Comparative Analysis of DNA Methylation Reveals Specific Regulations on Ethylene Pathway in Tomato Fruit

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
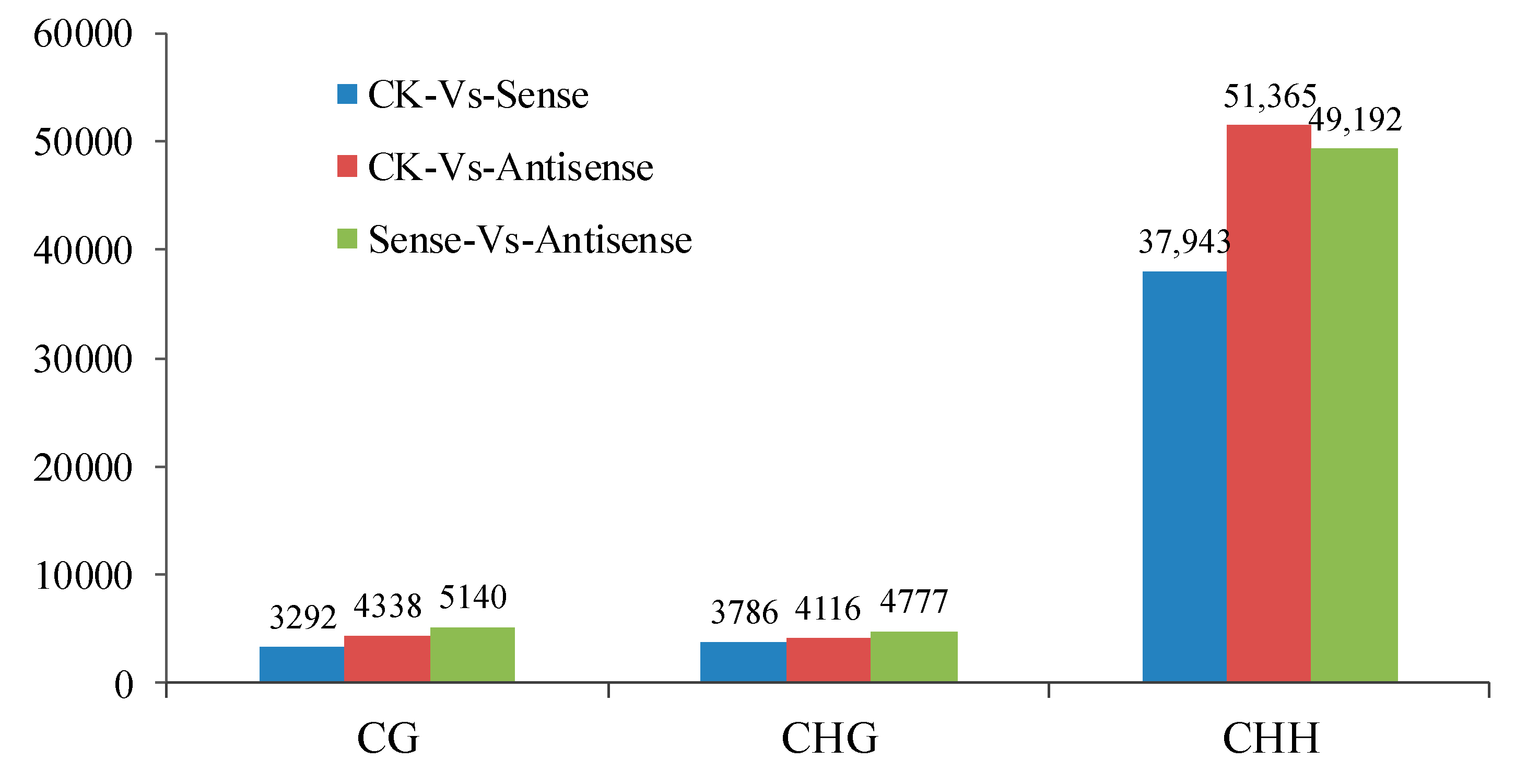{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Library Generation for Bisulfite-Seq
2.2. Quality Control
2.3. Methylation Calling
2.4. Methylation Level
2.5. Detection of Differentially Methylated Regions
2.6. Integrated Functional Analysis of Differentially Metilated Genes
2.7. DNA Methylation and MicroRNAs’ Targets Analysis
3. Results
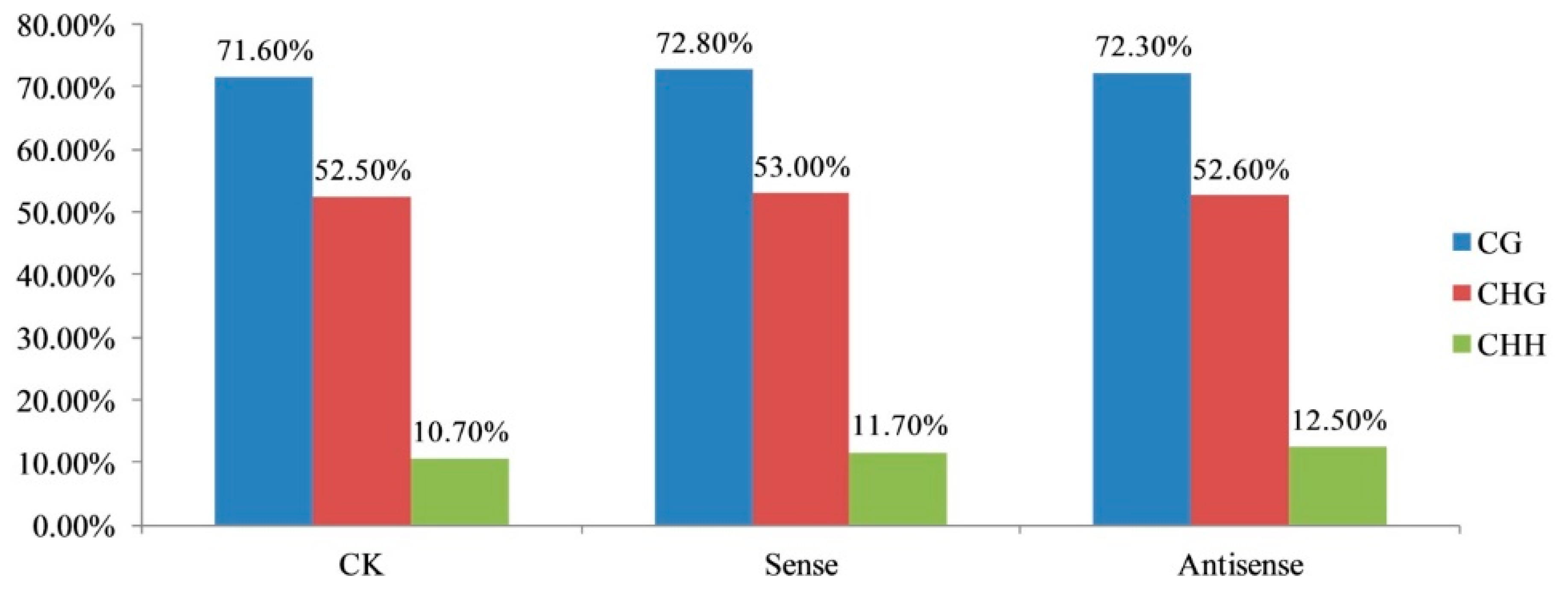
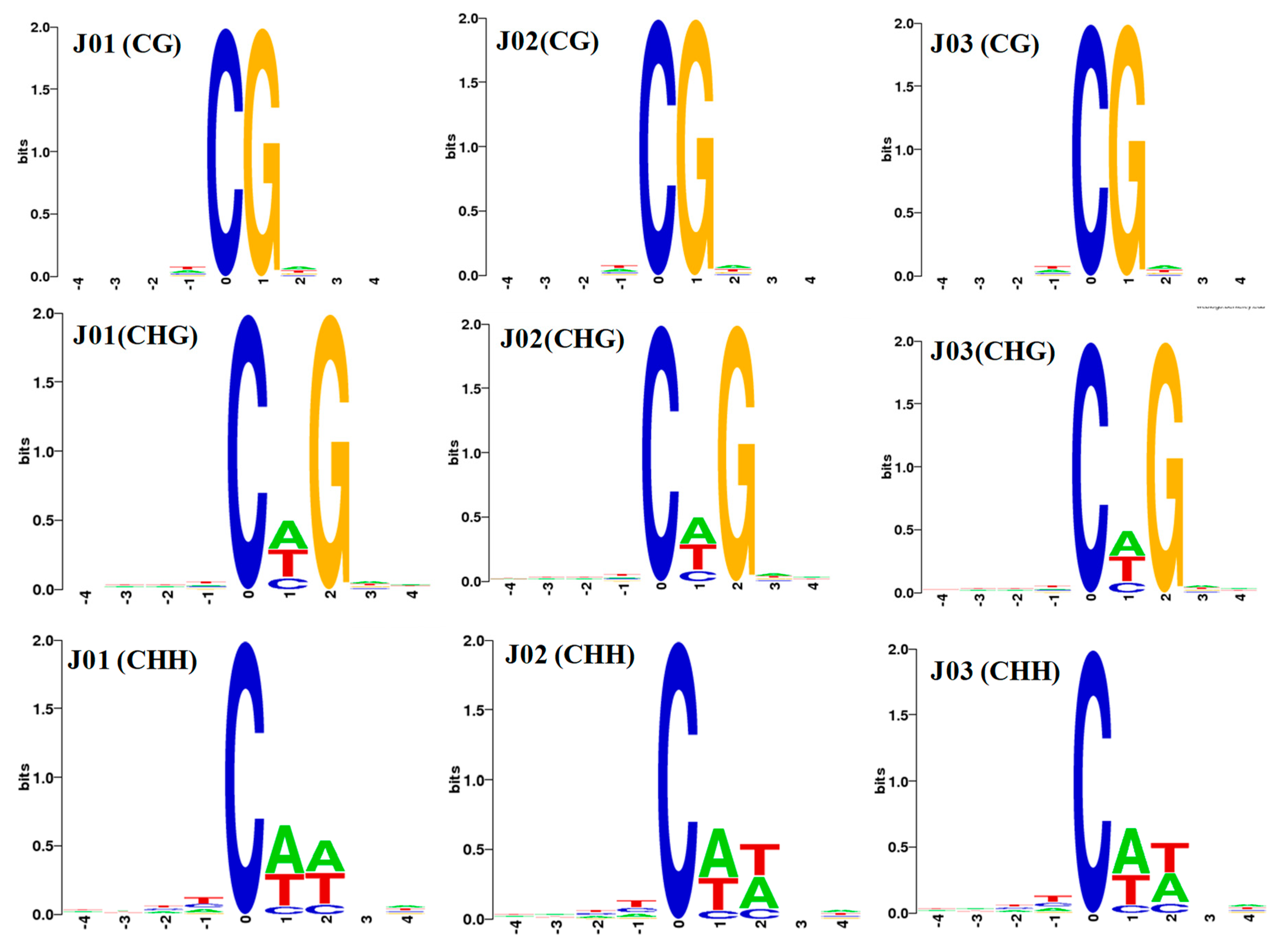
3.1. DNA Methylation Landscapes in Tomato Fruit
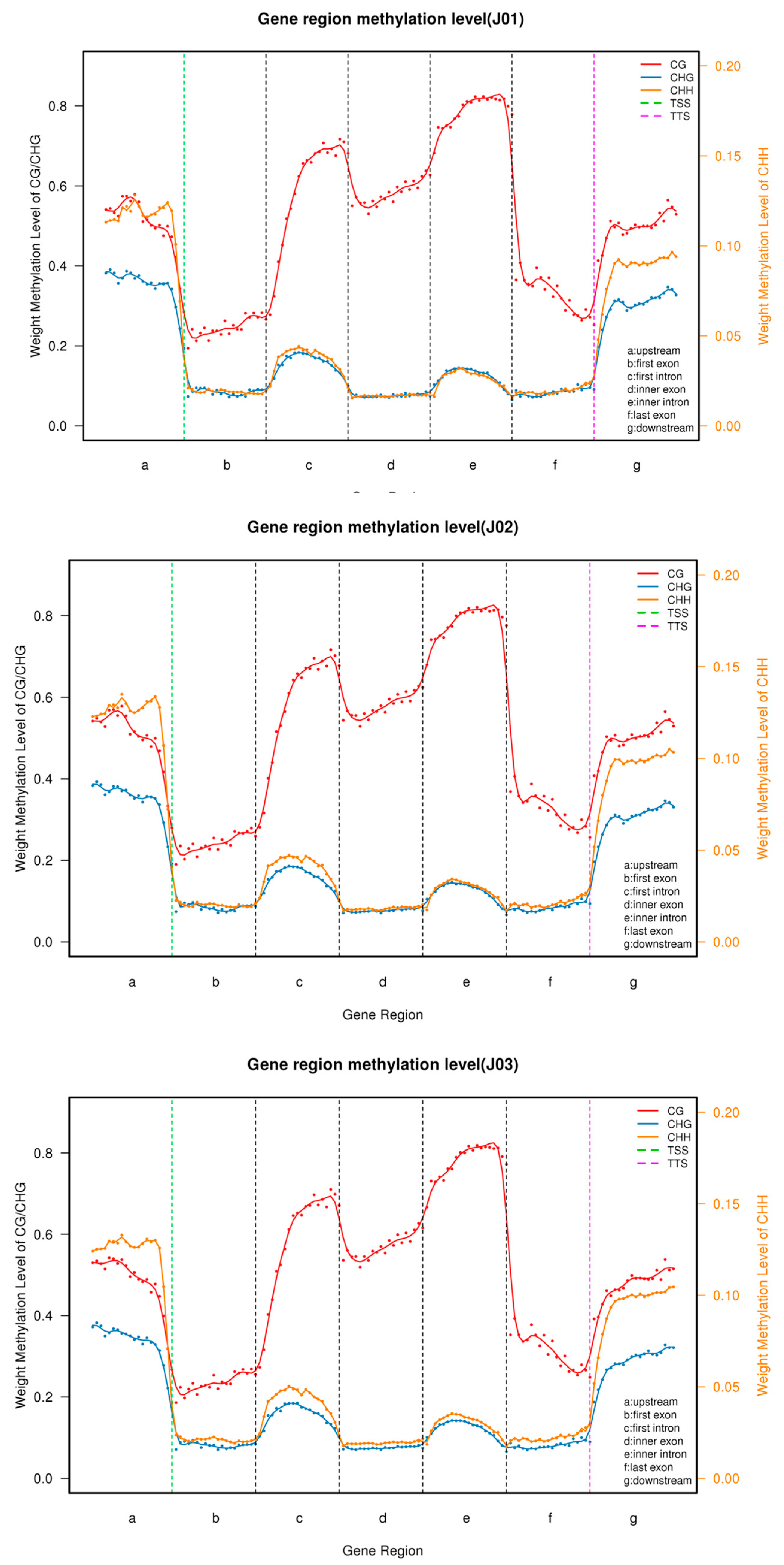
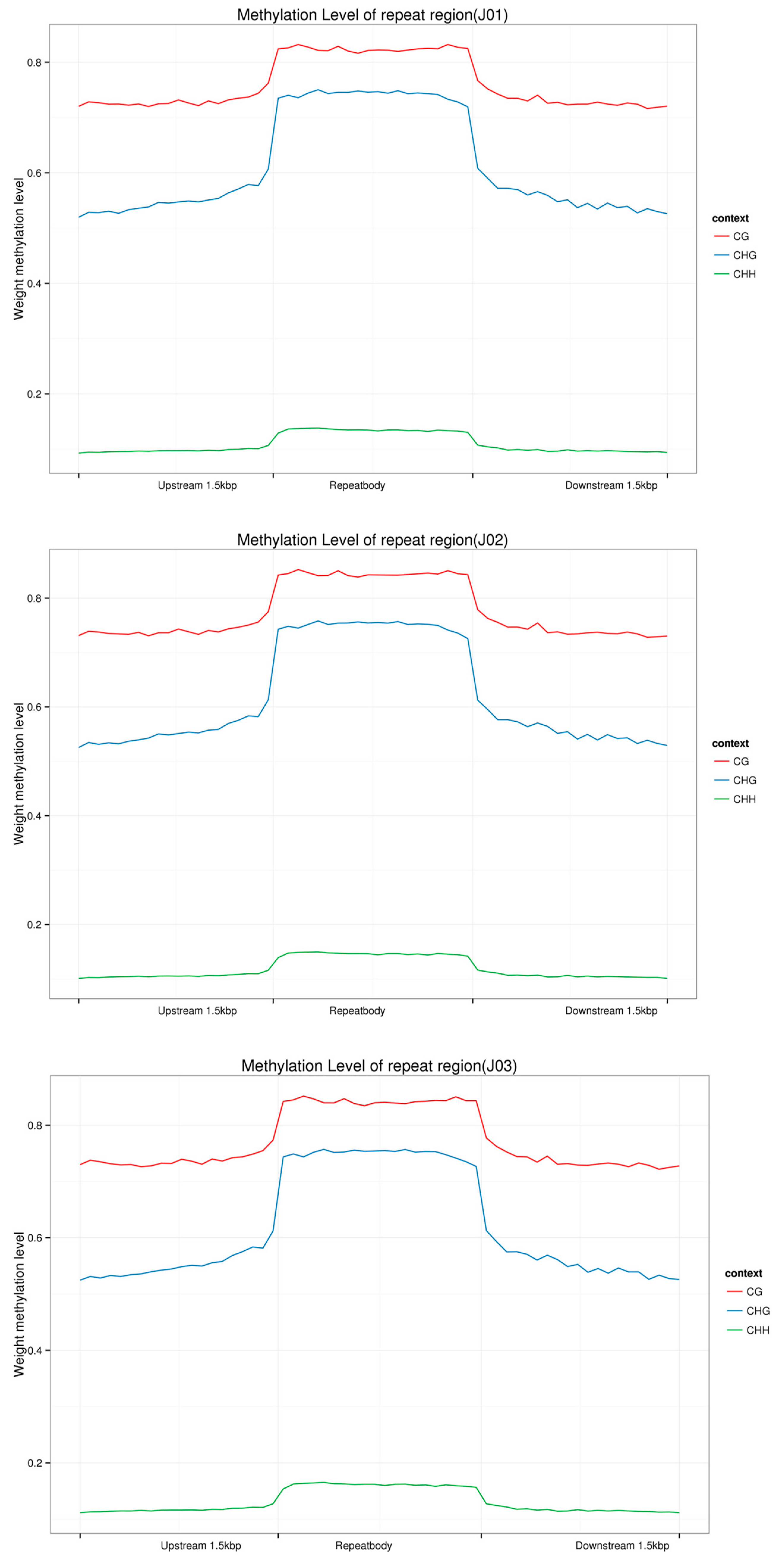
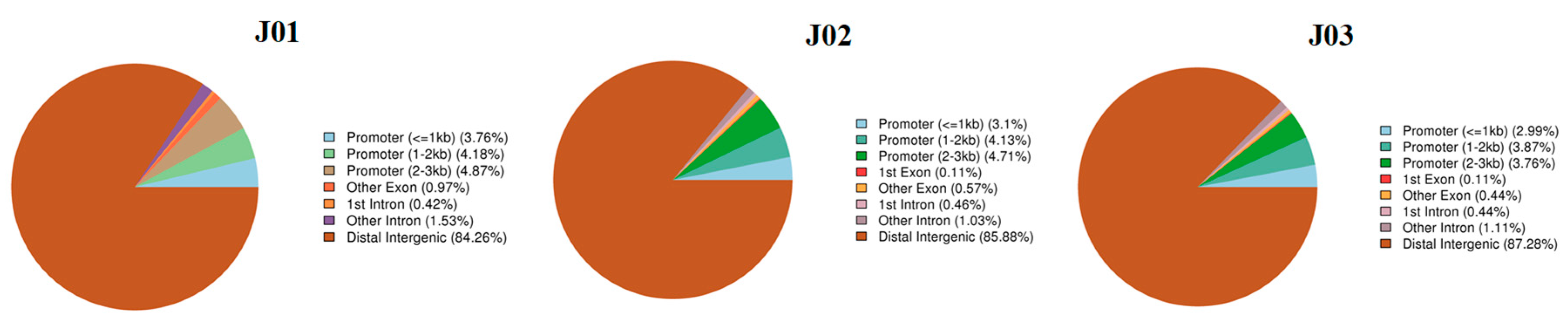
3.2. DNA Methylation Patterns in Different Tomato Genomic Regions
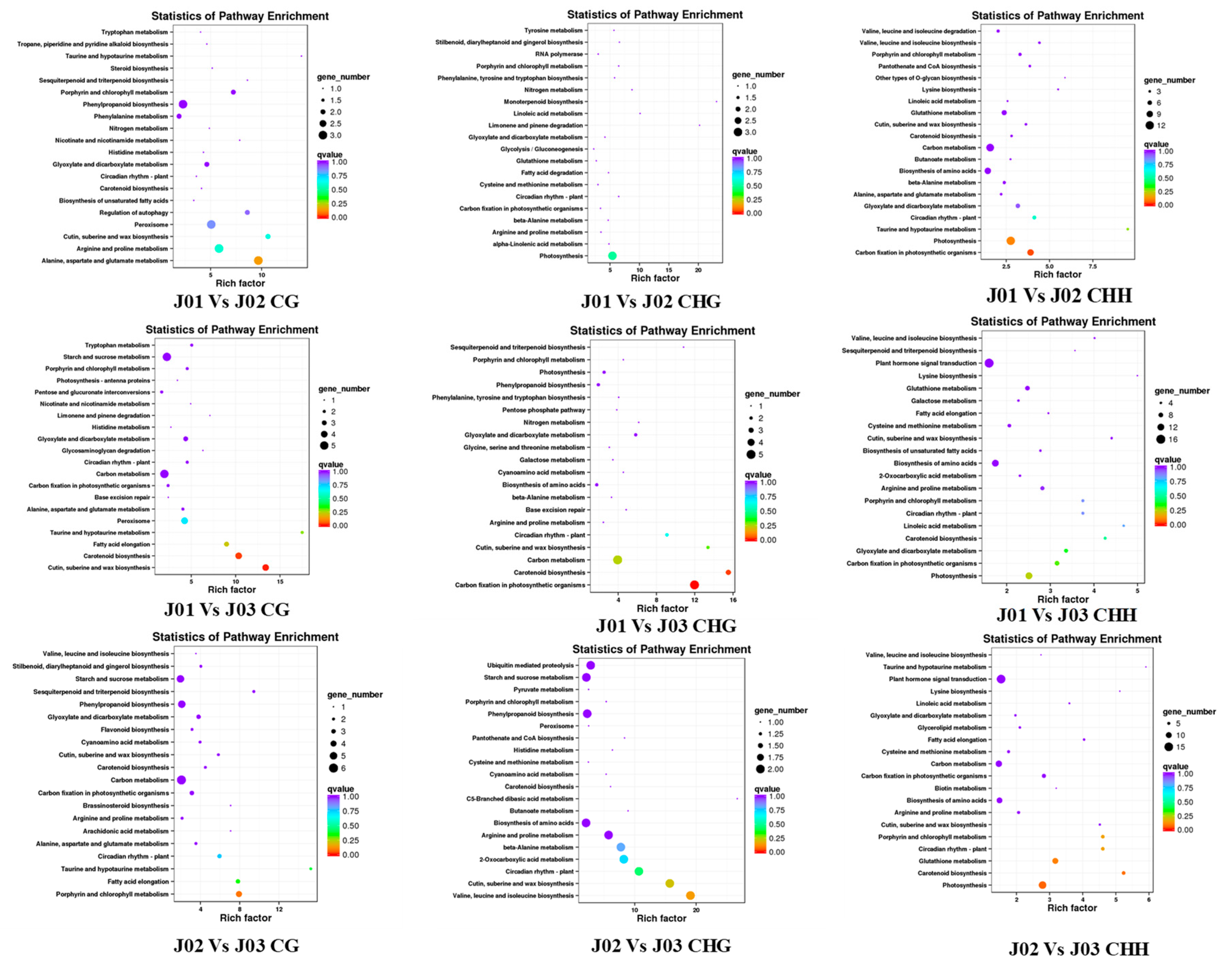
3.3. Differentially Methylated Regions Analysis and Function Parsing
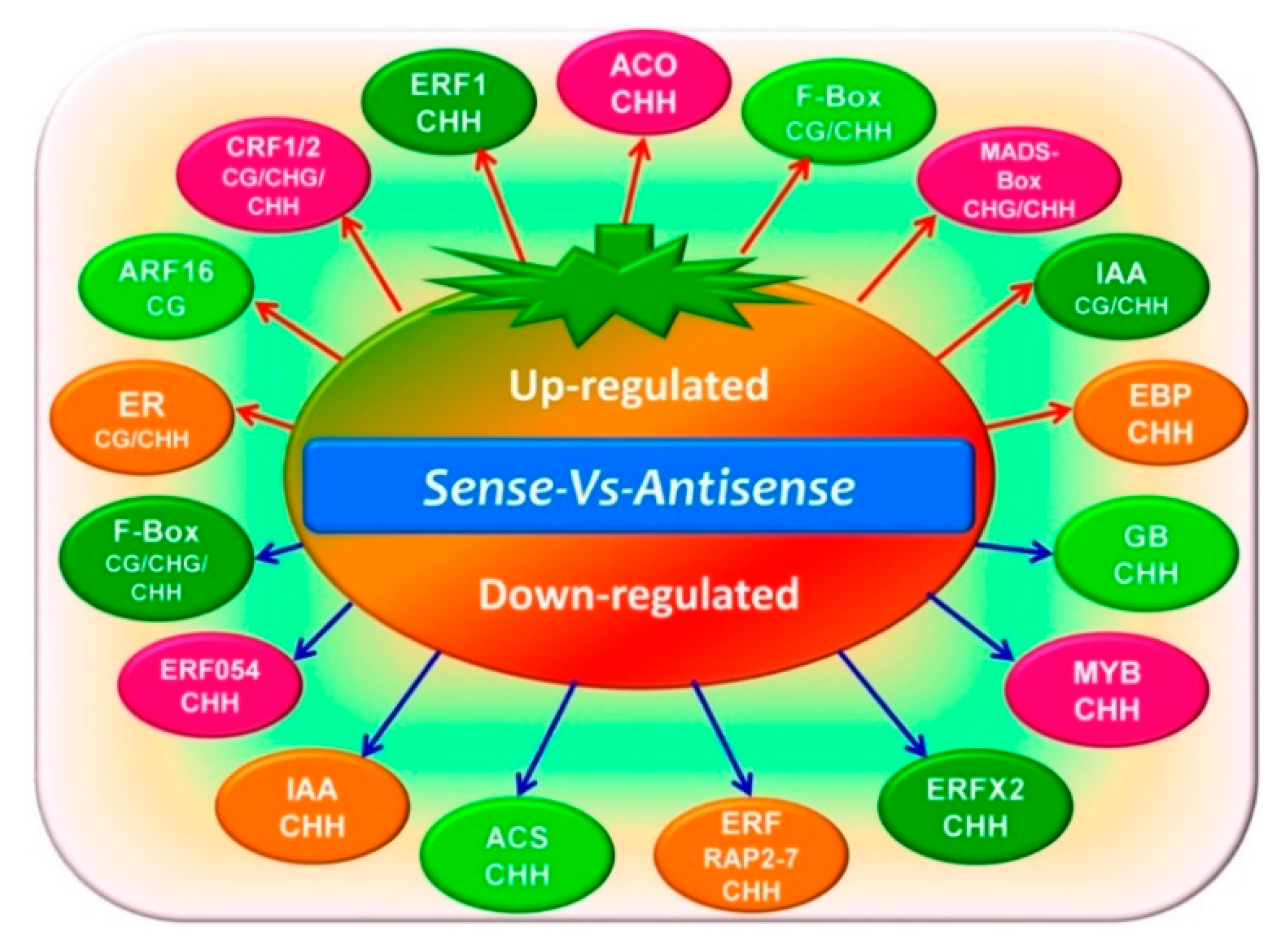
3.4. Differentially Methylated Regions and Differentially Expressed Genes Conjoint Analysis Involved in the Ethylene Pathway
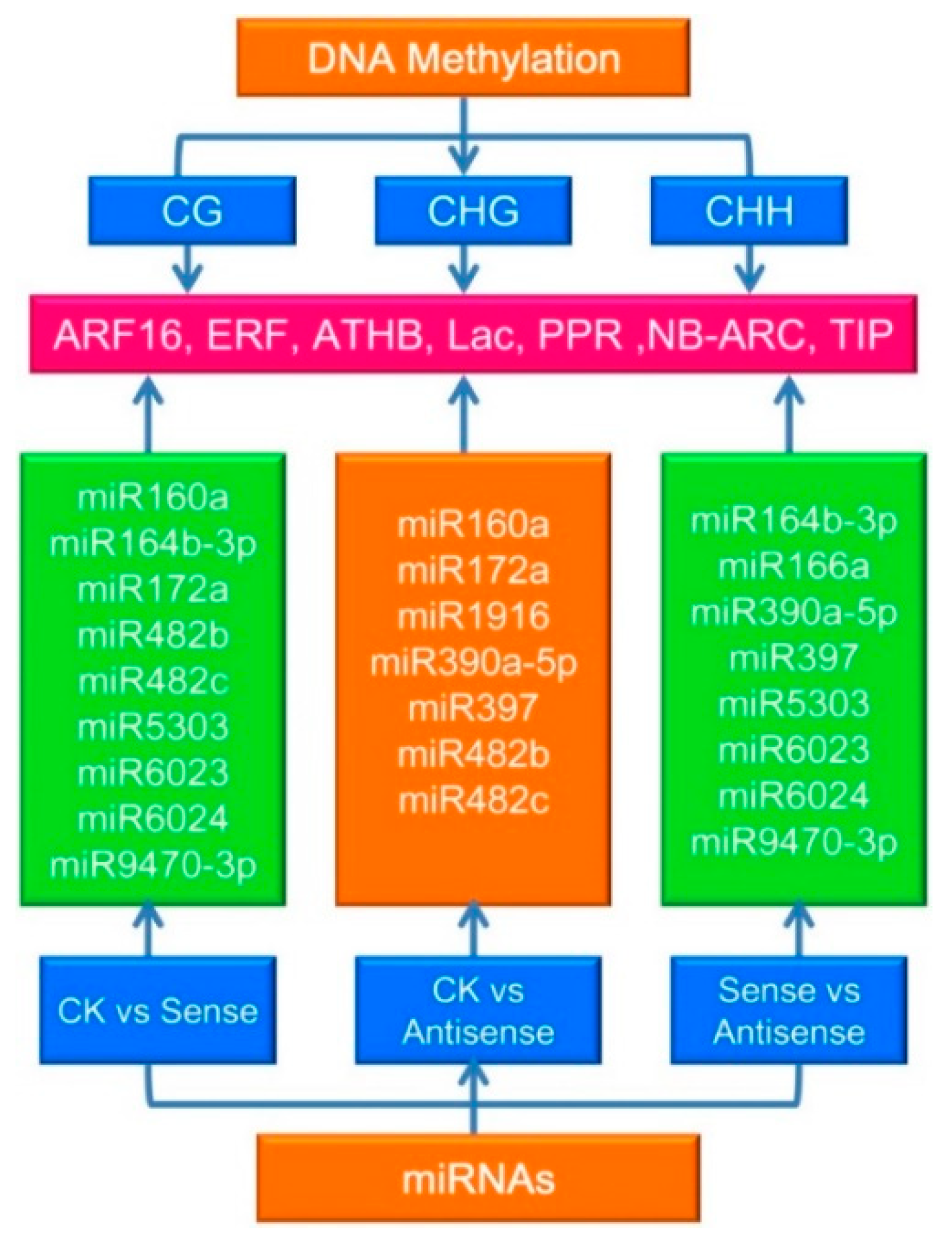
3.5. DNA Methylation and microRNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Gehring, M.; Henikoff, S. DNA methylation and demethylation in Arabidopsis. Arabidopsis Book 2008, 6, e0102. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Dalakouras, A.; Dadami, E.; Zwiebel, M.; Krczal, G.; Wassenegger, M. Transgenerational maintenance of transgene body CG but not CHG and CHH methylation. Epigenetics 2012, 7, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Podicheti, R.; Mishra, V.; Marasco, M.; Wang, J.; Rusch, D.; Tang, H.; Pikaard, C.S. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. eLife 2015, 4, e09591. [Google Scholar] [CrossRef] [PubMed]
- Reinders, J.; Wulff, B.B.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012, 81, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Eichten, S.R.; Schmitz, R.J.; Springer, N.M. Epigenetics: Beyond chromatin modifications and complex genetic regulation. Plant Physiol. 2014, 165, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Schmitz, R.J. Putting DNA methylation in context: From genomes to gene expression in plants. Biochim. Biophys. Acta 2017, 1860, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, J. Molecular biology of fruit maturation and ripening. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 725–749. [Google Scholar] [CrossRef] [PubMed]
- Klee, H.J.; Giovannoni, J.J. Genetics and control of tomato fruit ripening and quality attributes. Annu. Rev. Genet. 2011, 45, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Seymour, G.B.; Chapman, N.H.; Chew, B.L.; Rose, J.K. Regulation of ripening and opportunities for control in tomato and other fruits. Plant Biotechnol. J. 2013, 11, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Yuan, X.Y.; Meng, L.H.; Zhu, B.Z.; Zhu, H.L.; Luo, Y.B.; Fu, D.Q. Transcriptome analysis provides a preliminary regulation route of the ethylene signal transduction component, SlEIN2, during tomato ripening. PLoS ONE 2016, 11, e0168287. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, G.; Choudhary, D.; Singh, V.P.; Arora, A. Role of ethylene receptors during senescence and ripening in horticultural crops. Plant Signal Behav. 2012, 7, 827–846. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pirrello, J.; Chervin, C.; Roustan, J.P.; Bouzayen, M. Ethylene control of fruit ripening: revisiting the complex network of transcriptional regulation. Plant Physiol. 2015, 169, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Gallusci, P.; Dai, Z.; Génard, M.; Gauffretau, A.; Leblanc-Fournier, N.; Richard-Molard, C.; Vile, D.; Brunel-Muguet, S. Epigenetics for plant improvement: Current knowledge and modeling avenues. Trends Plant Sci. 2017, 22, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, J. Ripening activator turned repressor. Nat. Plants 2017, 3, 920–921. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gomes, B.L.; Mila, I.; Purgatto, E.; Peres, L.E.; Frasse, P.; Maza, E.; Zouine, M.; Roustan, J.P.; Bouzayen, M.; et al. Comprehensive profiling of ethylene response factor expression identifies ripening-associated ERF genes and their link to key regulators of fruit ripening in tomato. Plant Physiol. 2016, 170, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Liu, S.; Huang, L.; Hong, Y.; Li, X.; Huang, L.; Zhang, Y.; Zhang, H.; Li, D.; Song, F. Tomato SlERF.A1, SlERF.B4, SlERF.C3 and SlERF.A3, members of B3 group of ERF family, are required for resistance to Botrytis cinerea. Front. Plant Sci. 2016, 7, 1964. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.; Sanchez-Ballesta, M.T.; Jones, B.; Pesquet, E.; Regad, F.; Latché, A.; Pech, J.C.; Bouzayen, M. New members of the tomato ERF family show specific expression pattern and diverse DNA-binding capacity to the GCC box element. FEBS Lett. 2003, 550, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.Y.; Vrebalov, J.; Alba, R.; Lee, J.; McQuinn, R.; Chung, J.D.; Klein, P.; Giovannoni, J. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J. 2010, 64, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gupta, S.; Rashotte, A.M. Characterization of two tomato AP2/ERF genes, SlCRF1 and SlCRF2 in hormone and stress responses. Plant Cell. Rep. 2014, 33, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, A.; Zhang, Z.; Huang, Z.; Lu, P.; Zhang, D.; Liu, X.; Zhang, Z.F.; Huang, R. Ethylene response factor TERF1, regulated by ETHYLENE-INSENSITIVE3-like factors, functions in reactive oxygen species (ROS) scavenging in tobacco (Nicotiana tabacum L.). Sci. Rep. 2016, 6, 29948. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, A.; Ruggieri, V.; Sacco, A.; Rigano, M.M.; Carriero, F.; Bolger, A.; Fernie, A.R.; Frusciante, L.; Barone, A. Identification of candidate genes for phenolics accumulation in tomato fruit. Plant Sci. 2013, 205–206, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xu, Y.; Lu, P.; Wang, X.; Li, Z.; Cai, X.; Zhou, Z.; Wang, Y.; Zhang, Z.; Lin, Z.; et al. Salt stress responsiveness of a wild cotton species (Gossypium klotzschianum) based on transcriptomic analysis. PLoS ONE 2017, 12, e0178313. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Hu, P.; Wang, T.; Tao, J.; Zong, S. Differential transcriptome analysis reveals genes related to cold tolerance in seabuckthorn carpenter moth, Eogystia hippophaecolus. PLoS ONE 2017, 12, e0187105. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fei, B.; He, J.; Zhou, M.; Zhang, D.; Pan, L.; Li, S.; Liang, Y.; Wang, L.; Zhu, J.; et al. Transcriptome analysis reveals the host selection fitness mechanisms of the Rhizoctonia solani AG1IA pathogen. Sci. Rep. 2017, 7, 10120. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Pirrello, J.; Prasad, B.C.; Zhang, W.; Chen, K.; Mila, I.; Zouine, M.; Latché, A.; Pech, J.C.; Ohme-Takagi, M.; Regad, F.; et al. Functional analysis and binding affinity of tomato ethylene response factors provide insight on the molecular bases of plant differential responses to ethylene. BMC Plant Biol. 2012, 12, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Diretto, G.; Pirrello, J.; Roustan, J.P.; Li, Z.; Giuliano, G.; Regad, F.; Bouzayen, M. The chimeric repressor version of an ethylene response factor (ERF) family member, Sl-ERF.B3, shows contrasting effects on tomato fruit ripening. New Phytol. 2014, 203, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Q.; Gao, L.; Zhu, B.; Ju, Z.; Luo, Y.; Zuo, J. Parsing the regulatory network between small RNAs and target genes in ethylene pathway in tomato. Front. Plant Sci. 2017, 8, 527. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 9, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Gouil, Q.; Baulcombe, D.C. DNA methylation signatures of the plant chromomethyltransferases. PLoS Genet. 2016, 12, e1006526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Feng, X.; Yang, H.; Zhu, A.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F.; et al. Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genomics 2017, 18, 759. [Google Scholar] [CrossRef] [PubMed]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Cui, Y.; Gu, X. Genome-wide DNA methylation profiles changes associated with constant heat stress in pigs as measured by bisulfite sequencing. Sci. Rep. 2016, 6, 27507. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; An, S.M.; Kwon, S.; Park, D.H.; Kim, T.W.; Kang, D.G.; Yu, G.E.; Kim, I.S.; Park, H.C.; Ha, J.; et al. DNA methylation patterns and gene expression associated with litter size in Berkshire pig placenta. PLoS ONE 2017, 12, e0184539. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, B.; Xu, W.; Zhu, H.; Chen, A.; Xie, Y.; Shao, Y.; Luo, Y. LeERF1 positively modulated ethylene triple response on etiolated seedling, plant development and fruit ripening and softening in tomato. Plant Cell. Rep. 2007, 26, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yan, J.; Tang, G. MicroRNA-mediated DNA methylation in plants. Front. Biol. 2011, 6, 133–139. [Google Scholar] [CrossRef]
- Bao, N.; Lye, K.W.; Barton, M.K. MicroRNA binding sites in Arabidopsis class III HD-ZIP mRNAs are required for methylation of the template chromosome. Dev. Cell 2004, 7, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mao, L.; Qi, Y. Roles of DICER-LIKE and ARGONAUTE proteins in TAS-derived small interfering RNA-triggered DNA methylation. Plant Physiol. 2012, 160, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Yu, B. siRNA-directed DNA methylation in plants. Curr. Genomics 2015, 16, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, T.; Xu, J.; Li, H. MicroRNA mediates DNA methylation of target genes. Biochem. Biophys. Res. Commun. 2014, 444, 676–681. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, J.; Wang, Y.; Zhu, B.; Luo, Y.; Wang, Q.; Gao, L. Comparative Analysis of DNA Methylation Reveals Specific Regulations on Ethylene Pathway in Tomato Fruit. Genes 2018, 9, 266. https://doi.org/10.3390/genes9050266
Zuo J, Wang Y, Zhu B, Luo Y, Wang Q, Gao L. Comparative Analysis of DNA Methylation Reveals Specific Regulations on Ethylene Pathway in Tomato Fruit. Genes. 2018; 9(5):266. https://doi.org/10.3390/genes9050266
Chicago/Turabian StyleZuo, Jinhua, Yunxiang Wang, Benzhong Zhu, Yunbo Luo, Qing Wang, and Lipu Gao. 2018. "Comparative Analysis of DNA Methylation Reveals Specific Regulations on Ethylene Pathway in Tomato Fruit" Genes 9, no. 5: 266. https://doi.org/10.3390/genes9050266