; Pons, G.; Rey, E.; Treluyer, J.; Cresteil, T.; Thiroux, G.; D’Athis, P.; Olive, G. Biotransformation of caffeine in human liver microsomes from foetuses, neonates, infants and adults. Br. J. Clin. Pharmacol. 1994, 37, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Sonnier, M.; Cresteil, T. Delayed ontogenesis of CYP1A2 in the human liver. Eur. J. Biochem. 1998, 251, 893–898. [Google Scholar] [CrossRef] [PubMed]
- De Wildt, S.N.; Kearns, G.L.; Leeder, J.S.; van den Anker, J.N. Glucuronidation in humans. pharmacogenetic and developmental aspects. Clin. Pharmacokinet. 1999, 36, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, C.P.; Strassburg, A.; Kneip, S.; Barut, A.; Tukey, R.H.; Rodeck, B.; Manns, M.P. Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut 2002, 50, 259–265. [Google Scholar] [CrossRef]
- Anderson, B.J.; McKee, A.D.; Holford, N.H.G. Size, myths and the clinical pharmacokinetics of analgesia in paediatric patients. Clin. Pharmacokinet. 1997, 33, 313–327. [Google Scholar] [CrossRef]
- Chen, F.-C.M.; Yamamura, H.I.; Roeske, W.R. Ontogeny of mammalian myocardial β-adrenergic receptors. Eur. J. Pharmacol. 1979, 58, 255–264. [Google Scholar] [CrossRef]
- Chen, F.C.; I Yamamura, H.; Roeske, W.R. Adenylate cyclase and β adrenergic receptor development in the mouse heart. J. Pharmacol. Exp. Ther. 1982, 222, 7–13. [Google Scholar]
- Kojima, M.; Ishima, T.; Taniguchi, N.; Kimura, K.; Sada, H.; Sperelakis, N. Developmental changes in β-adrenoceptors, muscarinic cholinoceptors and Ca2+ channels in rat ventricular muscles. Br. J. Pharmacol. 1990, 99, 334–339. [Google Scholar] [CrossRef]
- Miyamoto, S.D.; Stauffer, B.L.; Nakano, S.; Sobus, R.; Nunley, K.; Nelson, P.; Sucharov, C.C. β-adrenergic adaptation in paediatric idiopathic dilated cardiomyopathy. Eur. Heart J. 2012, 35, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lefkowitz, R.; Hausdorff, W. β-adrenergic receptor sequestration. A potential mechanism of receptor resensitization. J. Biol. Chem. 1993, 268, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.-P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef] [PubMed]
- Sucharov, C.C.; Hijmans, J.G.; Sobus, R.D.; Melhado, W.F.A.; Miyamoto, S.D.; Stauffer, B.L. β-Adrenergic receptor antagonism in mice: A model for pediatric heart disease. J. Appl. Physiol. (1985) 2013, 115, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Bathe-Peters, M.; Gmach, P.; Boltz, H.-H.; Einsiedel, J.; Gotthardt, M.; Hübner, H.; Gmeiner, P.; Lohse, M.J.; Annibale, P. Visualization of beta-adrenergic receptor dynamics and differential localization in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2101119118. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
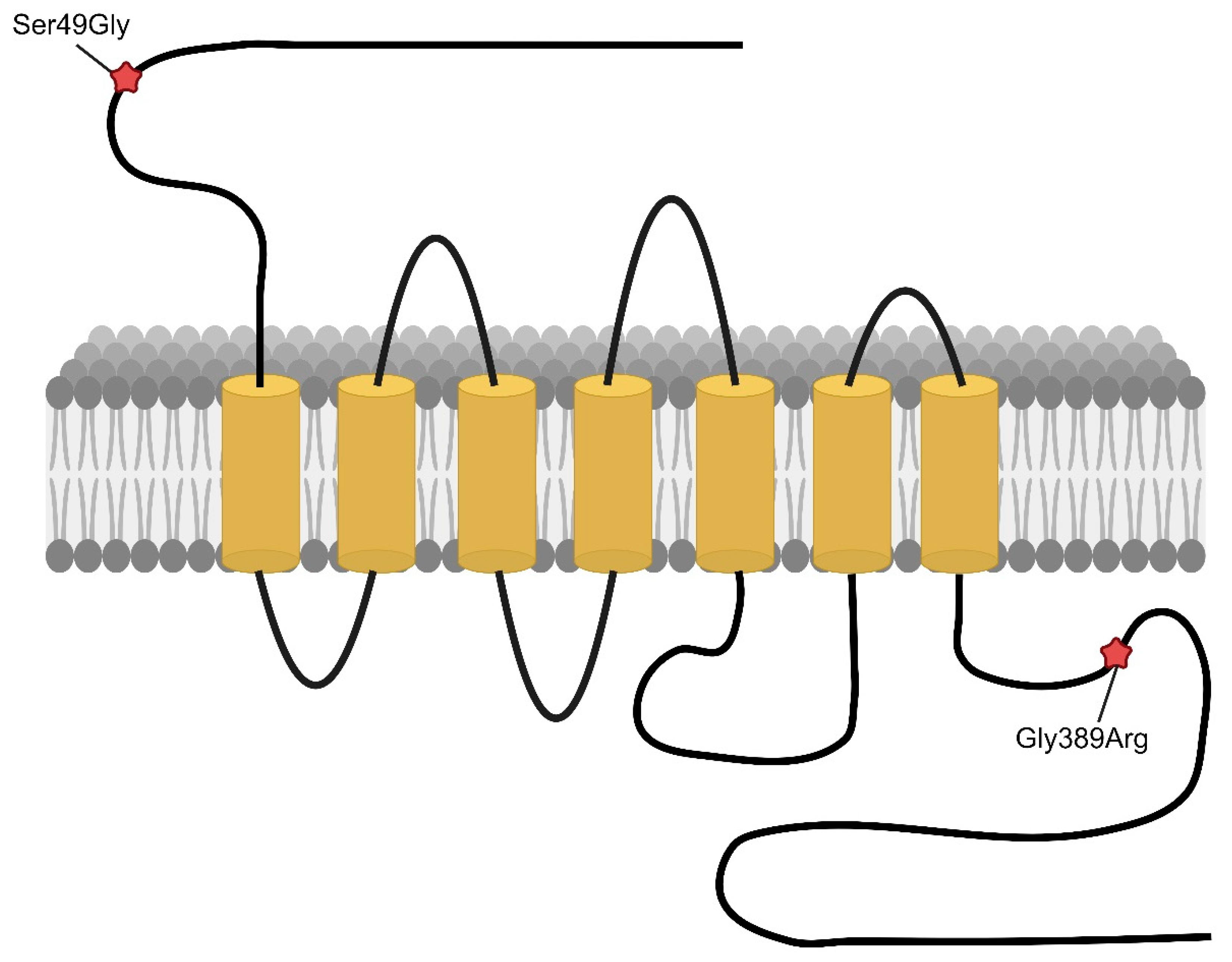{kind=link}
| Beta Blocker (Year of Approval) | Absorption | Distribution: Hepatic/Renal Uptake | Metabolism Phase 1 | Metabolism Phase 2 | Excretion: Efflux |
|---|---|---|---|---|---|
| Atenolol (1975) | OATP1A2 OATP2B1 | OCT1 (liver) OCT2 (kidney) | Minor | None | MATE1 MATE2 MDR1 |
| Carvedilol (1995) | Passive diffusion | Passive diffusion | CYP2D6 (major R+ enantiomer) CYP1A2 (major S− enantiomer) CYP3A4 (minor both R+ and S−) CYP2C9 (minor R+ enantiomer) | UGT1A1 UGT2B4 UGT2B7 | MDR1 (P-gp) MRP2 |
| Metoprolol (1978) | Passive diffusion | Passive diffusion | CYP2D6 CYP2B6 (minor) CYP2C9 (minor) CYP3A4 (minor) | Unknown | Unknown |
| Propranolol (1964) | Passive diffusion | Passive diffusion | CYP1A2 (major: side chain oxidation; minor: ring oxidation) CYP2D6 (major: ring oxidation; (minor: side chain oxidation) | UGT1A9 UGT2B4 UGT2B7 UGT1A10 (enterocyte) SULT1A3 | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, M.; Wagner, J.B. Pediatric Beta Blocker Therapy: A Comprehensive Review of Development and Genetic Variation to Guide Precision-Based Therapy in Children, Adolescents, and Young Adults. Genes 2024, 15, 379. https://doi.org/10.3390/genes15030379
Walton M, Wagner JB. Pediatric Beta Blocker Therapy: A Comprehensive Review of Development and Genetic Variation to Guide Precision-Based Therapy in Children, Adolescents, and Young Adults. Genes. 2024; 15(3):379. https://doi.org/10.3390/genes15030379
Chicago/Turabian StyleWalton, Mollie, and Jonathan B. Wagner. 2024. "Pediatric Beta Blocker Therapy: A Comprehensive Review of Development and Genetic Variation to Guide Precision-Based Therapy in Children, Adolescents, and Young Adults" Genes 15, no. 3: 379. https://doi.org/10.3390/genes15030379