Genes Involved in Type 1 Diabetes: An Update

Abstract
:1. Introduction
2. Genetic Component in Type 1 Diabetes
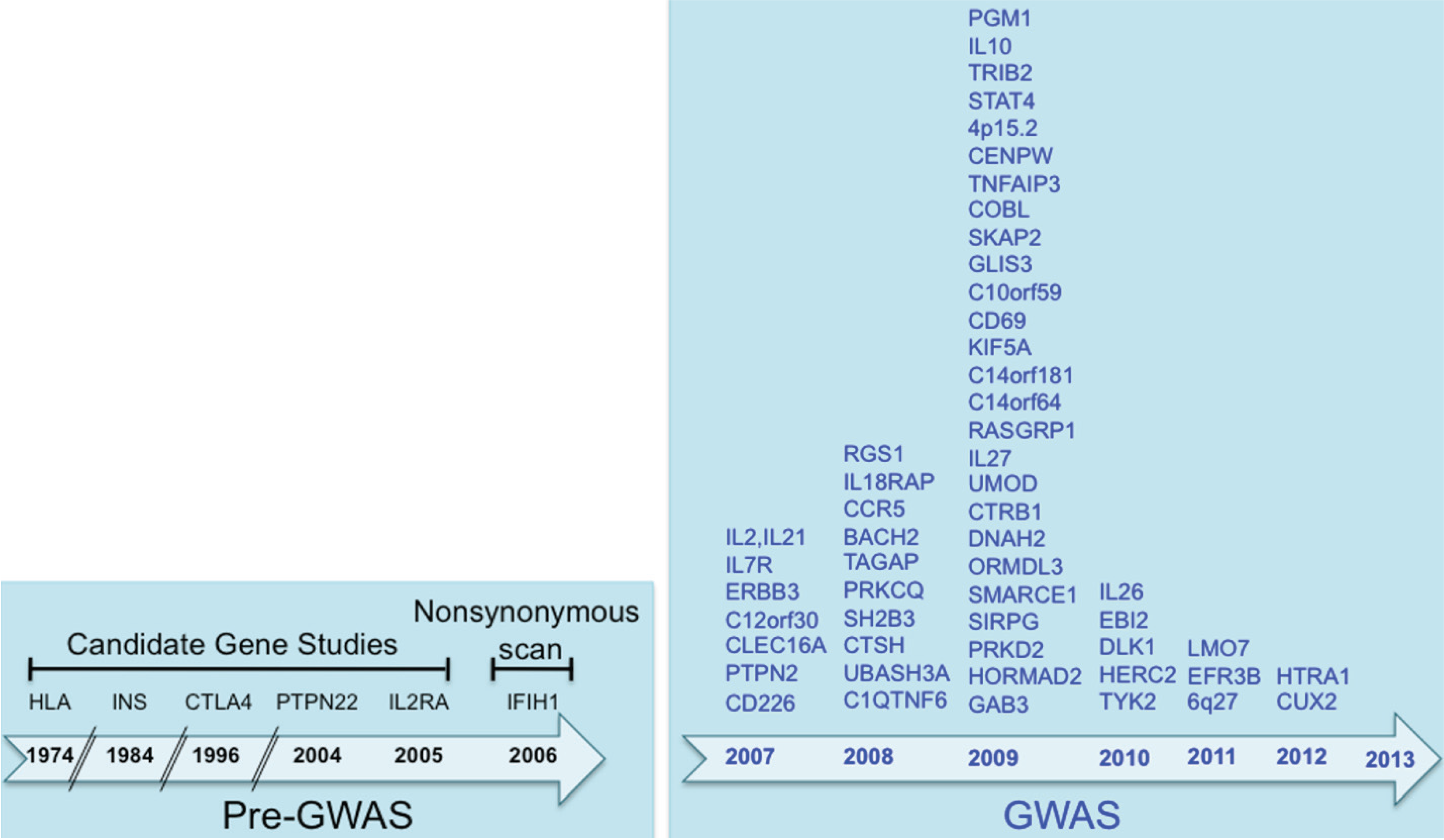
2.1. Before Genome-Wide Association Studies

2.2. GWAS of T1D
2.3. Meta-Analyses of T1D GWAS Datasets

{kind=link}
{kind=link}
{kind=link}
| Reference | Study Type | Main Findings | Sample Size | Replication Sample Size | Ethnic Group | |
|---|---|---|---|---|---|---|
| Hakonarson et al., 2007 [18] | GWAS | HLA-DRB1, HLA-DQA2, CLEC16A, INS, PTPN22 | 467 trios, 561 cases, 1,143 controls | 2,350 individuals in 549 families; 390 trios | European ancestry | |
| WTCCC 2007 [19] | GWAS | HLA-DRB1, INS, CTLA4, PTPN22, IL2RA, IFIH1, PPARG, KCNJ11, TCF7L2 | 1,963 cases, 2,938 controls | see Todd et al., 2007 | European, British | |
| Todd et al., 2007 [20] | GWAS | PHTF1-PTPN22, ERBB3, CLEC16A, C12orf30 | see WTCCC 2007 | 2,997 trios, 4,000 cases, 5,000 controls | European, British | |
| Hakonarson et al., 2008 [21] | GWAS | SUOX-IKZF4 | 467 trios, 561 cases, 1,143 controls | 549 families, 364 trios | European ancestry | |
| Concannon et al., 2008 [22] | GWAS | INS, IFIH1, CLEC16A, UBASH3A | 2,496 families | 2,214 trios, 7,721 cases, 9,679 controls | European ancestry | |
| Cooper et al., 2008 [29] | GWAS meta-analysis | PTPN22, CTLA4, HLA, IL2RA, ERRB3, C12orf30, CLEC16A, PTPN2 | 3,561 cases, 4,646 controls | 6,225 cases, 6,946 controls, 3,064 trios | European ancestry | |
| Grant et al., 2009 [35] | GWAS | EDG7, BACH2, GLIS3, UBASH3A, RASGRP1 | 563 cases, 1,146 controls, 483 case-parents trios | 636 families, 3,303 cases, 4,673 controls | European ancestry | |
| Awata et al., 2009 [39] | TaqMan genotyping | ERBB3, CLEC16A | 735 cases, 621 controls | − | Japanese | |
| Zoledziewska et al., 2009 [440] | TaqMan genotyping | CLEC16A | 1037 cases, 1706 controls | − | European, Sardinian | |
| Fung et al., 2009 [33] | TaqMan genotyping | STAT4, STAT3, ERAP1, TNFAIP3, KIF5A/PIP4K2C | 8010 cases, 9733 controls | − | European, British | |
| Wu et al., 2009 [41] | TaqMan genotyping | CLEC16A | 205 cases, 422 controls | − | Han Chinese | |
| Barrett et al., 2009 [31] | GWAS meta-analysis | MHC, PTPN22, INS, C10orf59, SH2B3, ERBB3, CLEC16A, CTLA4, PTPN2, IL2RA, IL27, C6orf173, IL2, ORMDL3, GLIS3, CD69, IL10, IFIH1, UBASH3A, COBL, BACH2, CTSH, PRKCQ, C1QTNF6, PGM1 | 7,514 cases, 9,045 controls | 4,267 cases, 4,670 controls, 4,342 trios | European | |
| Wallace et al., 2010 [42] | GWAS meta-analysis | DLK1, TYK2 | 7,514 cases, 9,045 controls | 4,840 cases, 2,670 controls, 4,152 trios | European ancestry | |
| Wang et al., 2010 [43] | GWAS | PTPN22, IL10, IFIH1, KIAA0746, BACH2, C6orf173, TAGAP, GLIS3, L2R, INS, ERBB3, C14orf181, IL27, PRKD2, HERC2, CLEC16A, IFNG, IL26 | 989 cases, 6,197 controls | − | European ancestry | |
| Reddy et al., 2011 [44] | TaqMan genotyping | PTPN22, INS, IFIH1, SH2B3, ERBB3, CTLA4, C14orf181, CTSH, CLEC16A, CD69, ITPR3, CENPW, SKAP2, PRKCQ, RNLS, IL27, SIRPG, CTRB2 | 1,434 cases, 1,864 controls | − | European ancestry, southeast USA | |
| Bradfield et al., 2011 [38] | GWAS meta-analysis | LMO7, EFR3B, 6q27, TNFRSF11B, LOC100128081, FOSL2 | 9,934 cases, 16,956 controls | 1,120 trios | European ancestry | |
| Asad et al., 2012 [45] | Genotyping andsequencing | HTR1A, RFN180 | 424 families, 3,078 cases, 1,363 controls | − | European, Scandinavians | |
| Huang et al., 2012 [46] | Genomes-based imputation | CUX2, IL2RA | 16,179 individuals | − | European ancestry | |
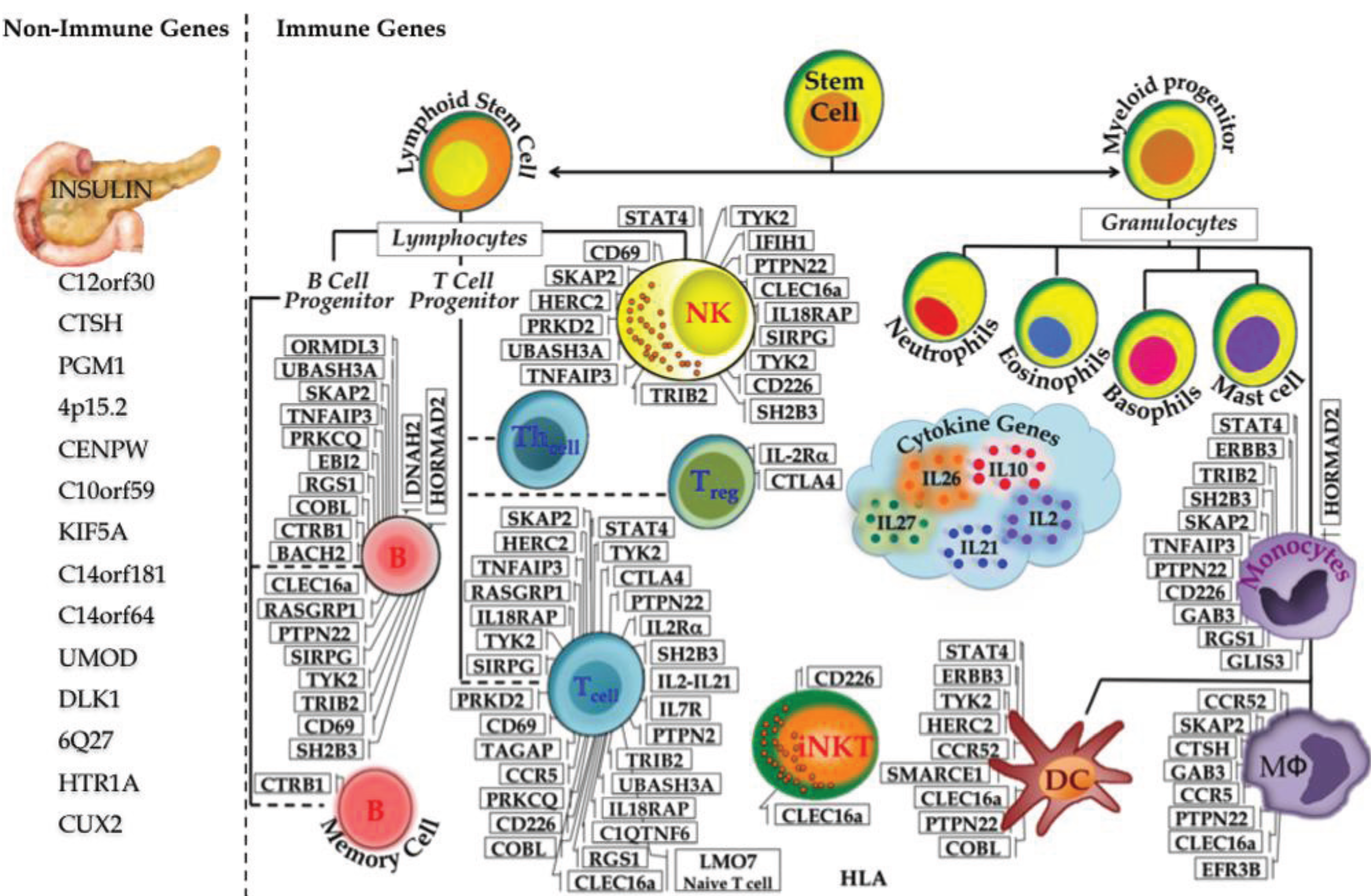
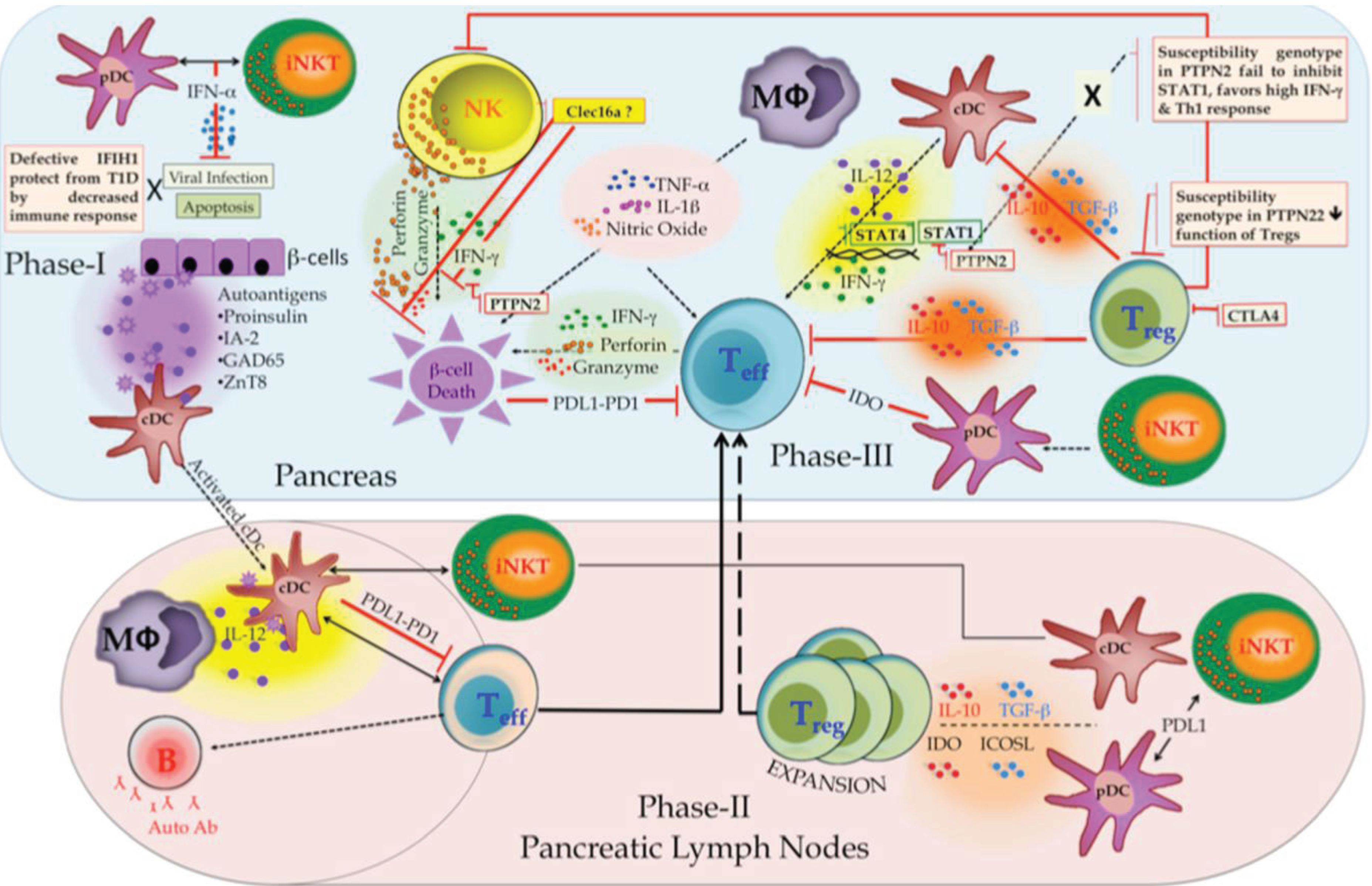
2.4. Immune Components in T1D


2.5. Insights from T1D Specific Loci
2.5.1. CLEC16A (16p13)
2.5.2. Latest Novel T1D Susceptibility Loci (2011–2013)
2.5.2.1. Region 13q22
2.5.2.2. Region 2q23
2.5.2.3. Intergenic Region 6q27
2.5.2.4. Region 12q24
2.5.2.5. Region 5p13-q13
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Steyn, N.P.; Lambert, E.V.; Tabana, H. Conference on “multidisciplinary approaches to nutritional problems”. Symposium on “diabetes and health”. Nutrition interventions for the prevention of type 2 diabetes. Proc. Nutr. Soc. 2009, 68, 55–70. [Google Scholar] [CrossRef]
- International Diabetes Federation. Available online: http://www.idf.org/diabetesatlas/ (accessed on 11 July 2013).
- Eurodiab ACE study group. Variation and trends in incidence of childhood diabetes in europe. Lancet 2000, 355, 873–876. [CrossRef]
- Onkamo, P.; Vaananen, S.; Karvonen, M.; Tuomilehto, J. Worldwide increase in incidence of type Ι diabetes—The analysis of the data on published incidence trends. Diabetologia 1999, 42, 1395–1403. [Google Scholar] [CrossRef]
- Redondo, M.J.; Yu, L.; Hawa, M.; Mackenzie, T.; Pyke, D.A.; Eisenbarth, G.S.; Leslie, R.D. Heterogeneity of type Ι diabetes: Analysis of monozygotic twins in great britain and the united states. Diabetologia 2001, 44, 354–362. [Google Scholar] [CrossRef]
- Clayton, D.G. Prediction and interaction in complex disease genetics: Experience in type 1 diabetes. PLoS Genet. 2009, 5, e1000540. [Google Scholar] [CrossRef]
- Rich, S.S. Mapping genes in diabetes. Genetic epidemiological perspective. Diabetes 1990, 39, 1315–1319. [Google Scholar]
- Cudworth, A.G.; Woodrow, J.C. Evidence for hl-a-linked genes in “juvenile” diabetes mellitus. Br. Med. J. 1975, 3, 133–135. [Google Scholar] [CrossRef]
- Nerup, J.; Platz, P.; Andersen, O.O.; Christy, M.; Lyngsoe, J.; Poulsen, J.E.; Ryder, L.P.; Nielsen, L.S.; Thomsen, M.; Svejgaard, A. Hl-a antigens and diabetes mellitus. Lancet 1974, 2, 864–866. [Google Scholar]
- Singal, D.P.; Blajchman, M.A. Histocompatibility (hl-a) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 1973, 22, 429–432. [Google Scholar]
- Bell, G.I.; Horita, S.; Karam, J.H. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 1984, 33, 176–183. [Google Scholar]
- Nistico, L.; Buzzetti, R.; Pritchard, L.E.; van der Auwera, B.; Giovannini, C.; Bosi, E.; Larrad, M.T.; Rios, M.S.; Chow, C.C.; Cockram, C.S.; et al. The ctla-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian diabetes registry. Hum. Mol. Genet. 1996, 5, 1075–1080. [Google Scholar] [CrossRef]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type Ι diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a type 1 diabetes locus in the il2ra/cd25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous snps identifies a type 1 diabetes locus in the interferon-induced helicase (ifih1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef]
- International HapMap Consortium. The international hapmap project. Nature 2003, 426, 789–796. [CrossRef]
- International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [CrossRef]
- Hakonarson, H.; Grant, S.F.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A genome-wide association study identifies kiaa0350 as a type 1 diabetes gene. Nature 2007, 448, 591–594. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661–678. [CrossRef]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef]
- Hakonarson, H.; Qu, H.Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A novel susceptibility locus for type 1 diabetes on chr12q13 identified by a genome-wide association study. Diabetes 2008, 57, 1143–1146. [Google Scholar] [CrossRef]
- Concannon, P.; Onengut-Gumuscu, S.; Todd, J.A.; Smyth, D.J.; Pociot, F.; Bergholdt, R.; Akolkar, B.; Erlich, H.A.; Hilner, J.E.; Julier, C.; et al. A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 2008, 57, 2858–2861. [Google Scholar] [CrossRef]
- Tsygankov, A.Y. Multidomain sts/tula proteins are novel cellular regulators. IUBMB Life 2008, 60, 224–231. [Google Scholar] [CrossRef]
- Cohen, S.; Dadi, H.; Shaoul, E.; Sharfe, N.; Roifman, C.M. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, lyp. Blood 1999, 93, 2013–2024. [Google Scholar]
- Cooper, J.D.; Walker, N.M.; Smyth, D.J.; Downes, K.; Healy, B.C.; Todd, J.A. Follow-up of 1715 snps from the wellcome trust case control consortium genome-wide association study in type Ι diabetes families. Genes Immun. 2009, 10, S85–S94. [Google Scholar] [CrossRef]
- Johnson, K.; Wong, R.; Barriga, K.J.; Klingensmith, G.; Ziegler, A.G.; Rewers, M.J.; Steck, A.K. Rs11203203 is associated with type 1 diabetes risk in population pre-screened for high-risk hla-dr,dq genotypes. Pediatr. Diabetes 2012, 13, 611–615. [Google Scholar] [CrossRef]
- Manolio, T.A.; Rodriguez, L.L.; Brooks, L.; Abecasis, G.; Ballinger, D.; Daly, M.; Donnelly, P.; Faraone, S.V.; Frazer, K.; Gabriel, S.; et al. New models of collaboration in genome-wide association studies: The genetic association information network. Nat. Genet. 2007, 39, 1045–1051. [Google Scholar] [CrossRef]
- Mueller, P.W.; Rogus, J.J.; Cleary, P.A.; Zhao, Y.; Smiles, A.M.; Steffes, M.W.; Bucksa, J.; Gibson, T.B.; Cordovado, S.K.; Krolewski, A.S.; et al. Genetics of kidneys in diabetes (gokind) study: A genetics collection available for identifying genetic susceptibility factors for diabetic nephropathy in type 1 diabetes. J. Am. Soc. Nephrol. JASN 2006, 17, 1782–1790. [Google Scholar] [CrossRef]
- Cooper, J.D.; Smyth, D.J.; Smiles, A.M.; Plagnol, V.; Walker, N.M.; Allen, J.E.; Downes, K.; Barrett, J.C.; Healy, B.C.; Mychaleckyj, J.C.; et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet. 2008, 40, 1399–1401. [Google Scholar] [CrossRef]
- Espino-Paisan, L.; de La Calle, H.; Fernandez-Arquero, M.; Figueredo, M.A.; de La Concha, E.G.; Urcelay, E.; Santiago, J.L. Study of polymorphisms in 4q27, 10p15, and 22q13 regions in autoantibodies stratified type 1 diabetes patients. Autoimmunity 2011, 44, 624–630. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.D.; Walker, N.M.; Healy, B.C.; Smyth, D.J.; Downes, K.; Todd, J.A. Analysis of 55 autoimmune disease and type ii diabetes loci: Further confirmation of chromosomes 4q27, 12q13.2 and 12q24.13 as type i diabetes loci, and support for a new locus, 12q13.3-q14.1. Genes Immun. 2009, 10, S95–S120. [Google Scholar] [CrossRef]
- Fung, E.Y.; Smyth, D.J.; Howson, J.M.; Cooper, J.D.; Walker, N.M.; Stevens, H.; Wicker, L.S.; Todd, J.A. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/tnfaip3 as a susceptibility locus. Genes Immun. 2009, 10, 188–191. [Google Scholar] [CrossRef]
- Smyth, D.J.; Plagnol, V.; Walker, N.M.; Cooper, J.D.; Downes, K.; Yang, J.H.; Howson, J.M.; Stevens, H.; McManus, R.; Wijmenga, C.; et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Eng. J. Med. 2008, 359, 2767–2777. [Google Scholar] [CrossRef]
- Grant, S.F.; Qu, H.Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Taback, S.P.; Frackelton, E.C.; Eckert, A.W.; et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes 2009, 58, 290–295. [Google Scholar] [CrossRef]
- Qu, H.Q.; Bradfield, J.P.; Li, Q.; Kim, C.; Frackelton, E.; Grant, S.F.; Hakonarson, H.; Polychronakos, C. In silico replication of the genome-wide association results of the type 1 diabetes genetics consortium. Hum. Mol. Genet. 2010, 19, 2534–2538. [Google Scholar] [CrossRef]
- Cooper, J.D.; Howson, J.M.; Smyth, D.; Walker, N.M.; Stevens, H.; Yang, J.H.; She, J.X.; Eisenbarth, G.S.; Rewers, M.; Todd, J.A.; et al. Confirmation of novel type 1 diabetes risk loci in families. Diabetologia 2012, 55, 996–1000. [Google Scholar] [CrossRef]
- Bradfield, J.P.; Qu, H.Q.; Wang, K.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Mentch, F.D.; Qiu, H.; Glessner, J.T.; Thomas, K.A.; et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011, 7, e1002293. [Google Scholar] [CrossRef]
- Awata, T.; Kawasaki, E.; Tanaka, S.; Ikegami, H.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Kobayashi, T.; Iizuka, H.; Uga, M.; et al. Association of type 1 diabetes with two loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in japanese. J. Clin. Endocrinol. Metab. 2009, 94, 231–235. [Google Scholar]
- Zoledziewska, M.; Costa, G.; Pitzalis, M.; Cocco, E.; Melis, C.; Moi, L.; Zavattari, P.; Murru, R.; Lampis, R.; Morelli, L.; et al. Variation within the clec16a gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in sardinia. Genes Immun. 2009, 10, 15–17. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, X.; Wang, X.; Ma, J.; Zhu, S.; Li, J.; Liu, Y. Intron polymorphism in the kiaa0350 gene is reproducibly associated with susceptibility to type 1 diabetes (t1d) in the han chinese population. Clin. Endocrinol. 2009, 71, 46–49. [Google Scholar] [CrossRef]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted dlk1-meg3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef]
- Wang, K.; Baldassano, R.; Zhang, H.; Qu, H.Q.; Imielinski, M.; Kugathasan, S.; Annese, V.; Dubinsky, M.; Rotter, J.I.; Russell, R.K.; et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum. Mol. Genet. 2010, 19, 2059–2067. [Google Scholar] [CrossRef]
- Reddy, M.V.; Wang, H.; Liu, S.; Bode, B.; Reed, J.C.; Steed, R.D.; Anderson, S.W.; Steed, L.; Hopkins, D.; She, J.X. Association between type 1 diabetes and gwas snps in the southeast us caucasian population. Genes Immun. 2011, 12, 208–212. [Google Scholar] [CrossRef]
- Asad, S.; Nikamo, P.; Gyllenberg, A.; Bennet, H.; Hansson, O.; Wierup, N.; Carlsson, A.; Forsander, G.; Ivarsson, S.A.; Larsson, H.; et al. Htr1a a novel type 1 diabetes susceptibility gene on chromosome 5p13-q13. PLoS One 2012, 7, e35439. [Google Scholar]
- Huang, J.; Ellinghaus, D.; Franke, A.; Howie, B.; Li, Y. 1,000 genomes-based imputation identifies novel and refined associations for the wellcome trust case control consortium phase 1 data. Eur. J. Hum. Genet. 2012, 20, 801–805. [Google Scholar] [CrossRef]
- Litman, G.W.; Cannon, J.P.; Dishaw, L.J. Reconstructing immune phylogeny: New perspectives. Nat. Rev. Immunol. 2005, 5, 866–879. [Google Scholar] [CrossRef]
- Storling, J.; Brorsson, C.A. Candidate genes expressed in human islets and their role in the pathogenesis of type 1 diabetes. Curr. Diab. Rep. 2013, in press. [Google Scholar]
- Van Belle, T.L.; Coppieters, K.T.; von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef]
- Peakman, M. Immunological pathways to β-cell damage in type 1 diabetes. Diabet. Med. 2013, 30, 147–154. [Google Scholar] [CrossRef]
- Diana, J.; Gahzarian, L.; Simoni, Y.; Lehuen, A. Innate immunity in type 1 diabetes. Discov. Med. 2011, 11, 513–520. [Google Scholar]
- Robinson, M.J.; Sancho, D.; Slack, E.C.; LeibundGut-Landmann, S.; Reis e Sousa, C. Myeloid c-type lectins in innate immunity. Nat. Immunol. 2006, 7, 1258–1265. [Google Scholar] [CrossRef]
- Cambi, A.; Figdor, C.G. Dual function of c-type lectin-like receptors in the immune system. Curr. Opin. Cell Biol. 2003, 15, 539–546. [Google Scholar] [CrossRef]
- Martinez, A.; Perdigones, N.; Cenit, M.C.; Espino, L.; Varade, J.; Lamas, J.R.; Santiago, J.L.; Fernandez-Arquero, M.; de la Calle, H.; Arroyo, R.; et al. Chromosomal region 16p13: Further evidence of increased predisposition to immune diseases. Ann. Rheum. Dis. 2010, 69, 309–311. [Google Scholar] [CrossRef]
- Sang, Y.; Zong, W.; Yan, J.; Liu, M. The correlation between the clec16a gene and genetic susceptibility to type 1 diabetes in chinese children. Int. J. Endocrinol. 2012, 2012, Article ID 245384. [Google Scholar]
- Yamashita, H.; Awata, T.; Kawasaki, E.; Ikegami, H.; Tanaka, S.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Takahashi, K.; Kobayashi, T.; et al. Analysis of the hla and non-hla susceptibility loci in japanese type 1 diabetes. Diabet. Metab. Res. Rev. 2011, 27, 844–848. [Google Scholar]
- Howson, J.M.; Rosinger, S.; Smyth, D.J.; Boehm, B.O.; Todd, J.A. Genetic analysis of adult-onset autoimmune diabetes. Diabetes 2011, 60, 2645–2653. [Google Scholar] [CrossRef]
- Nischwitz, S.; Cepok, S.; Kroner, A.; Wolf, C.; Knop, M.; Muller-Sarnowski, F.; Pfister, H.; Rieckmann, P.; Hemmer, B.; Ising, M.; et al. More clec16a gene variants associated with multiple sclerosis. Acta Neurol. Scand. 2011, 123, 400–406. [Google Scholar] [CrossRef]
- Zuvich, R.L.; Bush, W.S.; McCauley, J.L.; Beecham, A.H.; de Jager, P.L.; Ivinson, A.J.; Compston, A.; Hafler, D.A.; Hauser, S.L.; Sawcer, S.J.; et al. Interrogating the complex role of chromosome 16p13.13 in multiple sclerosis susceptibility: Independent genetic signals in the ciita-clec16a-socs1 gene complex. Hum. Mol. Genet. 2011, 20, 3517–3524. [Google Scholar] [CrossRef]
- Skinningsrud, B.; Husebye, E.S.; Pearce, S.H.; McDonald, D.O.; Brandal, K.; Wolff, A.B.; Lovas, K.; Egeland, T.; Undlien, D.E. Polymorphisms in clec16a and ciita at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 2008, 93, 3310–3317. [Google Scholar]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies tnip1, prdm1, jazf1, uhrf1bp1 and il10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, Y.; Zhou, X.; Li, Y.; Gao, J.; Han, J.; Quan, C.; He, S.; Lv, Y.; Hu, D.; et al. Polymorphisms at 16p13 are associated with systemic lupus erythematosus in the chinese population. J. Med. Genet. 2011, 48, 69–72. [Google Scholar] [CrossRef]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adany, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Marquez, A.; Varade, J.; Robledo, G.; Martinez, A.; Mendoza, J.L.; Taxonera, C.; Fernandez-Arquero, M.; Diaz-Rubio, M.; Gomez-Garcia, M.; Lopez-Nevot, M.A.; et al. Specific association of a clec16a/kiaa0350 polymorphism with nod2/card15(-) crohn’s disease patients. Eur. J. Hum. Genet. 2009, 17, 1304–1308. [Google Scholar] [CrossRef]
- Jagielska, D.; Redler, S.; Brockschmidt, F.F.; Herold, C.; Pasternack, S.M.; Garcia Bartels, N.; Hanneken, S.; Eigelshoven, S.; Refke, M.; Barth, S.; et al. Follow-up study of the first genome-wide association scan in alopecia areata: Il13 and kiaa0350 as susceptibility loci supported with genome-wide significance. J. Invest. Dermatol. 2012, 132, 2192–2197. [Google Scholar] [CrossRef]
- Skinningsrud, B.; Lie, B.A.; Husebye, E.S.; Kvien, T.K.; Forre, O.; Flato, B.; Stormyr, A.; Joner, G.; Njolstad, P.R.; Egeland, T.; et al. A clec16a variant confers risk for juvenile idiopathic arthritis and anti-cyclic citrullinated peptide antibody negative rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1471–1474. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Xie, G.; Lu, E.; Sun, Y.; Juran, B.D.; Chellappa, V.; Coltescu, C.; Mason, A.L.; Milkiewicz, P.; Myers, R.P; et al. Association of primary biliary cirrhosis with variants in the clec16a, socs1, spib and siae immunomodulatory genes. Genes Immun. 2012, 13, 328–335. [Google Scholar] [CrossRef]
- Mells, G.F.; Floyd, J.A.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2011, 43, 329–332. [Google Scholar] [CrossRef]
- Davison, L.J.; Wallace, C.; Cooper, J.D.; Cope, N.F.; Wilson, N.K.; Smyth, D.J.; Howson, J.M.; Saleh, N.; Al-Jeffery, A.; Angus, K.L.; et al. Long-range DNA looping and gene expression analyses identify dexi as an autoimmune disease candidate gene. Hum. Mol. Genet. 2012, 21, 322–333. [Google Scholar] [CrossRef]
- Kim, S.; Wairkar, Y.P.; Daniels, R.W.; DiAntonio, A. The novel endosomal membrane protein ema interacts with the class c vps-hops complex to promote endosomal maturation. J. Cell Biol. 2010, 188, 717–734. [Google Scholar] [CrossRef]
- Kim, S.; Naylor, S.A.; DiAntonio, A. Drosophila golgi membrane protein ema promotes autophagosomal growth and function. Proc. Natl. Acad. Sci. USA 2012, 109, E1072–E1081. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Chen, C.; Yan, Y.; Jiang, S.; Shao, B.; Xu, J.; Kang, L.; Huang, Y.; Zhu, L.; et al. Involvement of clec16a in activation of astrocytes after lps treated. Neurochem. Res. 2012, 37, 5–14. [Google Scholar] [CrossRef]
- Ooshio, T.; Irie, K.; Morimoto, K.; Fukuhara, A.; Imai, T.; Takai, Y. Involvement of lmo7 in the association of two cell-cell adhesion molecules, nectin and e-cadherin, through afadin and alpha-actinin in epithelial cells. J. Biol. Chem. 2004, 279, 31365–31373. [Google Scholar]
- Yamada, A.; Irie, K.; Fukuhara, A.; Ooshio, T.; Takai, Y. Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in mdck cells. Genes Cells 2004, 9, 843–855. [Google Scholar] [CrossRef]
- Furuya, M.; Tsuji, N.; Endoh, T.; Moriai, R.; Kobayashi, D.; Yagihashi, A.; Watanabe, N. A novel gene containing pdz and lim domains, pcd1, is overexpressed in human colorectal cancer. Anticancer Res. 2002, 22, 4183–4186. [Google Scholar]
- Kang, S.; Xu, H.; Duan, X.; Liu, J.J.; He, Z.; Yu, F.; Zhou, S.; Meng, X.Q.; Cao, M.; Kennedy, G.C. Pcd1, a novel gene containing pdz and lim domains, is overexpressed in several human cancers. Cancer Res. 2000, 60, 5296–5302. [Google Scholar]
- Lindvall, J.M.; Blomberg, K.E.; Wennborg, A.; Smith, C.I. Differential expression and molecular characterisation of lmo7, myo1e, sash1, and mcoln2 genes in btk-defective b-cells. Cell. Immunol. 2005, 235, 46–55. [Google Scholar] [CrossRef]
- Semenova, E.; Wang, X.; Jablonski, M.M.; Levorse, J.; Tilghman, S.M. An engineered 800 kilobase deletion of uchl3 and lmo7 on mouse chromosome 14 causes defects in viability, postnatal growth and degeneration of muscle and retina. Hum. Mol. Genet. 2003, 12, 1301–1312. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Nakamura, H.; Mukai, M.; Komatsu, K.; Tanaka-Okamoto, M.; Itoh, Y.; Ishizaki, H.; Tatsuta, M.; Inoue, M.; Miyoshi, J. Transforming growth factor-beta1 induces lmo7 while enhancing the invasiveness of rat ascites hepatoma cells. Cancer Lett. 2005, 220, 95–99. [Google Scholar] [CrossRef]
- Kutlu, B.; Burdick, D.; Baxter, D.; Rasschaert, J.; Flamez, D.; Eizirik, D.L.; Welsh, N.; Goodman, N.; Hood, L. Detailed transcriptome atlas of the pancreatic beta cell. BMC Med. Genomics 2009, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.B.; Salen, G.; Hidaka, H.; Kwiterovich, P.O.; Stalenhoef, A.F.; Miettinen, T.A.; Grundy, S.M.; Lee, M.H.; Rubenstein, J.S.; Polymeropoulos, M.H.; et al. Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J. Clin. Invest. 1998, 102, 1041–1044. [Google Scholar] [CrossRef]
- Shulenin, S.; Schriml, L.M.; Remaley, A.T.; Fojo, S.; Brewer, B.; Allikmets, R.; Dean, M. An atp-binding cassette gene (abcg5) from the abcg (white) gene subfamily maps to human chromosome 2p21 in the region of the sitosterolemia locus. Cytogenet. Cell Genet. 2001, 92, 204–208. [Google Scholar] [CrossRef]
- Zumsteg, U.; Muller, P.Y.; Miserez, A.R. Alstrom syndrome: Confirmation of linkage to chromosome 2p12-13 and phenotypic heterogeneity in three affected sibs. J. Med. Genet. 2000, 37, E8. [Google Scholar] [CrossRef]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar]
- Torchia, J.; Rose, D.W.; Inostroza, J.; Kamei, Y.; Westin, S.; Glass, C.K.; Rosenfeld, M.G. The transcriptional co-activator p/cip binds cbp and mediates nuclear-receptor function. Nature 1997, 387, 677–684. [Google Scholar] [CrossRef]
- Okada, M.; Cheeseman, I.M.; Hori, T.; Okawa, K.; McLeod, I.X.; Yates, J.R., 3rd; Desai, A.; Fukagawa, T. The cenp-h-i complex is required for the efficient incorporation of newly synthesized cenp-a into centromeres. Nat. Cell Biol. 2006, 8, 446–457. [Google Scholar] [CrossRef]
- Ludwig, M.G.; Seuwen, K. Characterization of the human adenylyl cyclase gene family: Cdna, gene structure, and tissue distribution of the nine isoforms. J. Recept. Signal Transduct. Res. 2002, 22, 79–110. [Google Scholar] [CrossRef]
- Wong, S.T.; Trinh, K.; Hacker, B.; Chan, G.C.; Lowe, G.; Gaggar, A.; Xia, Z.; Gold, G.H.; Storm, D.R. Disruption of the type iii adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron 2000, 27, 487–497. [Google Scholar] [CrossRef]
- Nepomuceno-Silva, J.L.; de Melo, L.D.; Mendonca, S.M.; Paixao, J.C.; Lopes, U.G. Rjls: A new family of ras-related gtp-binding proteins. Gene 2004, 327, 221–232. [Google Scholar] [CrossRef]
- Hung, C.N.; Poon, W.T.; Lee, C.Y.; Law, C.Y.; Chan, A.Y. A case of early-onset obesity, hypocortisolism, and skin pigmentation problem due to a novel homozygous mutation in the proopiomelanocortin (pomc) gene in an indian boy. J. Pediatr. Endocrinol. Metab. 2012, 25, 175–179. [Google Scholar]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by pomc mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Martin, R.J.; Savage, D.A.; Carson, D.J.; McKnight, A.J.; Maxwell, A.P.; Patterson, C.C. Association analysis of proopiomelanocortin (pomc) haplotypes in type 1 diabetes in a uk population. Diabetes Metab. 2011, 37, 298–304. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Ito, E.; Yuasa, Y.; Maruyama, K. The human DNA methyltransferases dnmt3a and dnmt3b have two types of promoters with different cpg contents. Biochim. Biophys. Acta 2002, 1577, 457–465. [Google Scholar] [CrossRef]
- Yoo, A.S.; Staahl, B.T.; Chen, L.; Crabtree, G.R. Microrna-mediated switching of chromatin-remodelling complexes in neural development. Nature 2009, 460, 642–646. [Google Scholar]
- Dontje, W.; Schotte, R.; Cupedo, T.; Nagasawa, M.; Scheeren, F.; Gimeno, R.; Spits, H.; Blom, B. Delta-like1-induced notch1 signaling regulates the human plasmacytoid dendritic cell versus t-cell lineage decision through control of gata-3 and spi-b. Blood 2006, 107, 2446–2452. [Google Scholar] [CrossRef]
- Santos, M.A.; Sarmento, L.M.; Rebelo, M.; Doce, A.A.; Maillard, I.; Dumortier, A.; Neves, H.; Radtke, F.; Pear, W.S.; Parreira, L.; et al. Notch1 engagement by delta-like-1 promotes differentiation of b lymphocytes to antibody-secreting cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15454–15459. [Google Scholar] [CrossRef]
- Su, Y.; Buchler, P.; Gazdhar, A.; Giese, N.; Reber, H.A.; Hines, O.J.; Giese, T.; Buchler, M.W.; Friess, H. Pancreatic regeneration in chronic pancreatitis requires activation of the notch signaling pathway. J. Gastrointest. Surg. 2006, 10, 1230–1241. [Google Scholar] [CrossRef]
- Li, D.; Kang, Q.; Wang, D.M. Constitutive coactivator of peroxisome proliferator-activated receptor (ppargamma), a novel coactivator of ppargamma that promotes adipogenesis. Mol. Endocrinol. 2007, 21, 2320–2333. [Google Scholar] [CrossRef]
- Trachtulec, Z.; Hamvas, R.M.; Forejt, J.; Lehrach, H.R.; Vincek, V.; Klein, J. Linkage of tata-binding protein and proteasome subunit c5 genes in mice and humans reveals synteny conserved between mammals and invertebrates. Genomics 1997, 44, 1–7. [Google Scholar] [CrossRef]
- Keutgens, A.; Zhang, X.; Shostak, K.; Robert, I.; Olivier, S.; Vanderplasschen, A.; Chapelle, J.P.; Viatour, P.; Merville, M.P.; Bex, F.; et al. Bcl-3 degradation involves its polyubiquitination through a fbw7-independent pathway and its binding to the proteasome subunit psmb1. J. Biol. Chem. 2010, 285, 25831–25840. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the pd-1 antigen on the surface of stimulated mouse t and b lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Cohen, D.R.; Ferreira, P.C.; Gentz, R.; Franza, B.R., Jr.; Curran, T. The product of a fos-related gene, fra-1, binds cooperatively to the ap-1 site with jun: Transcription factor ap-1 is comprised of multiple protein complexs. Genes Dev. 1989, 3, 173–184. [Google Scholar] [CrossRef]
- Gingras, H.; Cases, O.; Krasilnikova, M.; Berube, G.; Nepveu, A. Biochemical characterization of the mammalian cux2 protein. Gene 2005, 344, 273–285. [Google Scholar] [CrossRef]
- Iulianella, A.; Sharma, M.; Durnin, M.; Vanden Heuvel, G.B.; Trainor, P.A. Cux2 (cutl2) integrates neural progenitor development with cell-cycle progression during spinal cord neurogenesis. Development 2008, 135, 729–741. [Google Scholar] [CrossRef]
- Iwata, I.; Nagafuchi, S.; Nakashima, H.; Kondo, S.; Koga, T.; Yokogawa, Y.; Akashi, T.; Shibuya, T.; Umeno, Y.; Okeda, T.; et al. Association of polymorphism in the neurod/beta2 gene with type 1 diabetes in the japanese. Diabetes 1999, 48, 416–419. [Google Scholar] [CrossRef]
- Kavvoura, F.K.; Ioannidis, J.P. Ala45thr polymorphism of the neurod1 gene and diabetes susceptibility: A meta-analysis. Hum. Genet. 2005, 116, 192–199. [Google Scholar] [CrossRef]
- Nerup, J.; Pociot, F. A genomewide scan for type 1-diabetes susceptibility in scandinavian families: Identification of new loci with evidence of interactions. Am. J. Hum. Genet. 2001, 69, 1301–1313. [Google Scholar] [CrossRef]
- Barnes, N.M.; Sharp, T. A review of central 5-ht receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Lesurtel, M.; Soll, C.; Graf, R.; Clavien, P.A. Role of serotonin in the hepato-gastrointestinal tract: An old molecule for new perspectives. Cell. Mol. life Sci. 2008, 65, 940–952. [Google Scholar] [CrossRef]
- Sundler, F.; Hakanson, R.; Loren, I.; Lundquist, I. Amine storage and function in peptide hormone-producing cells. Invest. Cell Pathol. 1980, 3, 87–103. [Google Scholar]
- Zawalich, W.S.; Tesz, G.J.; Zawalich, K.C. Effects of prior 5-hydroxytryptamine exposure on rat islet insulin secretory and phospholipase c responses. Endocrine 2004, 23, 11–16. [Google Scholar] [CrossRef]
- Coulie, B.; Tack, J.; Bouillon, R.; Peeters, T.; Janssens, J. 5-hydroxytryptamine-1 receptor activation inhibits endocrine pancreatic secretion in humans. Am. J. Physiol. 1998, 274, E317–E320. [Google Scholar]
- Mohanan, V.V.; Khan, R.; Paulose, C.S. Hypothalamic 5-ht functional regulation through 5-ht1a and 5-ht2c receptors during pancreatic regeneration. Life Sci. 2006, 78, 1603–1609. [Google Scholar] [CrossRef]
- Aune, T.M.; McGrath, K.M.; Sarr, T.; Bombara, M.P.; Kelley, K.A. Expression of 5ht1a receptors on activated human t cells. Regulation of cyclic amp levels and t cell proliferation by 5-hydroxytryptamine. J. Immunol. 1993, 151, 1175–1183. [Google Scholar]
- Wei, Z.; Wang, K.; Qu, H.Q.; Zhang, H.; Bradfield, J.; Kim, C.; Frackleton, E.; Hou, C.; Glessner, J.T.; Chiavacci, R.; et al. From disease association to risk assessment: An optimistic view from genome-wide association studies on type 1 diabetes. PLoS Genet. 2009, 5, e1000678. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bakay, M.; Pandey, R.; Hakonarson, H. Genes Involved in Type 1 Diabetes: An Update. Genes 2013, 4, 499-521. https://doi.org/10.3390/genes4030499
Bakay M, Pandey R, Hakonarson H. Genes Involved in Type 1 Diabetes: An Update. Genes. 2013; 4(3):499-521. https://doi.org/10.3390/genes4030499
Chicago/Turabian StyleBakay, Marina, Rahul Pandey, and Hakon Hakonarson. 2013. "Genes Involved in Type 1 Diabetes: An Update" Genes 4, no. 3: 499-521. https://doi.org/10.3390/genes4030499
APA StyleBakay, M., Pandey, R., & Hakonarson, H. (2013). Genes Involved in Type 1 Diabetes: An Update. Genes, 4(3), 499-521. https://doi.org/10.3390/genes4030499