Validation of Ion TorrentTM Inherited Disease Panel with the PGMTM Sequencing Platform for Rapid and Comprehensive Mutation Detection

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
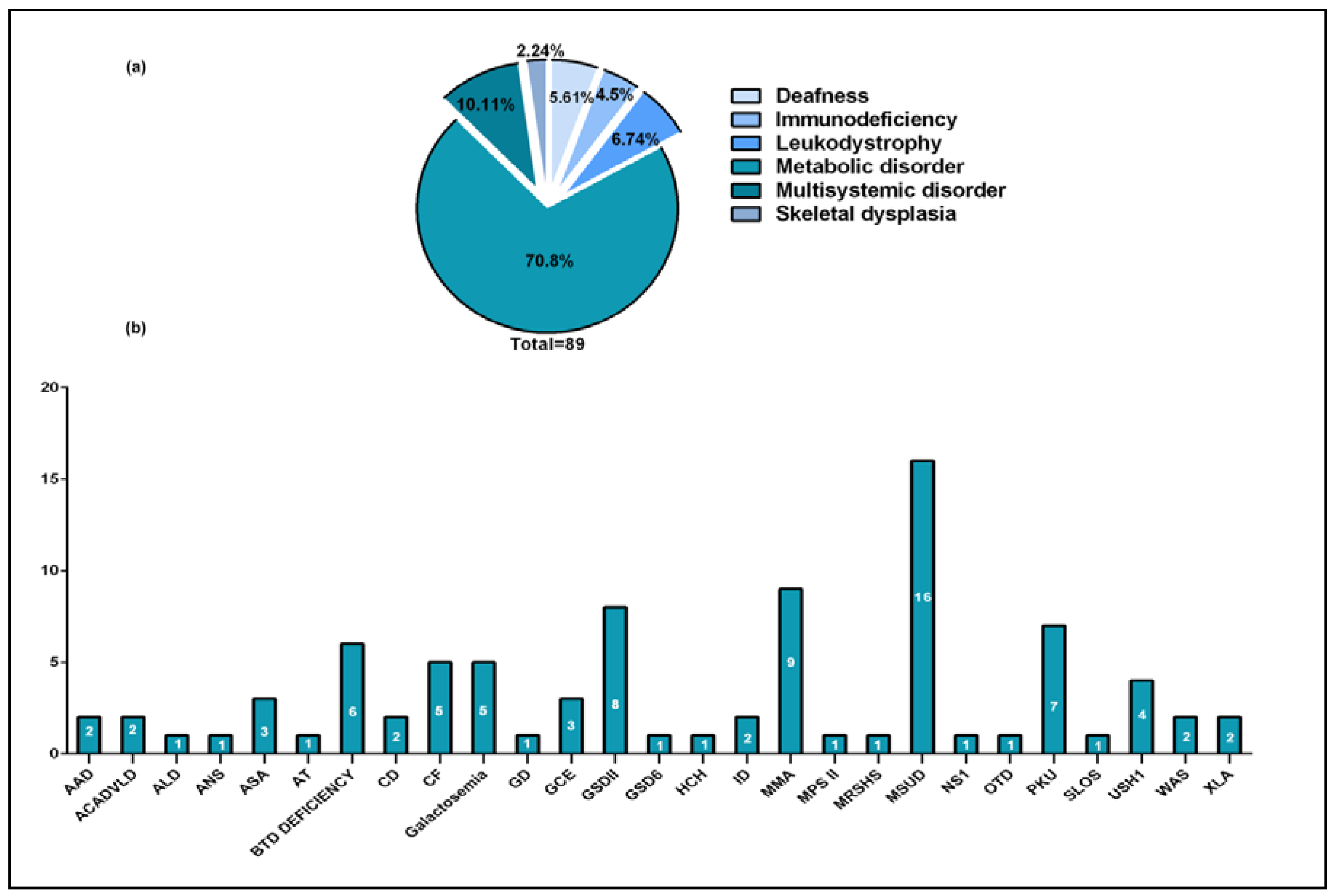
2.1. Samples
2.2. Library Building and Sequencing
2.3. Data Analysis
2.4. Tertiary Analysis and Variant Validation
3. Results
3.1. Sequencing Quality, Coverage and Overall Panel Performance
3.2. Variant Calling
3.3. Variant Detection Yield
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, S.B.; Hood, L.E. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Kwitek, A.E.; Cogen, P.H.; Metzger, A.K.; Duyk, G.M.; Sheffield, V.C. A denaturing gradient gel electrophoresis assay for sensitive detection of p53 mutations. Hum. Genet. 1993, 91, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Brahmandam, M.; Yan, L.; Cantor, M.; Namgyal, C.; Mino-Kenudson, M.; Lauwers, G.Y.; Loda, M.; Fuchs, C.S. Sensitive sequencing method for KRAS mutation detection by pyrosequencing. J. Mol. Diagn. 2005, 7, 413–421. [Google Scholar] [CrossRef]
- Kumazawa, R.; Tomiyama, H.; Li, Y.; Imamichi, Y.; Funayama, M.; Yoshino, H.; Yokochi, F.; Fukusako, T.; Takehisa, Y.; Kashihara, K.; et al. Mutation analysis of the PINK1 gene in 391 patients with parkinson disease. Arch. Neurol. 2008, 65, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Quesada, M.A.; Mathies, R.A. DNA sequencing using capillary array electrophoresis. Anal. Chem. 1992, 64, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Takahashi, S. Multiple-sheathflow capillary array DNA analyser. Nature 1993, 361, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ning, B.; Fang, H.; Hong, H.; Perkins, R.; Tong, W.; Shi, L. Next-generation sequencing and its applications in molecular diagnostics. Expert. Rev. Mol. Diagn. 2011, 11, 333–343. [Google Scholar] [PubMed]
- Nemeth, A.H.; Kwasniewska, A.C.; Lise, S.; Parolin Schnekenberg, R.; Becker, E.B.; Bera, K.D.; Shanks, M.E.; Gregory, L.; Buck, D.; Zameel Cader, M.; et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013, 136, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- Sikkema-Raddatz, B.; Johansson, L.F.; de Boer, E.N.; Almomani, R.; Boven, L.G.; van den Berg, M.P.; van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; Sijmons, R.H.; Jongbloed, J.D.; et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum. Mutat. 2013, 34, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Z.; Wang, Z.; Chen, D.; Chai, Y.; Pang, X.; Sun, L.; Wang, X.; Yang, T.; Wu, H. Targeted next-generation sequencing in Uyghur families with non-syndromic sensorineural hearing loss. PLoS ONE 2015, 10, e0127879. [Google Scholar] [CrossRef] [PubMed]
- Nijman, I.J.; van Montfrans, J.M.; Hoogstraat, M.; Boes, M.L.; van de Corput, L.; Renner, E.D.; van Zon, P.; van Lieshout, S.; Elferink, M.G.; van der Burg, M.; et al. Targeted next-generation sequencing: A novel diagnostic tool for primary immunodeficiencies. J. Allergy Clin. Immunol. 2014, 133, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Besnard, T.; Garcia-Garcia, G.; Baux, D.; Vache, C.; Faugere, V.; Larrieu, L.; Leonard, S.; Millan, J.M.; Malcolm, S.; Claustres, M.; et al. Experience of targeted usher exome sequencing as a clinical test. Mol. Genet. Genom. Med. 2014, 2, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.R.; Riesch, E.; Scheurenbrand, T.; Schubach, M.; Wilhelm, C.; Steiner, I.; Hansen, J.; Courage, C.; Gallati, S.; Burki, S.; et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012, 53, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-San Jose, P.; Corton, M.; Blanco-Kelly, F.; Avila-Fernandez, A.; Lopez-Martinez, M.A.; Sanchez-Navarro, I.; Sanchez-Alcudia, R.; Perez-Carro, R.; Zurita, O.; Sanchez-Bolivar, N.; et al. Targeted next-generation sequencing improves the diagnosis of autosomal dominant retinitis pigmentosa in Spanish patients. Invest. Ophthalmol. Vis. Sci. 2015, 56, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Yohe, S.; Hauge, A.; Bunjer, K.; Kemmer, T.; Bower, M.; Schomaker, M.; Onsongo, G.; Wilson, J.; Erdmann, J.; Zhou, Y.; et al. Clinical validation of targeted next-generation sequencing for inherited disorders. Arch. Pathol. Lab. Med. 2015, 139, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Vasli, N.; Bohm, J.; Le Gras, S.; Muller, J.; Pizot, C.; Jost, B.; Echaniz-Laguna, A.; Laugel, V.; Tranchant, C.; Bernard, R.; et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol. 2012, 124, 273–283. [Google Scholar] [CrossRef] [PubMed]
- O'Sullivan, J.; Mullaney, B.G.; Bhaskar, S.S.; Dickerson, J.E.; Hall, G.; O'Grady, A.; Webster, A.; Ramsden, S.C.; Black, G.C. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J. Med. Genet. 2012, 49, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, H.; Zhang, L.; Tang, C.; Jones, L.; Ye, H.; Ban, L.; Wang, A.; Liu, Z.; Lou, F.; et al. Rapid detection of genetic mutations in individual breast cancer patients by next-generation DNA sequencing. Hum. Genom. 2015, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Saudi Mendeliome, G. Comprehensive gene panels provide advantages over clinical exome sequencing for mendelian diseases. Genome Biol. 2015, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Al-Mousa, H.; Abouelhoda, M.; Monies, D.M.; Al-Tassan, N.; Al-Ghonaium, A.; Al-Saud, B.; Al-Dhekri, H.; Arnaout, R.; Al-Muhsen, S.; Ades, N.; et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J. Allergy Clin. Immunol. 2016, 137, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. Sift web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using polyphen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.S.; Cooper, D.N.; Polychronakos, C.; Naidoo, N.; Wu, M.; Soong, R. Exome sequencing: Dual role as a discovery and diagnostic tool. Ann. Neurol. 2012, 71, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ruivenkamp, C.A.; Hoffer, M.J.; Vrijenhoek, T.; Kriek, M.; van Asperen, C.J.; den Dunnen, J.T.; Santen, G.W. Next-generation diagnostics: Gene panel, exome, or whole genome? Hum. Mutat. 2015, 36, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, H.L.; Schroeder, B.; Turner, C.; Shriver, C.D.; Ellsworth, D.L.; Ellsworth, R.E. Management of incidental findings in the era of next-generation sequencing. Curr. Genom. 2015, 16, 159–174. [Google Scholar] [CrossRef] [PubMed]
- ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Genet. Med. 2012, 14, 759–761. [Google Scholar]
- LaDuca, H.; Farwell, K.D.; Vuong, H.; Lu, H.M.; Mu, W.; Shahmirzadi, L.; Tang, S.; Chen, J.; Bhide, S.; Chao, E.C. Exome sequencing covers > 98% of mutations identified on targeted next generation sequencing panels. PLoS ONE 2017, 12, e0170843. [Google Scholar] [CrossRef] [PubMed]
- Alkuraya, F.S. Genetics and genomic medicine in saudi arabia. Mol. Genet. Genom. Med. 2014, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.; Butler, T.; Rhodes, K.; Quist, M.; Neff, T.L.; Moore, S.; Tomlins, S.A.; Reinig, E.; Beadling, C.; Andersen, M.; et al. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J. Mol. Diagn. 2015, 17, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Santani, A.; Murrell, J.; Funke, B.; Yu, Z.; Hegde, M.; Mao, R.; Ferreira-Gonzalez, A.; Voelkerding, K.V.; Weck, K.E. Development and validation of targeted next-generation sequencing panels for detection of germline variants in inherited diseases. Arch. Pathol. Lab. Med. 2017, 141, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Broendberg, A.K.; Christiansen, M.K.; Nielsen, J.C.; Pedersen, L.N.; Jensen, H.K. Targeted next generation sequencing in a young population with suspected inherited malignant cardiac arrhythmias. Eur. J. Hum. Genet. 2018, 26, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Malone, C.P.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-based population next-generation sequencing for inherited retinal degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hosomichi, K.; Chiga, M.; Mandai, S.; Nakaoka, H.; Sohara, E.; Okado, T.; Rai, T.; Sasaki, S.; Inoue, I.; et al. Comprehensive genetic testing approach for major inherited kidney diseases, using next-generation sequencing with a custom panel. Clin. Exp. Nephrol. 2017, 21, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Pua, C.J.; Bhalshankar, J.; Miao, K.; Walsh, R.; John, S.; Lim, S.Q.; Chow, K.; Buchan, R.; Soh, B.Y.; Lio, P.M.; et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J. Cardiovasc. Transl. Res. 2016, 9, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Benmeakel, M.; Hossain, M.A.; Al Mutairi, F.; Al Othaim, A.; Alfares, A.A.; Al Balwi, M.; Alzaben, A.; Eyaid, W. Thirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia. Orphanet J. Rare Dis. 2016, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Afifi, A.M.; Abdul-Jabbar, M.A. Saudi newborn screening. A national public health program: Needs, costs, and challenges. Saudi. Med. J. 2007, 28, 1167–1170. [Google Scholar] [PubMed]
- Memish, Z.A.; Saeedi, M.Y. Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and β-thalassemia in saudi arabia. Ann. Saudi. Med. 2011, 31, 229–235. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical Diagnosis | Abbreviation | Category | Genes Included in the Panel | OMIM# |
|---|---|---|---|---|
| Agammaglobulinemia, X-linked, Type I | XLA | Immunodeficiency | BTK | 300755 |
| Argininosuccinate Lyase Deficiency | ASA | Metabolic disorder | ASL | 207900 |
| Arylsulfatase A Deficiency | AAD | Leukodystrophy | ARSA | 250100 |
| Ataxia Neuropathy Spectrum/Alpers Syndrome | ANS | Multisystemic disorder | POLG | 203700 |
| Ataxia-telangiectasia | AT | Multisystemic disorder | ATM | 208900 |
| Biotinidase Deficiency | BTD deficiency | Metabolic disorder | BTD | 253260 |
| Canavan Disease | CD | Leukodystrophy | ASPA | 271900 |
| Cystic Fibrosis | CF | Multisystemic disorder | CFTR | 219700 |
| Galactosemia | Galactosemia | Metabolic disorder | GALT | 230400 |
| Gaucher Disease | GD | Multisystemic disorder | GBA | 230800 |
| Glycine Encephalopathy | GCE | Metabolic disorder | GCSH, GLDC, AMT | 605899 |
| Glycogen Storage Disease Type VI | GSD6 | Metabolic disorder | GBE1 | 232700 |
| Hunter Syndrome/Mucopolysaccharidosis, Type II (MPS II) | MPS II | Metabolic disorder | IDS | 309900 |
| Hypochondroplasia | HCH | Skeletal dysplasia | FGFR3 | 146000 |
| Inherited Deafness | ID | Deafness | GJB2, GJB3, GJB6, COL11A2, KCNQ4 | 220290 |
| Maple syrup Urine Disease | MSUD | Metabolic disorder | BCKDHA, BCKDHB, DBT, DLD | 248600 |
| Marshall syndrome | MRSHS | Skeletal dysplasia | COL11A1 | 154780 |
| Methylmalonic Acidemia | MMA | Metabolic disorder | MMAA, MMAB, MMACHC, MUT | 251100 |
| Noonan Syndrome (Types 1, 3, 4, 5 ,6) | NS | Multisystemic disorder | KRAS, NRAS, PTPN11, RAF1, SOS1 | 163950/609942/610733/611533/613224 |
| Ornithine Transcarbamylase Deficiency | OTD | Metabolic disorder | OTC | 311250 |
| Phenylketonuria | PKU | Metabolic disorder | PAH | 261600 |
| Pompe Disease/Glycogen Storage Disease II (GSD II) | GSDII | Metabolic disorder | GAA | 232300 |
| Smith-Lemli-Optiz Syndrome | SLOS | Metabolic disorder | DHCR7 | 270400 |
| Usher Syndrome Type 1 | USH1 | Deafness | PCDH15, USH1C, CDH23, MYO7A | 276900/601067 |
| Very Long Chain Acyl-Coenzyme A Dehydrogenase Deficiency | ACADVLD | Metabolic disorder | ACADVL | 201475 |
| Wiskott-Aldrich Syndrome | WAS | Immunodeficiency | WAS | 301000 |
| X-Linked Adrenoleukodystrophy | ALD | Leukodystrophy | ABCD1 | 300100 |
| Original Mutation | IDP Panel | |||||
|---|---|---|---|---|---|---|
| Case ID | Gene | Transcript ID | cDNA | Protein | Original Mutation Found (Y/N) | Genotype Matching (Y/N) |
| 13-0045 | ABCD1 | NM_000033 | c.1581C>A | p.Y527X | Y | Y |
| 13-0051 | ACADVL | NM_000018 | c.65C>A | p.S22X | Y | Y |
| 14-3258 | ACADVL | NM_000018 | c.65C>A | p.S22X | Y | Y |
| 13-0097 | AMT | NM_001164710 | c.533G>A | p.R178H | Y | Y |
| 13-0098 | AMT | NM_001164710 | c.280C>T | p.R94W | Y | Y |
| 13-0059 | ARSA | NM_000487 | c.1055A>G | p.N352S | Y | Y |
| 13-0095 | ARSA | NM_000487 | #Missense | Y | Y | |
| 13-0040 | ASL | NM_001024943 | c.1000C>T | p.Q334X | Y | Y |
| 13-0131 | ASL | NM_001024943 | c.556C>T | p.R186W | Y | Y |
| 13-0132 | ASL | NM_001024943 | c.544C>T | p.R182X | Y | Y |
| 13-0133 | ASPA | NM_000049 | #Frameshift insertion | N | NA | |
| 14-3064 | ASPA | NM_000049 | #Frameshift deletion | N | NA | |
| 13-0020 | ATM | NM_000051 | c.381_381delA | p.V128* | Y | Y |
| 13-0009 | BCKDHA | NM_000709 | c.905A>C | p.D302A | Y | Y |
| 13-0013 | BCKDHA | NM_000709 | c.905A>C | p.D302A | Y | Y |
| 13-0093 | BCKDHA | NM_000709 | c.1270C>T | p.Q424X | Y | Y |
| 13-0094 | BCKDHA | NM_000709 | c.647-1G>C | NA | Y | Y |
| 13-0096 | BCKDHA | NM_000709 | c.347A>G | p.D116G | Y | Y |
| 13-0120 | BCKDHA | NM_000709 | c.808G>A | p.A270T | Y | Y |
| 13-0135 | BCKDHA | NM_000709 | c.659_662delCGTA | p.Y221Qfs*108 | Y | Y |
| 13-0091 | BCKDHB | NM_000056 | c.286_288delGAA | p.E96del | N | NA |
| 13-0092 | BCKDHB | NM_000056 | c.1A>T | p.M1L | N | NA |
| 13-0129 | BCKDHB | NM_000056 | c.1006G>A | p.G336S | Y | Y |
| 13-0053 | BTD | NM_000060 | #Missense | Y | Y | |
| 13-0061 | BTD | NM_000060 | #Frameshift deletion | Y | Y | |
| 13-0062 | BTD | NM_000060 | #Missense | Y | Y | |
| 13-0087 | BTD | NM_000060 | #Missense | Y | Y | |
| 13-0088 | BTD | NM_000060 | #Frameshift deletion | Y | Y | |
| 13-0019 | BTK | NM_000061 | c.763C>T | p.R255X | Y | Y |
| 13-0028 | BTK | NM_000061 | c.982C>T | p.Q328X | Y | Y |
| 13-0043 | CFTR | NM_000492 | c.1418_1418delG | p.G473Efs*54 | N | NA |
| 13-0054 | CFTR | NM_000492 | c.1520_1522delTCT | p.F508del | Y | Y |
| 14-3079 | CFTR | NM_000492 | c.3700A>G | p.I1234V | Y | Y |
| 14-3072 | CFTR | NM_000492 | c.416A>T | p.H139L | Y | Y |
| 14-3071 | CFTR | NM_000492 | c.3700A>G | p.I1234V | Y | Y |
| 13-0090 | CDH23 | NM_001171930 | #Missense | Y | Y | |
| 13-0102 | COL11A1 | NM_080630 | c.2354G>A | p.G785E | Y | Y |
| 13-0011 | DBT | NM_001918 | c.61_61delC | p.R21Afs*12 | Y | Y |
| 13-0046 | DBT | NM_001918 | c.773-2A>G | NA | Y | Y |
| 13-0099 | DBT | NM_001918 | c.1195T>C | p.S399P | Y | Y |
| 13-0104 | DBT | NM_001918 | c.1281+3A>G | NA | Y | Y |
| 13-0107 | DBT | NM_001918 | c.137A>G | p.K46R | Y | Y |
| 13-0127 | DBT | NM_001918 | c.773-2A>G | NA | Y | Y |
| 13-0108 | DHCR7 | NM_001163817 | c.861C>G | p.N287K | Y | Y |
| 13-0100 | FGFR3 | NM_000142 | c.1138G>A | p.G380R | Y | Y |
| 13-0010 | GAA | NM_000152 | #Nonsense | Y | Y | |
| 13-0012 | GAA | NM_000152 | #Nonsense | Y | Y | |
| 13-0055 | GAA | NM_000152 | #Nonsense | Y | Y | |
| 13-0103 | GAA | NM_000152 | #Nonsense | Y | Y | |
| 13-0109 | GAA | NM_000152 | #Missense | Y | Y | |
| 13-0110 | GAA | NM_000152 | #Missense | Y | Y | |
| 13-0124 | GAA | NM_000152 | c.655G>A | p.G219R | Y | Y |
| 13-0128 | GAA | NM_000152 | #Nonsense | Y | Y | |
| 13-0064 | GALT | NM_000155 | #Frameshift deletion | Y | Y | |
| 13-0125 | GALT | NM_001258332 | #Missense | Y | Y | |
| 13-0134 | GALT | NM_001258332 | #Missense | Y | Y | |
| 14-3078 | GALT | NM_001258332 | #Missense | Y | Y | |
| 14-3067 | GALT | NM_001258332 | #Missense | Y | Y | |
| 13-0101 | GBA | NM_000157 | c.152G>T | p.S51I | Y | Y |
| 13-0130 | GBE1 | NM_000158 | #Missense | Y | Y | |
| 13-0116 | GJB2 | NM_004004 | c.299T>C | p.W77R | Y | Y |
| 13-0116 | GJB2 | NM_004004 | c.506G>A | p.C169Y | Y | Y |
| 13-0111 | GLDC | NM_000170 | c.2113G>A | p.V705M | Y | Y |
| 13-0060 | IDS | NM_000202 | #Nonsense | Y | Y | |
| 13-0112 | MMAA | NM_172250 | #Nonsense | Y | Y | |
| 13-0126 | MMAA | NM_172250 | #Missense | Y | Y | |
| 13-0063 | MUT | NM_000255 | #Frameshift deletion | Y | Y | |
| 13-0105 | MUT | NM_000255 | c.329A>G | p.Y110C | Y | Y |
| 13-0121 | MUT | NM_000255 | c.278G>A | p.R93H | Y | Y |
| 14-3081 | MUT | NM_000255 | c.1160C>T | p.T387I | Y | Y |
| 14-3080 | MUT | NM_000255 | c.2200C>T | p.Q734X | Y | Y |
| 14-3070 | MUT | NM_000255 | c.2200C>T | p.Q734X | Y | Y |
| 14-3065 | MUT | NM_000255 | c.1677-1G>C | NA | Y | Y |
| 13-0044 | MYO7A | NM_000260 | c.5880-5882delCTT | p.F1961del | Y | Y |
| 13-0117 | MYO7A | NM_000260 | c.2005C>T | p.R669X | Y | Y |
| 13-0066 | OTC | NM_000531 | #Missense | Y | Y | |
| 13-0057 | PAH | NM_000277 | #Missense | Y | Y | |
| 13-0113 | PAH | NM_000277 | #Nonsense | Y | Y | |
| 13-0114 | PAH | NM_000277 | #Missense | N | NA | |
| 13-0122 | PAH | NM_000277 | #Missense | Y | Y | |
| 13-0123 | PAH | NM_000277 | #Nonsense | Y | Y | |
| 13-0136 | PAH | NM_000277 | #Missense | Y | Y | |
| 14-3073 | PAH | NM_000277 | #Missense | Y | Y | |
| 13-0058 | POLG | NM_001126131 | c.2419C>T | p.R807C | Y | Y |
| 13-0115 | PTPN11 | NM_002834 | c.188A>G | p.Y63C | Y | Y |
| 13-0026 | WAS | NM_000377 | c.91G>A | p.E31K | Y | Y |
| 13-0027 | WAS | NM_000377 | c.100C>T | p.R34X | Y | Y |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafa, A.E.; Faquih, T.; Baz, B.; Kattan, R.; Al-Issa, A.; Tahir, A.I.; Imtiaz, F.; Ramzan, K.; Al-Sayed, M.; Alowain, M.; et al. Validation of Ion TorrentTM Inherited Disease Panel with the PGMTM Sequencing Platform for Rapid and Comprehensive Mutation Detection. Genes 2018, 9, 267. https://doi.org/10.3390/genes9050267
Mustafa AE, Faquih T, Baz B, Kattan R, Al-Issa A, Tahir AI, Imtiaz F, Ramzan K, Al-Sayed M, Alowain M, et al. Validation of Ion TorrentTM Inherited Disease Panel with the PGMTM Sequencing Platform for Rapid and Comprehensive Mutation Detection. Genes. 2018; 9(5):267. https://doi.org/10.3390/genes9050267
Chicago/Turabian StyleMustafa, Abeer E., Tariq Faquih, Batoul Baz, Rana Kattan, Abdulelah Al-Issa, Asma I. Tahir, Faiqa Imtiaz, Khushnooda Ramzan, Moeenaldeen Al-Sayed, Mohammed Alowain, and et al. 2018. "Validation of Ion TorrentTM Inherited Disease Panel with the PGMTM Sequencing Platform for Rapid and Comprehensive Mutation Detection" Genes 9, no. 5: 267. https://doi.org/10.3390/genes9050267
APA StyleMustafa, A. E., Faquih, T., Baz, B., Kattan, R., Al-Issa, A., Tahir, A. I., Imtiaz, F., Ramzan, K., Al-Sayed, M., Alowain, M., Al-Hassnan, Z., Al-Zaidan, H., Abouelhoda, M., Al-Mubarak, B. R., & Al Tassan, N. A. (2018). Validation of Ion TorrentTM Inherited Disease Panel with the PGMTM Sequencing Platform for Rapid and Comprehensive Mutation Detection. Genes, 9(5), 267. https://doi.org/10.3390/genes9050267