Delivery Mode and the Transition of Pioneering Gut-Microbiota Structure, Composition and Predicted Metabolic Function

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Subjects
2.2. Sample Collection and 16S Ribosomal RNA Sequencing
2.3. 16S rRNA Gene Sequence Analysis
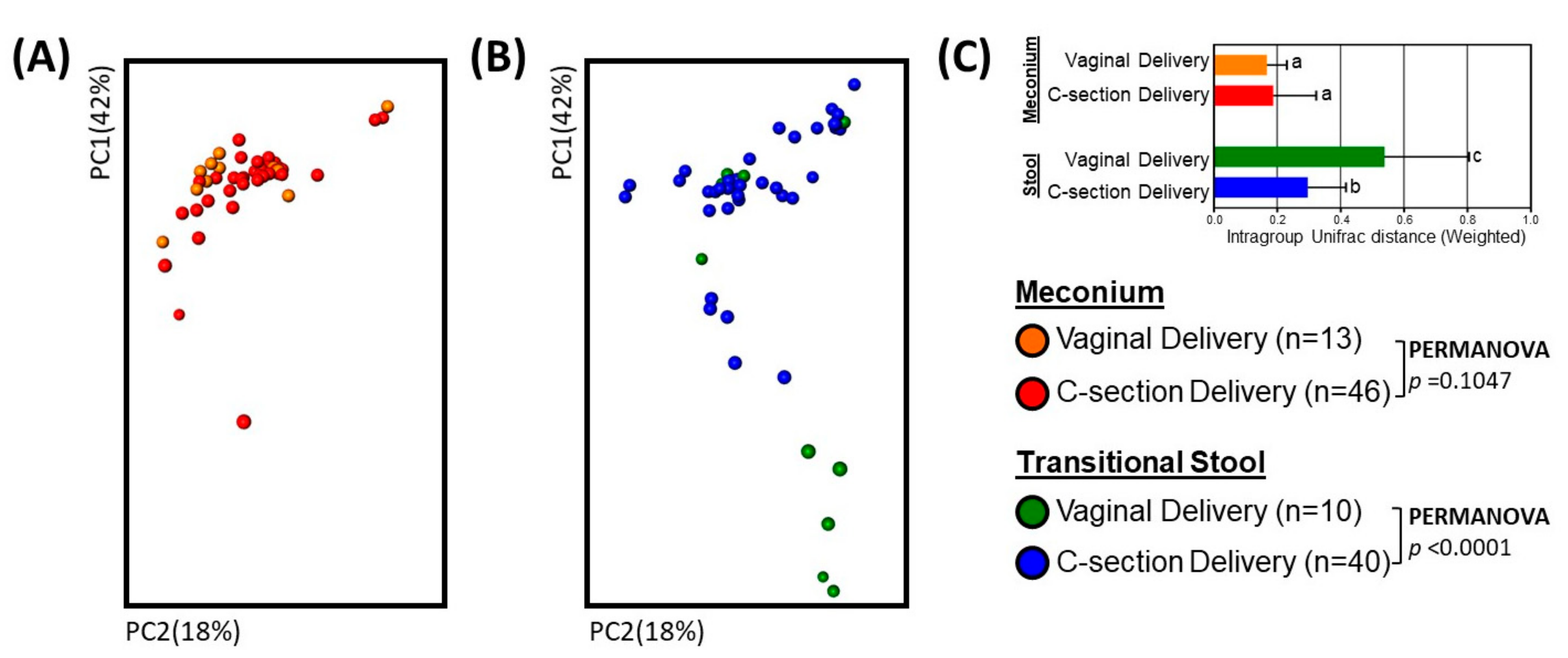
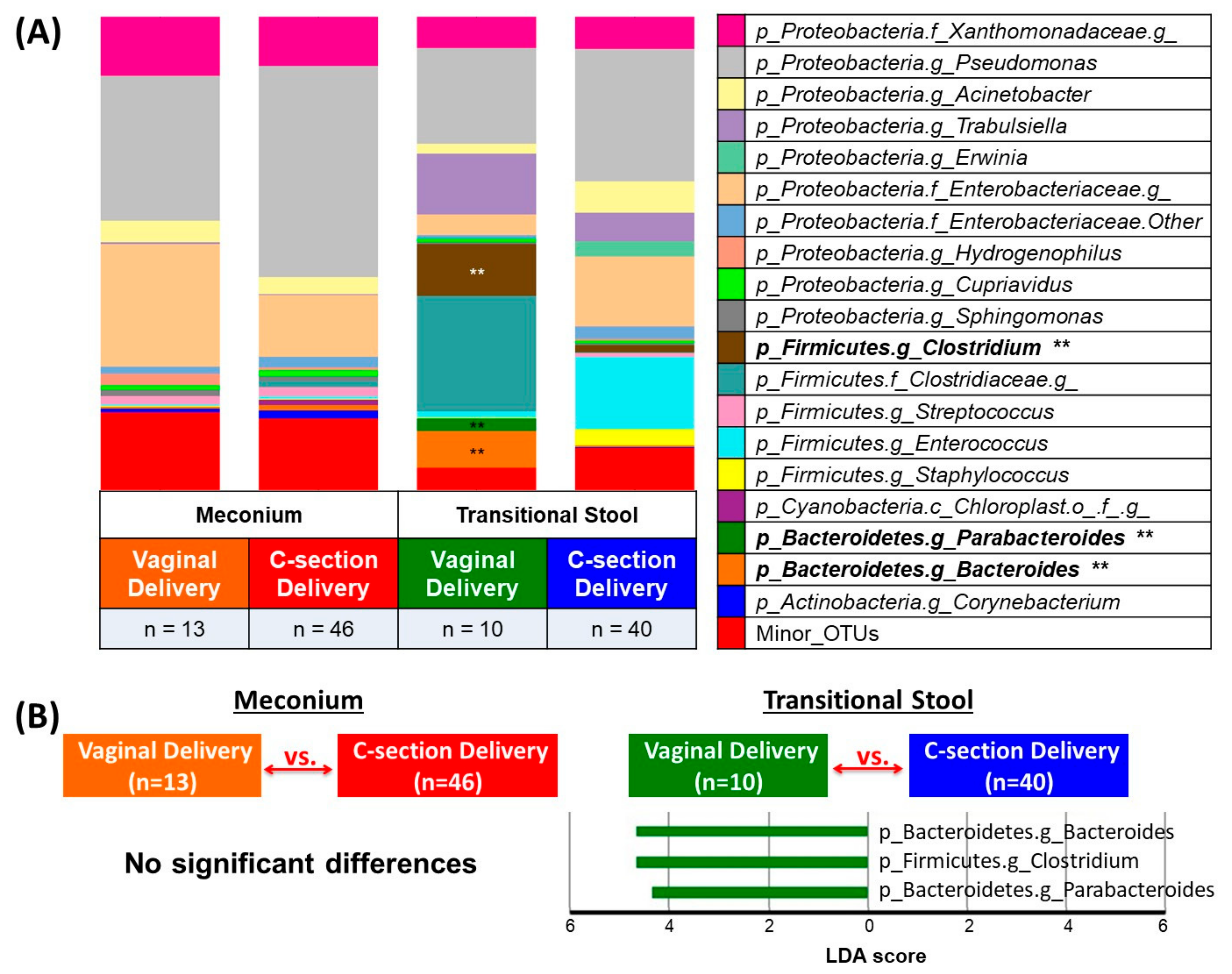
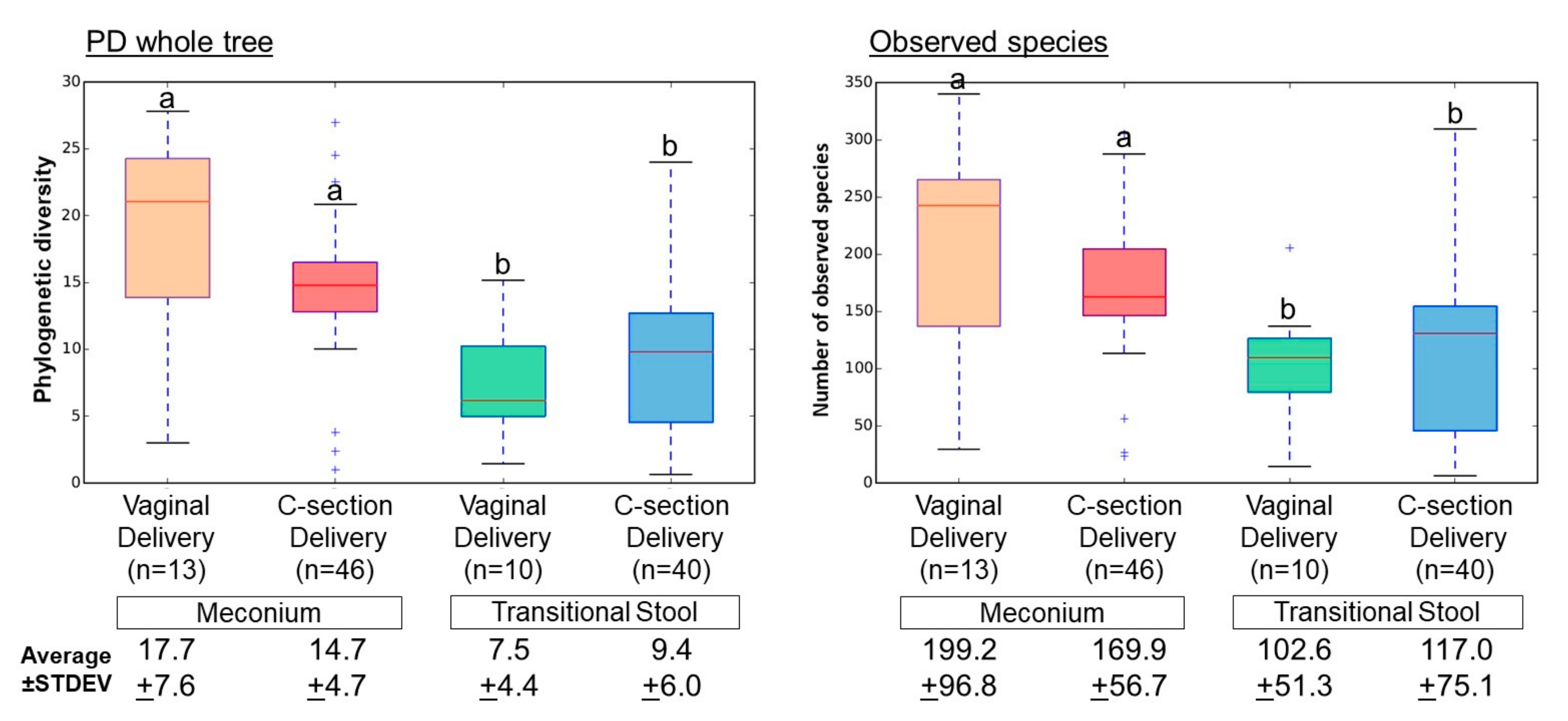
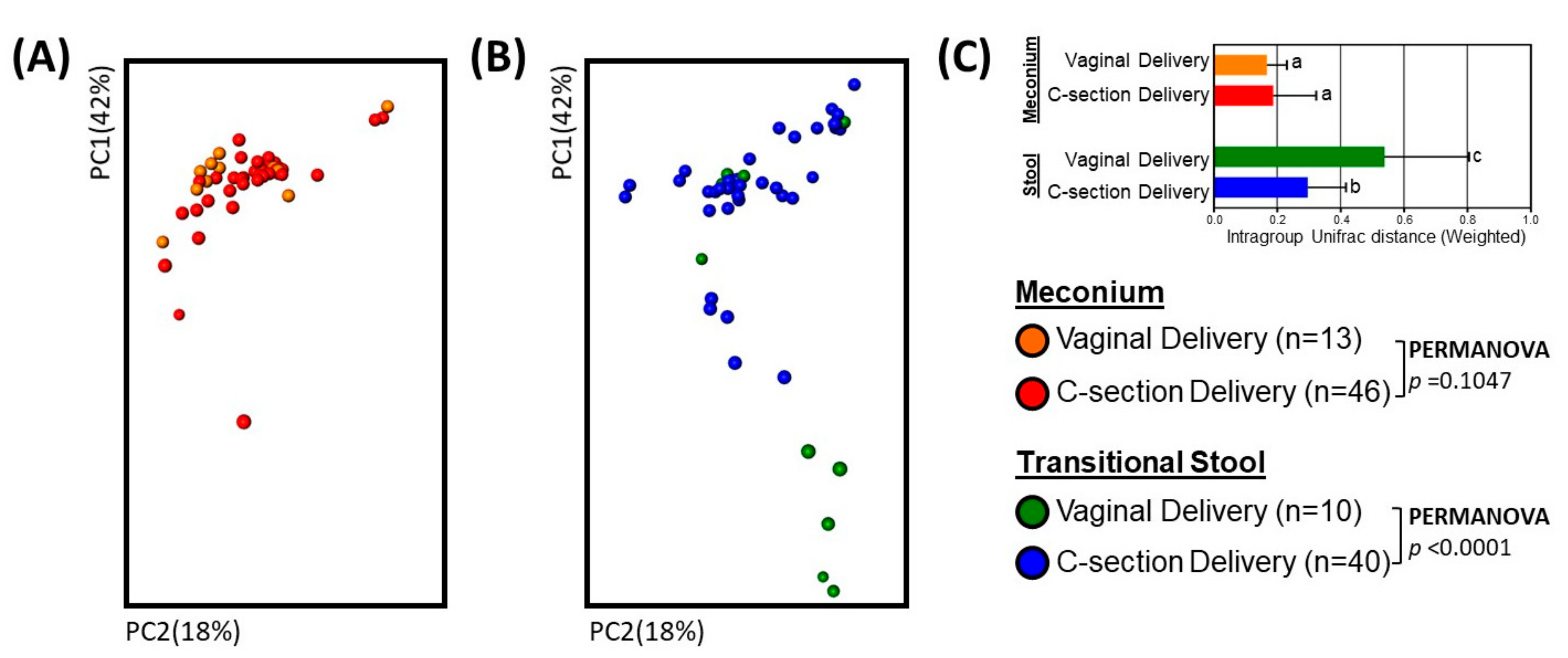
3. Results
3.1. Neonates and Samples
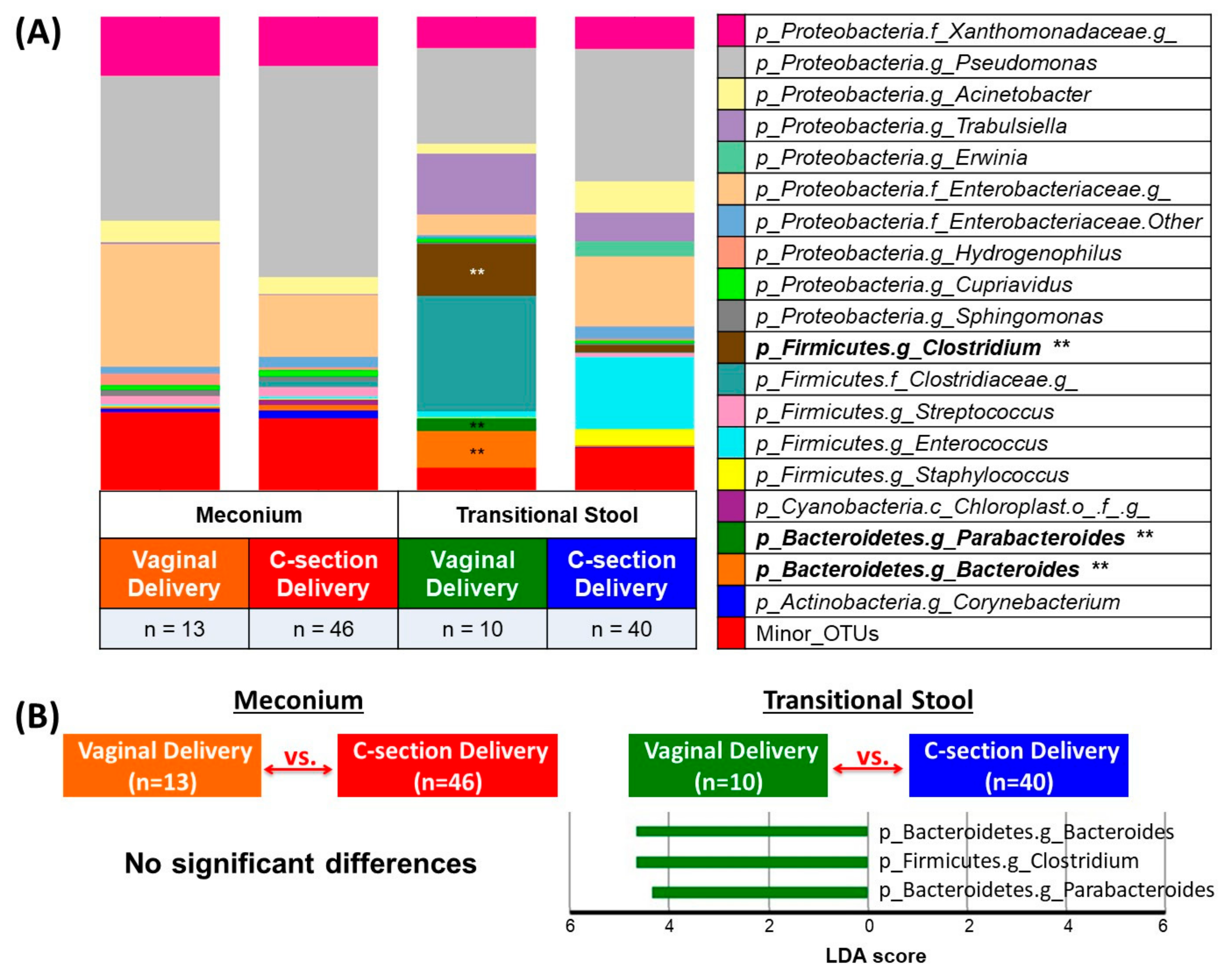
3.2. Microbial Community Diversity, Abundance and Predicted Function
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lavender, T.; Hofmeyr, G.J.; Neilson, J.P.; Kingdon, C.; Gyte, G.M. Caesarean section for non-medical reasons at term. Cochrane Database Syst. Rev. 2012, 14. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists (College); Society for Maternal-Fetal Medicine; Caughey, A.B.; Cahill, A.G.; Guise, J.M.; Rouse, D.J. Safe prevention of the primary cesarean delivery. Am. J. Obstet. Gynecol. 2014, 210, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, A.J.; Blondel, B.; Mohangoo, A.D.; Cuttini, M.; Nijhuis, J.; Novak, Z.; Olafsdottir, H.S.; Zeitlin, J.; Euro-Peristat Scientific Committee. Wide differences in mode of delivery within Europe: Risk-stratified analyses of aggregated routine data from the Euro-Peristat study. BJOG 2016, 123, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Vieira, G.O.; Fernandes, L.G.; de Oliveira, N.F.; Silva, L.R.; Vieira Tde, O. Factors associated with cesarean delivery in public and private hospitals in a city of northeastern Brazil: A cross-sectional study. BMC Pregnancy Childbirth 2015, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Mao, G.; Bennet, W.L.; Hourigan, S.K.; Dominguez-Bello, M.G.; Appel, L.J.; Wang, X. Does vaginal delivery mitigate or strengthen the intergenerational association of overweight and obesity? Findings from the Boston Birth Cohort. Int. J Obes. 2017, 41, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Whyatt, R.; Hoepner, L.; Oberfield, S.; Dominguez-Bello, M.G.; Widen, E.M.; Hassoun, A.; Perera, F.; Rundle, A. Prenatal exposure to antibiotics, cesarean section and risk of childhood obesity. Int. J. Obes. 2015, 39, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends Mol. Med. 2015, 21, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, F.; Zugna, D.; Annesi-Maesano, I.; Baiz, N.; Barros, H.; Correia, S.; Duijts, L.; Forastiere, F.; Inskip, H.; Kelleher, C.C.; et al. Mode of Delivery and Asthma at School Age in 9 European Birth Cohorts. Am. J. Epidemiol. 2017, 185, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.A.; Devlin, J.C.; Lacher, C.R.; Yin, Y.; Cai, Y.; Wang, J.; Dominguez-Bello, M.G. Increased weight gain by C-section: Functional significance of the primordial microbiome. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Kushiro, A.; Ishikawa, E.; Muylaert, D.; Kubota, H.; Sakai, T.; Oishi, K.; Martin, R.; Ben Amor, K.; Oozeer, R.; et al. Transmission of intestinal Bifidobacterium longum subsp. longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl. Environ. Microbiol. 2011, 77, 6788–6793. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Kushiro, A.; Ishikawa, E.; Kubota, H.; Gawad, A.; Sakai, T.; Oishi, K.; Martin, R.; Ben-Amor, K.; Knol, J.; et al. Mother-to-infant transmission of intestinal bifidobacterial strains has an impact on the early development of vaginally delivered infant’s microbiota. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Rochat, F.; Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 2014, 16, 2891–2904. [Google Scholar] [CrossRef] [PubMed]
- Funkhouser, L.J.; Bordenstein, S.R. Mom knows best: The universality of maternal microbial transmission. PLoS Biol. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Konya, T.; Persaud, R.R.; Guttman, D.S.; Chari, R.S.; Field, C.J.; Sears, M.R.; Mandhane, P.J.; Turvey, S.E.; Subbarao, P.; et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: A prospective cohort study. BJOG 2016, 123, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Makino, H.; Cetinyurek Yavuz, A.; Ben-Amor, K.; Roelofs, M.; Ishikawa, E.; Kubota, H.; Swinkels, S.; Sakai, T.; Oishi, K.; et al. Early-Life Events, Including Mode of Delivery and Type of Feeding, Siblings and Gender, Shape the Developing Gut Microbiota. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Pei, Z.; Martinez, K.A.; Rivera-Vinas, J.I.; Mendez, K.; Cavallin, H.; Dominguez-Bello, M.G. The first microbial environment of infants born by C-section: The operating room microbes. Microbiome 2015, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; Lieber, A.D.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Romano-Keeler, J.; Moore, D.J.; Wang, C.; Brucker, R.M.; Fonnesbeck, C.; Slaughter, J.C.; Li, H.; Curran, D.P.; Meng, S.; Correa, H.; et al. Early life establishment of site-specific microbial communities in the gut. Gut Microbes 2014, 5, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R.J.F. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr. Obes. 2017, 12 (Suppl. 1), 3–17. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA. 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Hamady, M.; Knight, R. UniFrac—An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 2006, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.J.; Gross, R.; Bittinger, K.; Sherrill-Mix, S.; Lewis, J.D.; Collman, R.G.; Bushman, F.D.; Li, H. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 2015, 31, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Gen. Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Pantoja-Feliciano, I.G.; Clemente, J.C.; Costello, E.K.; Perez, M.E.; Blaser, M.J.; Knight, R.; Dominguez-Bello, M.G. Biphasic assembly of the murine intestinal microbiota during early development. ISME J. 2013, 7, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; De Jesus-Laboy, K.M.; Shen, N.; Cox, L.M.; Amir, A.; Gonzalez, A.; Bokulich, N.A.; Song, S.J.; Hoashi, M.; Rivera-Vinas, J.I.; et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med. 2016, 22, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Kunji, E.R.; Mierau, I.; Hagting, A.; Poolman, B.; Konings, W.N. The proteolytic systems of lactic acid bacteria. Antonie Van Leeuwenhoek 1996, 70, 187–221. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Savaiano, D.A. In vitro lactose fermentation by human colonic bacteria is modified by Lactobacillus acidophilus supplementation. J. Nutr. 1997, 127, 1489–1495. [Google Scholar] [PubMed]
- Angelakis, E.; Armougom, F.; Million, M.; Raoult, D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012, 7, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Soergel, K.H. Colonic fermentation: Metabolic and clinical implications. Clin. Investig. 1994, 72, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Tsuji, H.; Takahashi, T.; Kawashima, K.; Nagata, S.; Nomoto, K.; Yamashiro, Y. Sensitive Quantitative Analysis of the Meconium Bacterial Microbiota in Healthy Term Infants Born Vaginally or by Cesarean Section. Front. Microbiol. 2016, 7, 1997. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.M.; Ma, J.; Prince, A.L.; Antony, K.M.; Seferovic, M.D.; Aagaard, K.M. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 2017, 23, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Shin, H.; Pizoni, A.; Werlang, I.C.; Matte, U.; Goldani, M.Z.; Goldani, H.A.; Dominguez-Bello, M.G. Birth mode-dependent association between pre-pregnancy maternal weight status and the neonatal intestinal microbiome. Sci. Rep. 2016, 6, 23133. [Google Scholar] [CrossRef] [PubMed]
- Perez-Munoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Heineck, I.; Ferreira, M.B.; Schenkel, E.P. Prescribing practice for antibiotic prophylaxis for cesarean section in a teaching hospital in Brazil. Am. J. Infect. Control. 2002, 30, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Farret, T.C.; Dalle, J.; Monteiro Vda, S.; Riche, C.V.; Antonello, V.S. Risk factors for surgical site infection following cesarean section in a Brazilian Women’s Hospital: A case-control study. Braz. J. Infect. Dis. 2015, 19, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Gerber, S.; Hohlfeld, P.; Sandrine, G.; Witkin, S.S. Mycoplasma hominis in mid-trimester amniotic fluid: Relation to pregnancy outcome. J. Perinat. Med. 2004, 32, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Cassell, G.H.; Davis, R.O.; Waites, K.B.; Brown, M.B.; Marriott, P.A.; Stagno, S.; Davis, J.K. Isolation of Mycoplasma hominis and Ureaplasma urealyticum from amniotic fluid at 16–20 weeks of gestation: Potential effect on outcome of pregnancy. Sex. Transm. Dis. 1983, 10, 294–302. [Google Scholar] [PubMed]
- Bearfield, C.; Davenport, E.S.; Sivapathasundaram, V.; Allaker, R.P. Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG 2002, 109, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.; Fernandez, L.; Marin, M.L.; Martin, R.; Odriozola, J.M.; Nueno-Palop, C.; Narbad, A.; Olivares, M.; Xaus, J.; Rodriguez, J.M. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr. Microbiol. 2005, 51, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.H.; Malatos, S.; Kennea, N.; Edwards, A.D.; Miles, L.; Duggan, P.; Reynolds, P.R.; Feldman, R.G.; Sullivan, M.H. Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr. Res. 2005, 57, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Gomez de Aguero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Koleva, P.T.; Kim, J.S.; Scott, J.A.; Kozyrskyj, A.L. Microbial programming of health and disease starts during fetal life. Birth Defects Res. C Embryo Today 2015, 105, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Scott, K.P.; Khan, S.; Martin, J.C.; Berry, S.H.; Stevenson, M.; Okpapi, A.; Munro, M.J.; Hold, G.L. First-Pass Meconium Samples from Healthy Term Vaginally-Delivered Neonates: An Analysis of the Microbiota. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunderarajan, K.; Kelkar, S.S. Bacterial flora of meconium and faeces during the first year of life. Indian J. Pediatr. 1979, 46, 92–95. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Vaginal Delivery (n = 13) | C-Section Delivery (n = 46) | |
|---|---|---|
| Maternal characteristics | ||
| Mothers age, y | 28.5 (5.9) | 30.5 (4.7) |
| Mothers race | ||
| White, n (%) | 12 (92) | 42 (91) |
| Non-white, n (%) | 1 (8) | 4 (9) |
| Urinary tract infection | ||
| Yes, n (%) | 4 (31) | 6 (13) |
| No, n (%) | 9 (69) | 40 (87) |
| Medications in pregnancy | ||
| Yes, n (%) | 4 (31) | 19 (41) |
| No, n (%) | 9 (69) | 27 (59) |
| Antibiotics in pregnancy | ||
| None, n (%) | 9 (69) | 36 (78) |
| 1st Trimester, n (%) | 1 (8) | 5 (11) |
| 2nd Trimester, n (%) | 3 (23) | 5 (11) |
| Ever smoke tobacco | ||
| Yes (%) | 0 (0) | 12 (26) |
| No (%) | 13 (100) | 34 (74) |
| Pre-pregnancy BMI, kg/m2 | 22.4 (3.2) | 25.3 (5.0) |
| Pregnancy weight gain, kg | 13.0 (3.0) | 13.7 (5.4) |
| Newborn characteristics | ||
| Sex | ||
| Boy, n (%) | 3 (23) | 30 (65) |
| Girl, n (%) | 10 (77) | 16 (35) |
| Birth weight, g | 3127.6 (334.5) | 3234.3 (427.4) |
| Birth length, cm | 48.7 (2.2) | 48.9 (1.6) |
| Head circumference, cm † | 35.9 (4.4) | 35.7 (2.8) |
| Placenta weight, g § | 674.8 (91.3) | 624.2 (131.7) |
| Breastfed 1st 24 h | ||
| Yes, n (%) | 13 (100) | 40 (87) |
| No, n (%) | 0 (0) | 6 (13) |
| Transitional Stool | Meconium | |||||||
|---|---|---|---|---|---|---|---|---|
| KEGG Functional Categories | Vaginal Delivery (n = 10) | Vs. ⇔ | C-Section Delivery (n = 40) | Vaginal Delivery (n = 13) | Vs. ⇔ | C-Section Delivery (n = 46) | ||
| Level 2 | Level 3 | LDA | P Value | LDA | ||||
| Amino Acid Metabolism | Tryptophan metabolism | - | 0.01635 | → 3.06 | No significant differences | |||
| Amino Acid Metabolism | Valine/leucine/isoleucine degradation | - | 0.0066 | → 3.12 | ||||
| Carbohydrate Metabolism | Amino sugar/nucleotide sugar metabolism | 3.16 ← | 0.03282 | - | ||||
| Enzyme Families | Peptidases | 3.20 ← | 0.0049 | - | ||||
| Lipid Metabolism | Fatty acid metabolism | - | 0.00946 | → 3.04 | ||||
| Metabolism of Cofactors/Vitamins | - | 3.22 ← | 0.03282 | - | ||||
| Metabolism of Cofactors/Vitamins | Porphyrin/chlorophyll metabolism | 3.03 ← | 0.02905 | - | ||||
| Metabolism of Terpenoids/Polyketides | - | - | 0.01864 | → 3.01 | ||||
| Xenobiotics Biodegradation and Metabolism | - | - | 0.00881 | → 3.67 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mueller, N.T.; Shin, H.; Pizoni, A.; Werlang, I.C.; Matte, U.; Goldani, M.Z.; Goldani, H.A.S.; Dominguez-Bello, M.G. Delivery Mode and the Transition of Pioneering Gut-Microbiota Structure, Composition and Predicted Metabolic Function. Genes 2017, 8, 364. https://doi.org/10.3390/genes8120364
Mueller NT, Shin H, Pizoni A, Werlang IC, Matte U, Goldani MZ, Goldani HAS, Dominguez-Bello MG. Delivery Mode and the Transition of Pioneering Gut-Microbiota Structure, Composition and Predicted Metabolic Function. Genes. 2017; 8(12):364. https://doi.org/10.3390/genes8120364
Chicago/Turabian StyleMueller, Noel T., Hakdong Shin, Aline Pizoni, Isabel C. Werlang, Ursula Matte, Marcelo Z. Goldani, Helena A. S. Goldani, and Maria G. Dominguez-Bello. 2017. "Delivery Mode and the Transition of Pioneering Gut-Microbiota Structure, Composition and Predicted Metabolic Function" Genes 8, no. 12: 364. https://doi.org/10.3390/genes8120364