The Asymmetry is Derived from Mechanical Interlocking of Achiral Axle and Achiral Ring Components –Syntheses and Properties of Optically Pure [2]Rotaxanes–

 ,
,
Abstract
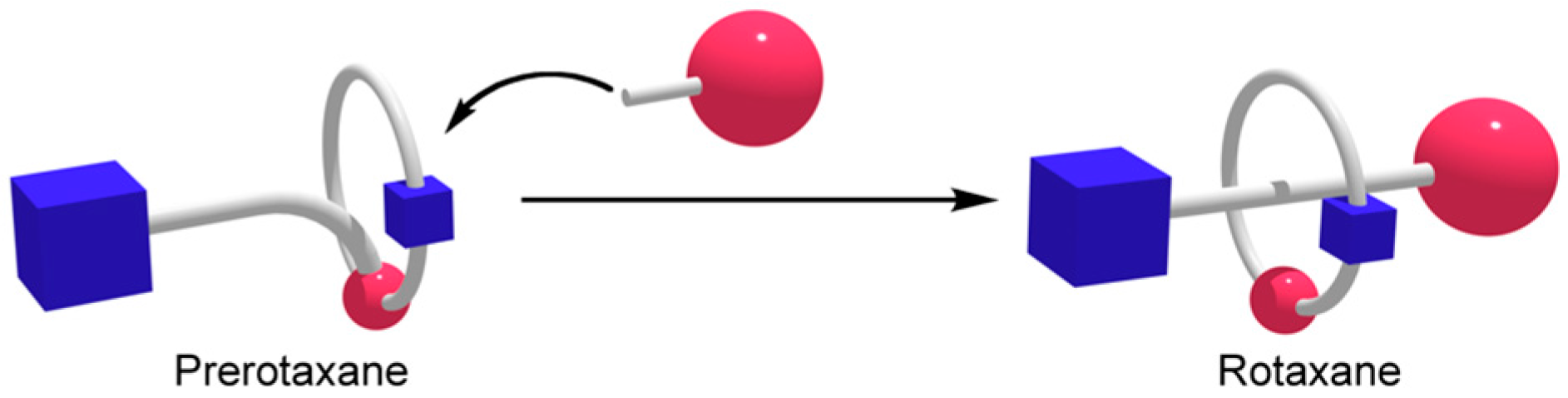
:1. Introduction
2. Materials and Methods
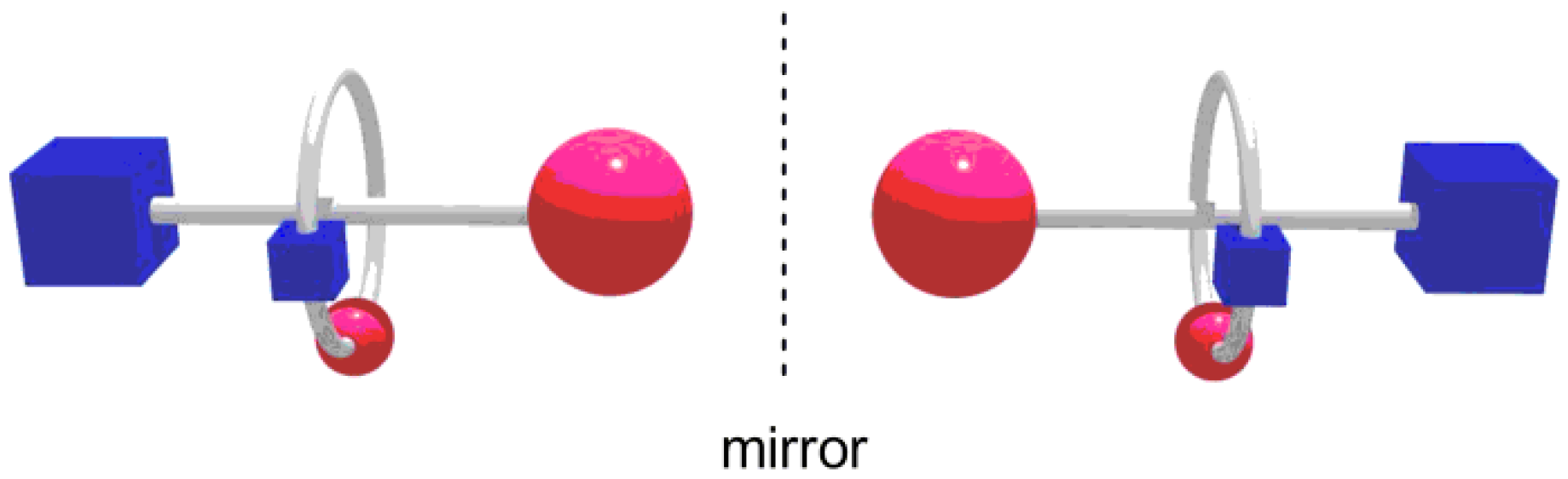
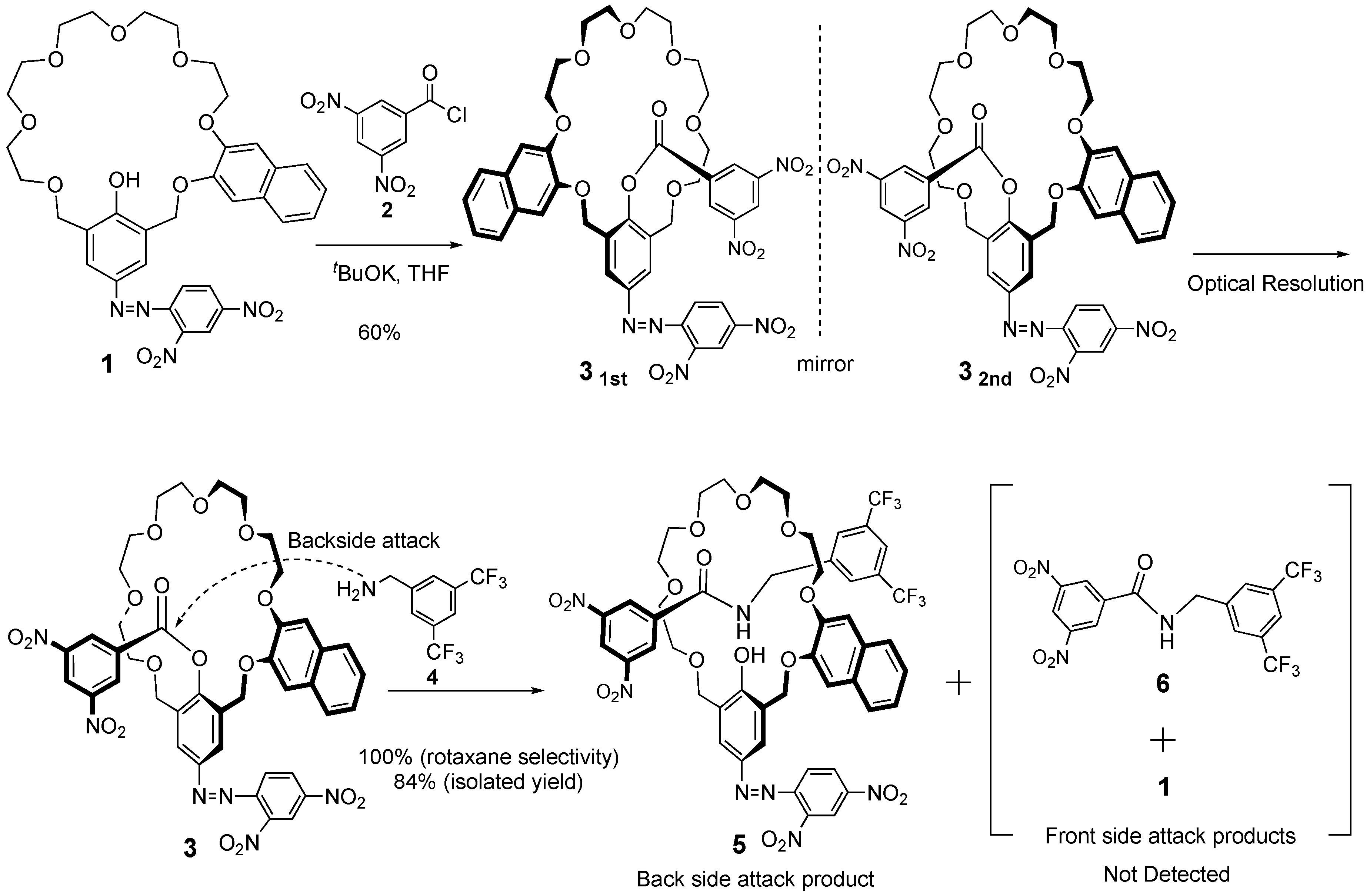
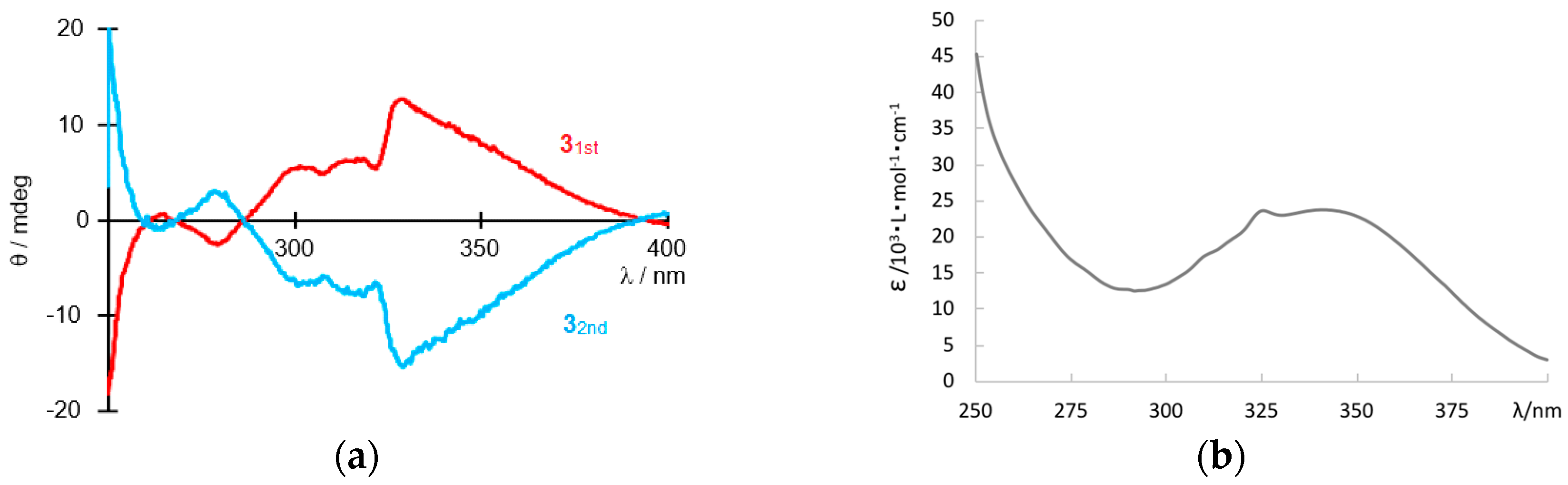
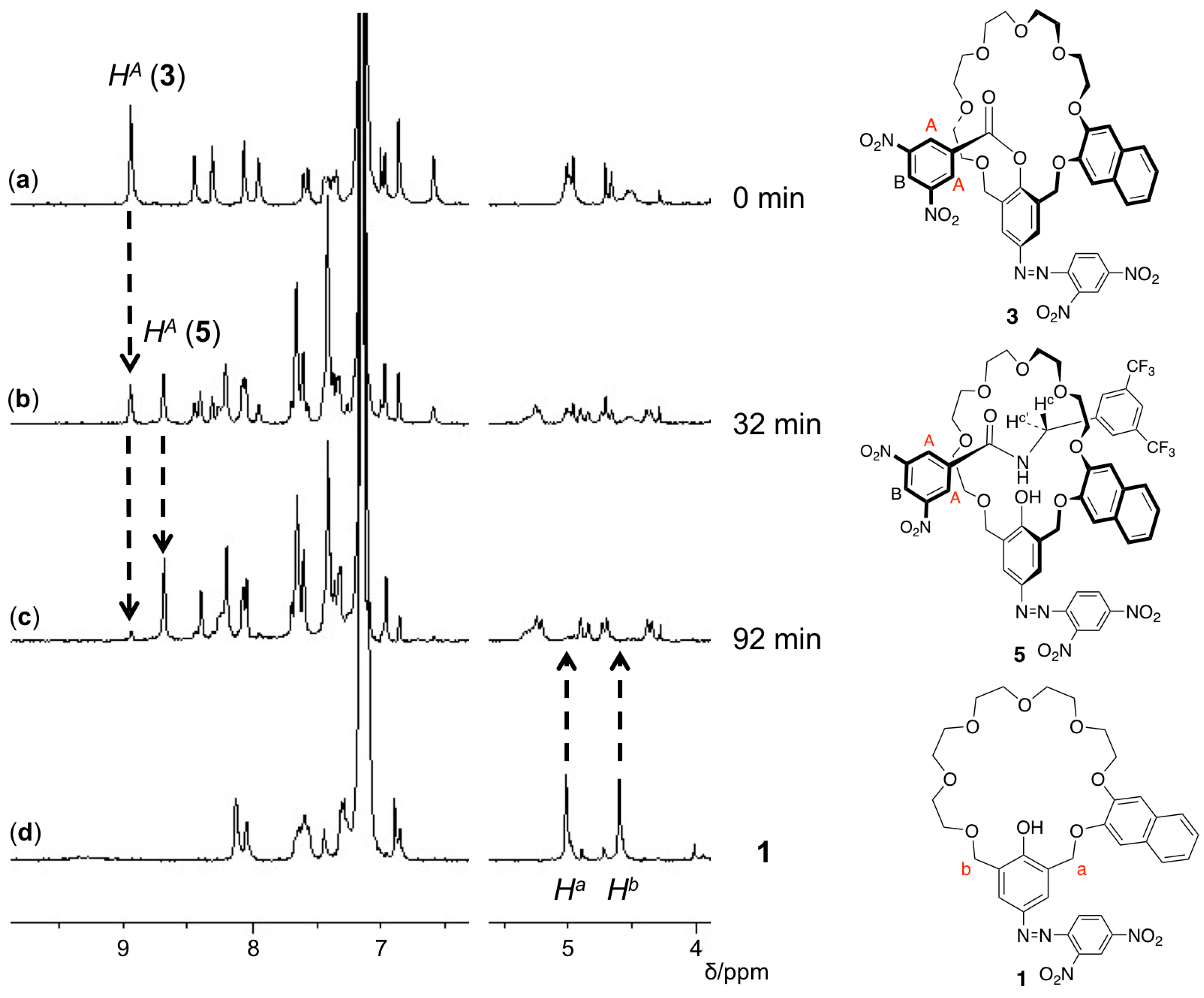
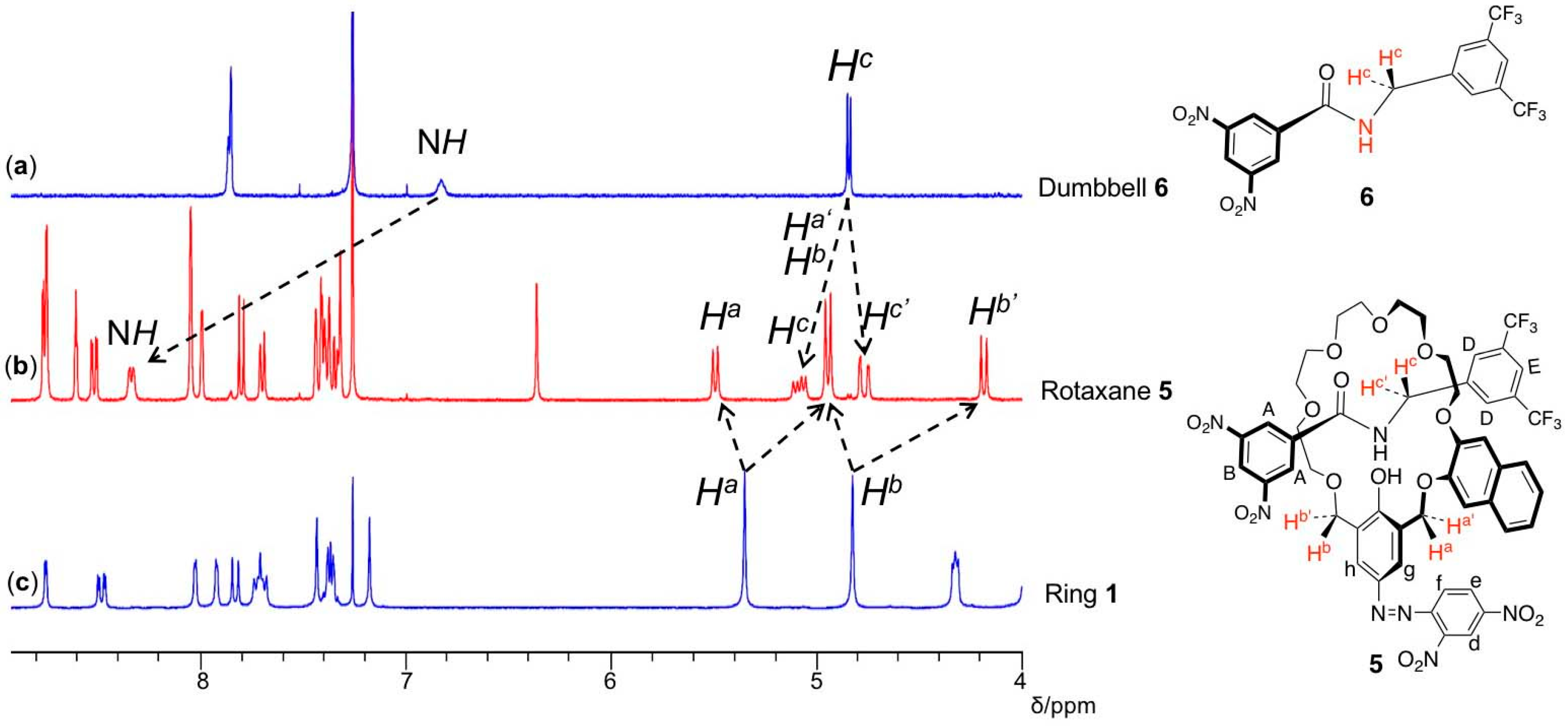
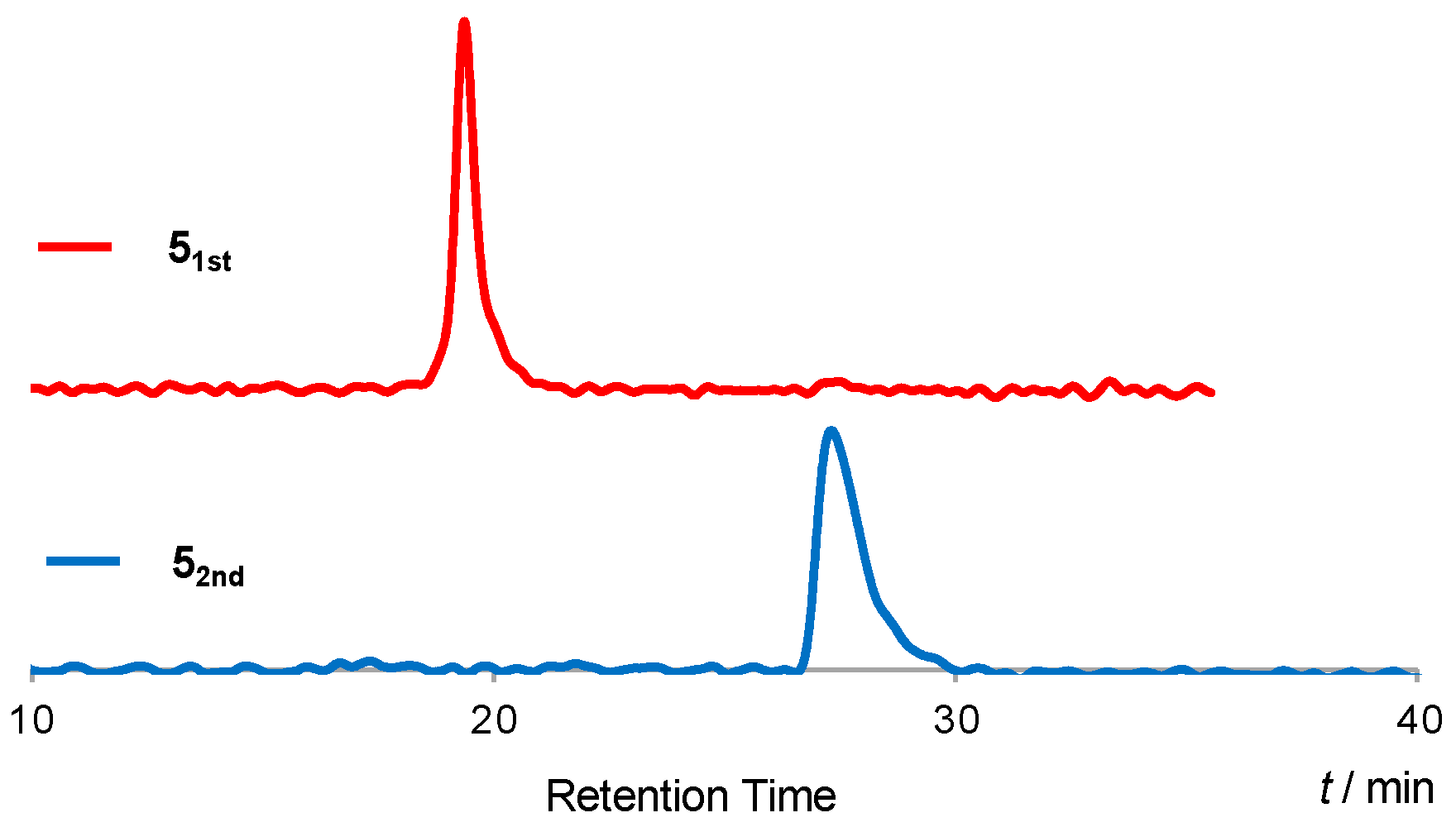
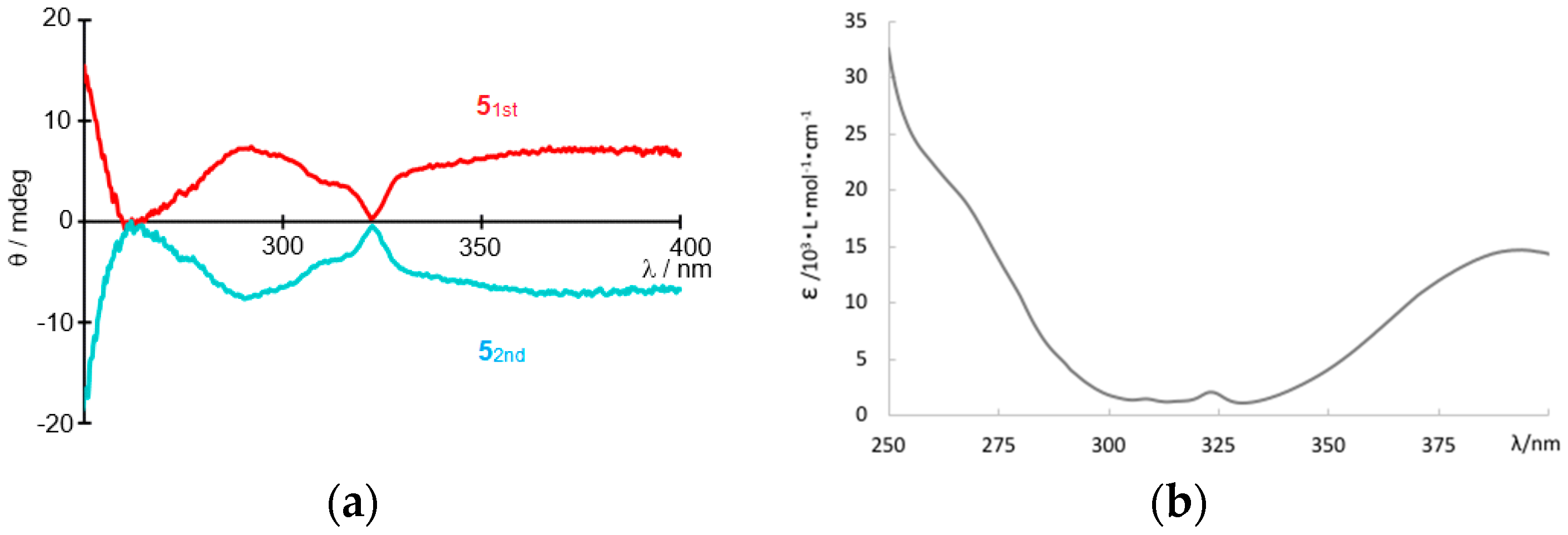
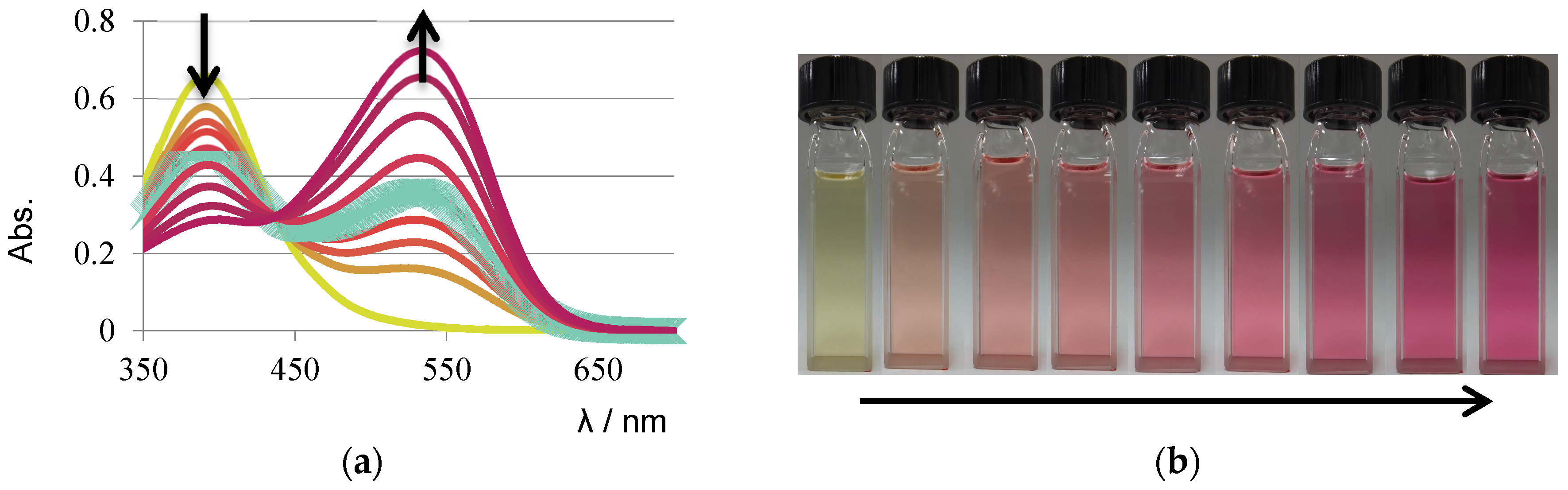
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. General
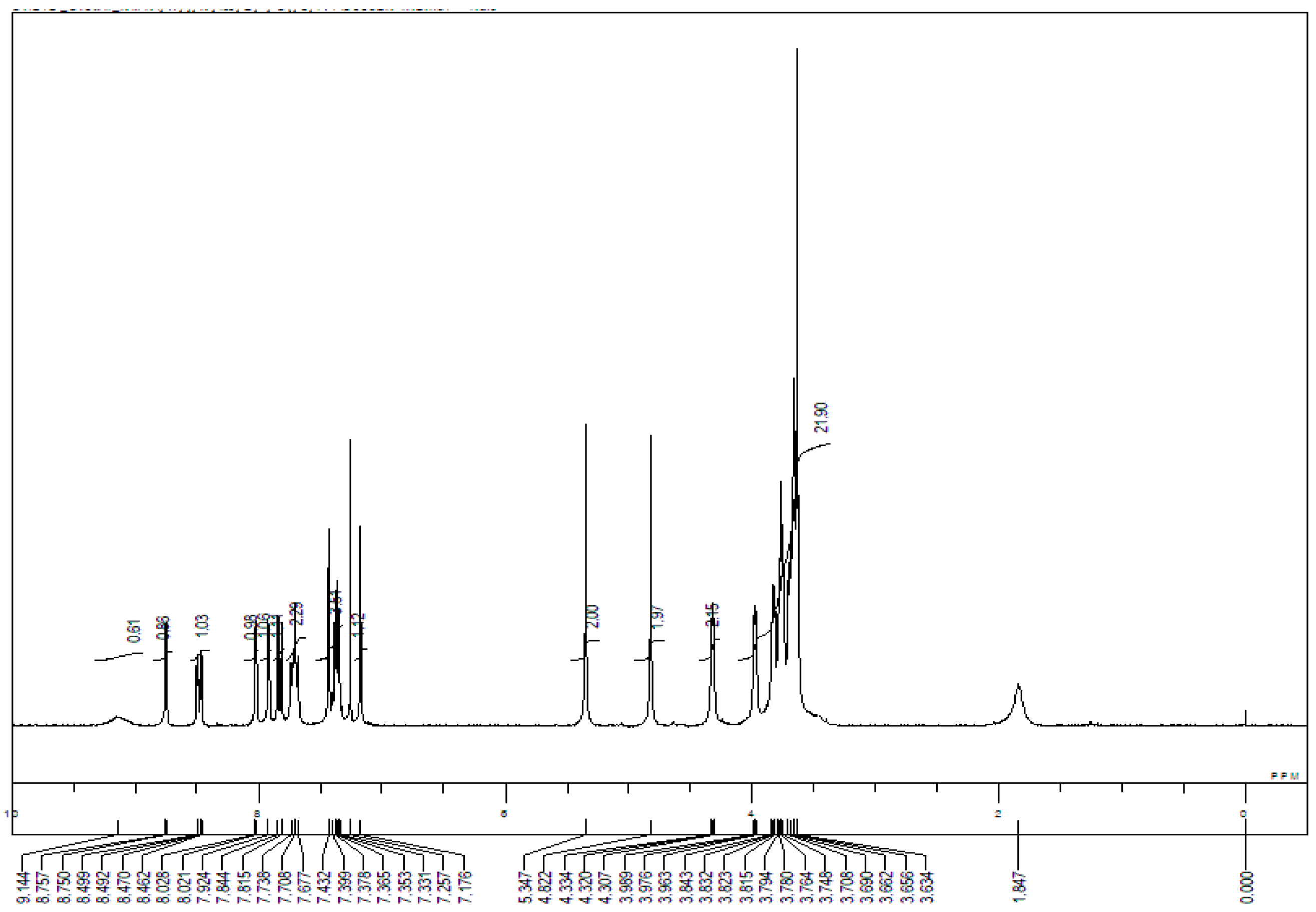
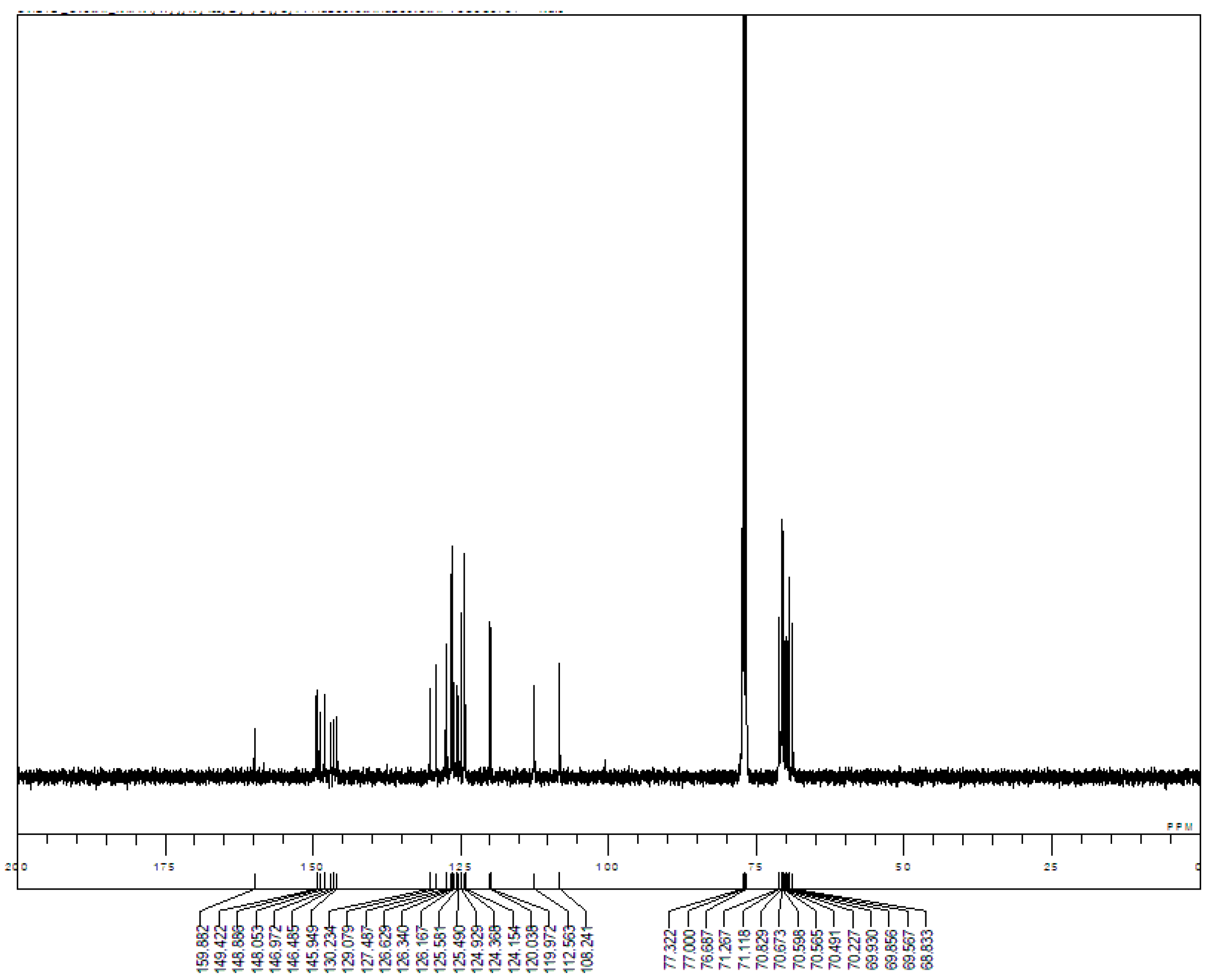
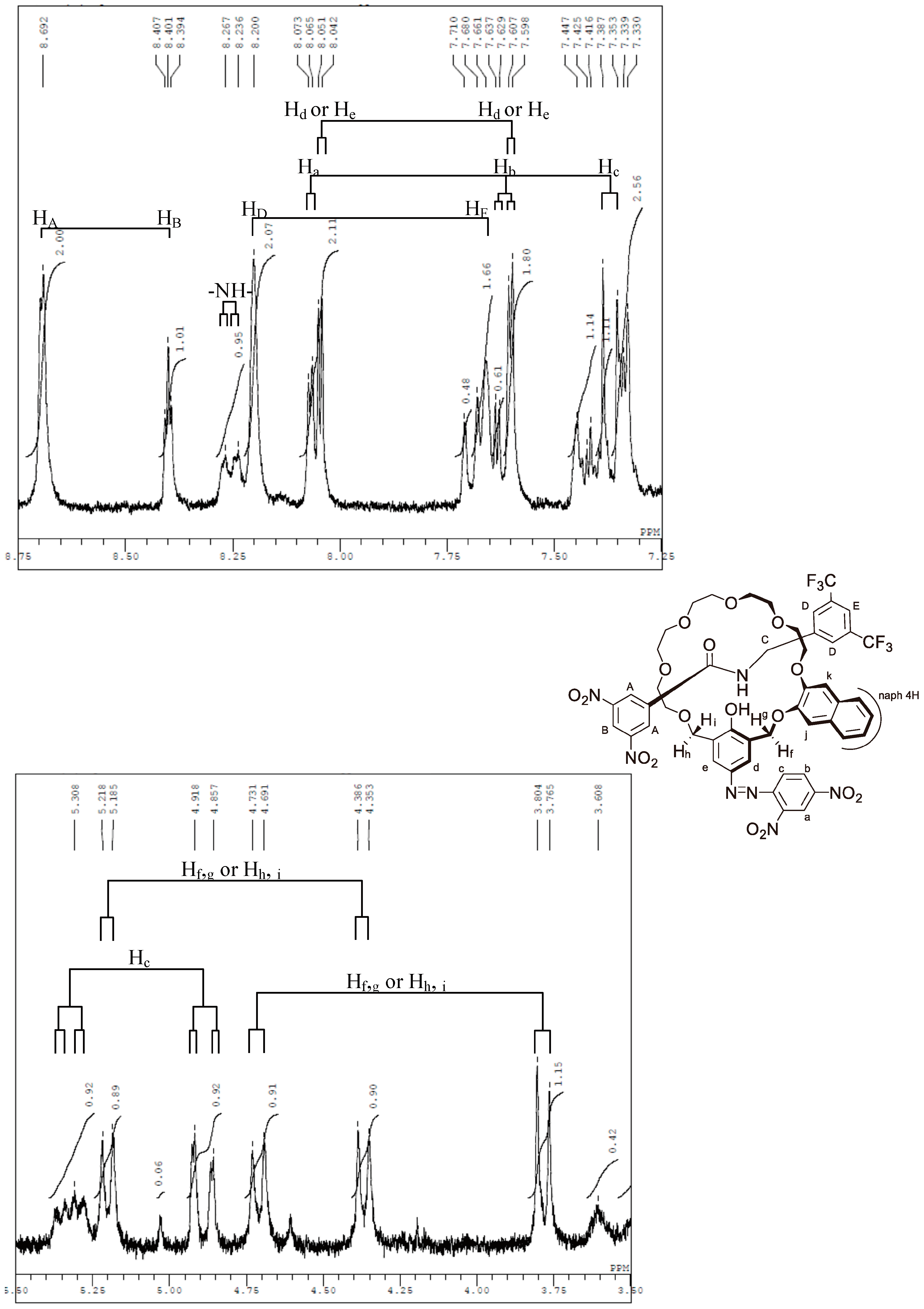
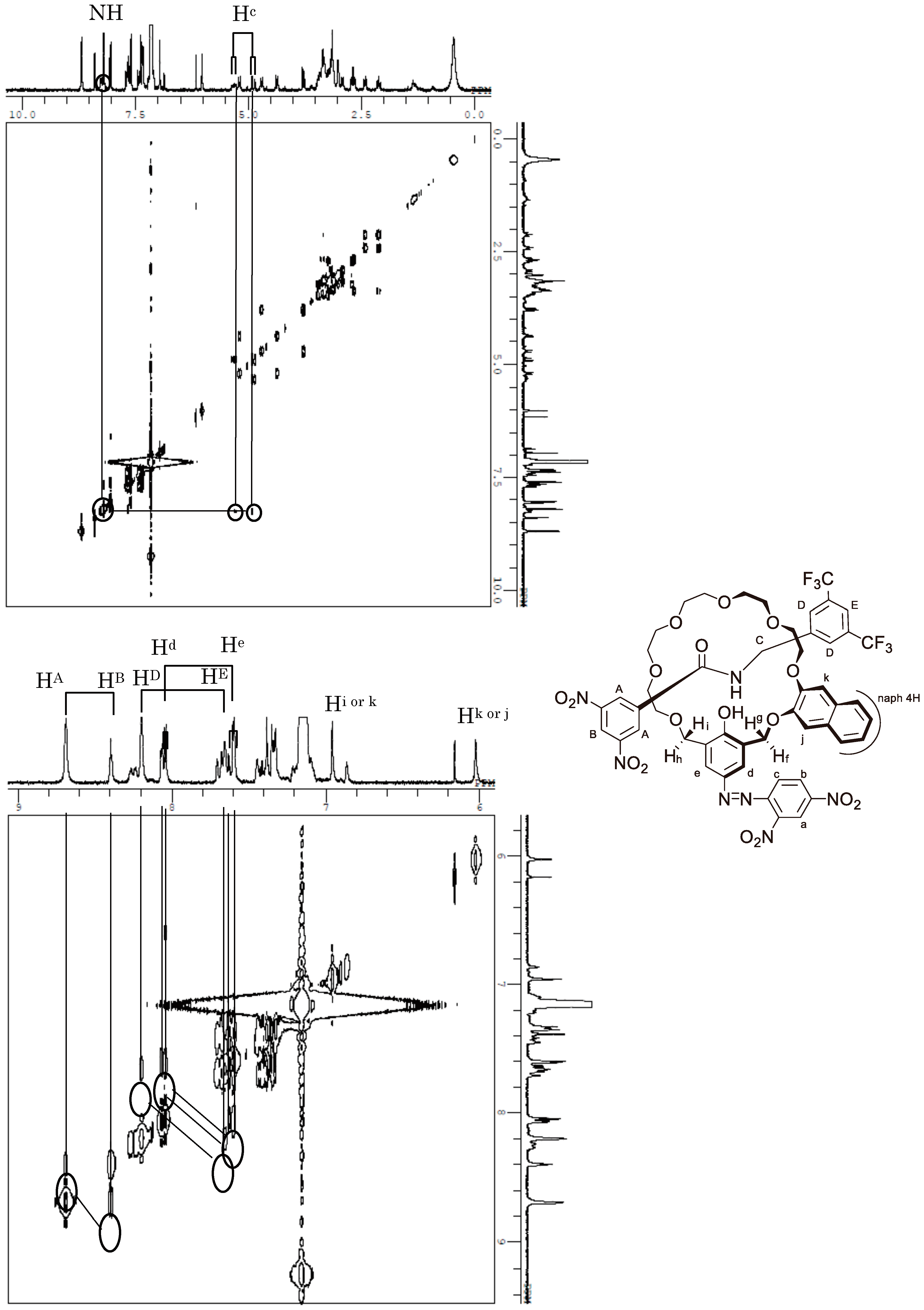
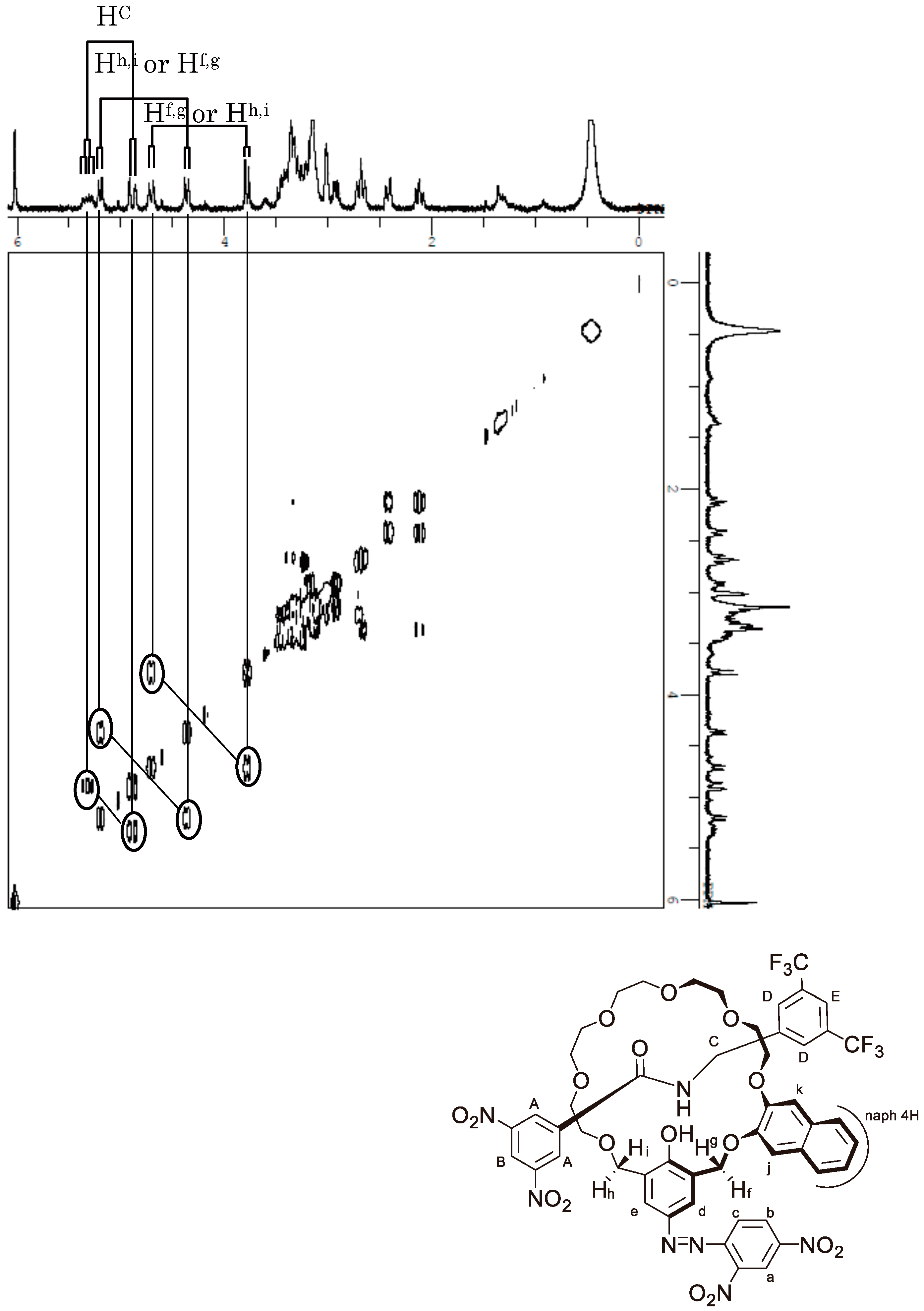
Appendix B. 1H-and 13C-NMR Spectra of Ring 1
Appendix C. 1H-and 13C-NMR Spectra of Prerotaxanes 3

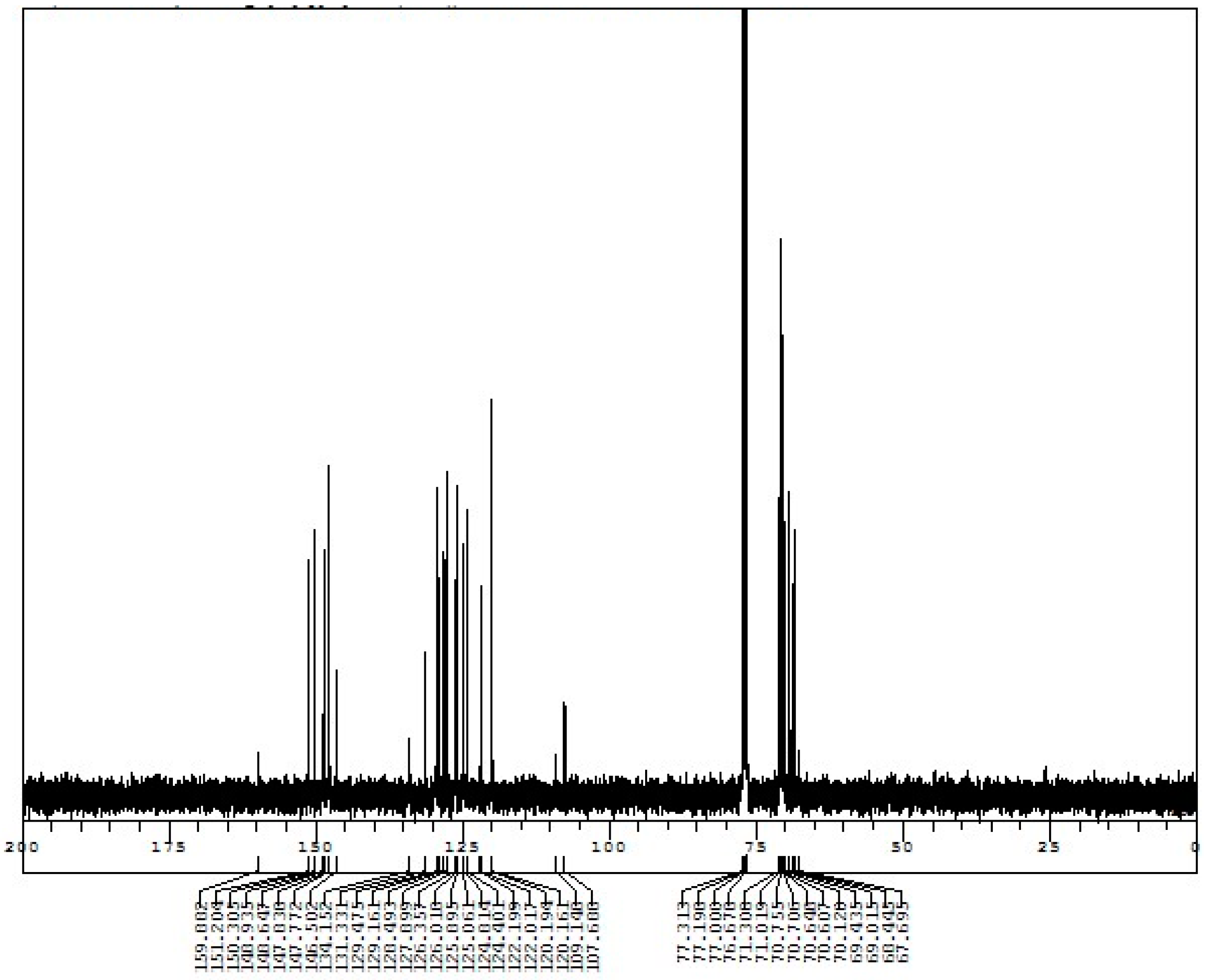
Appendix D. 1H-and 13C-NMR Spectra of Rotaxanes 5
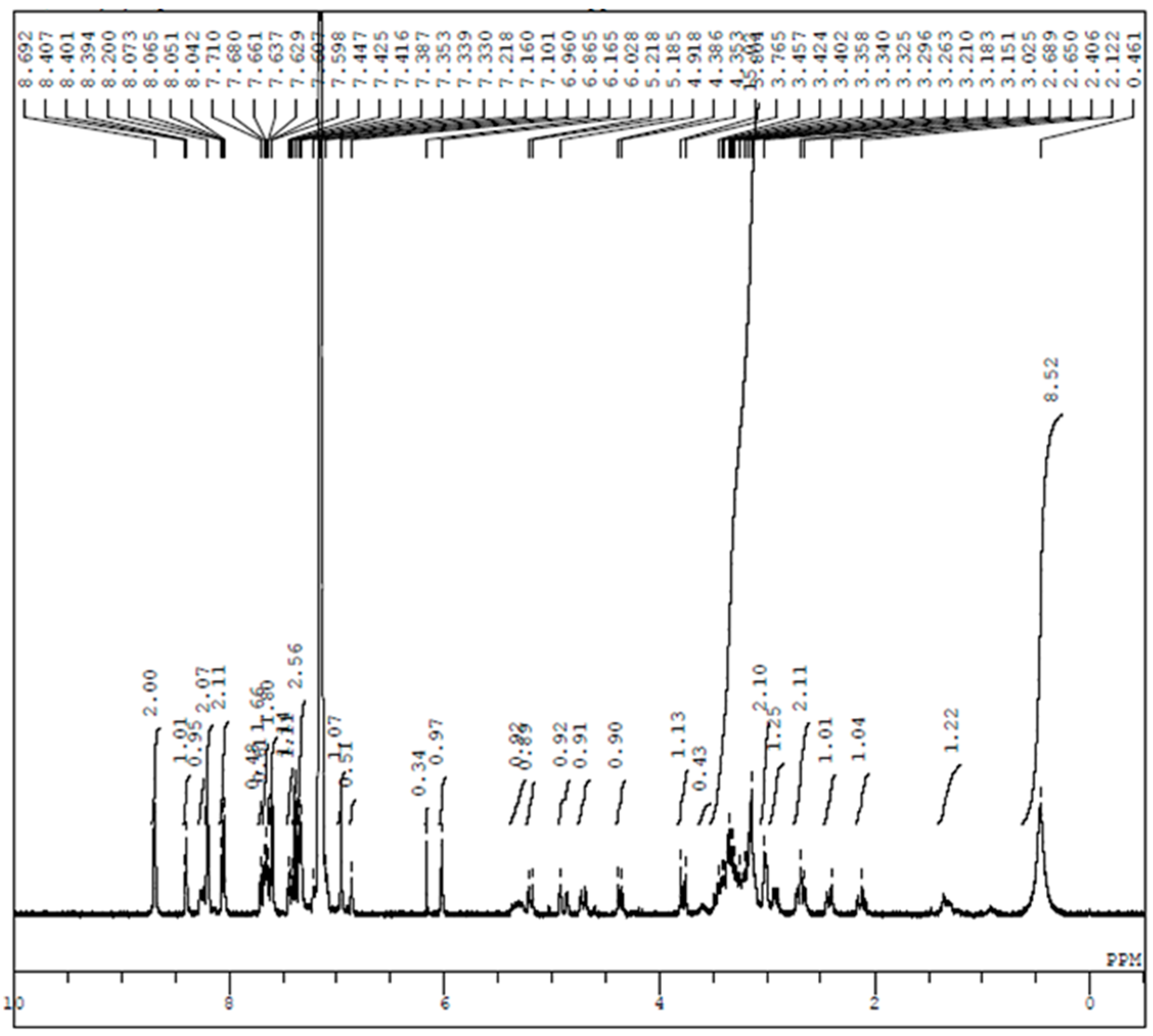

References and Note
- Pedersen, C.J. The discovery of crown ethers. Science 1988, 241, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Cram, D.J. The design of molecular hosts, guests, and their complexes. J. Incl. Phenom. 1988, 6, 397–413. [Google Scholar] [CrossRef]
- Lehn, J.M. Supramolecular chemistry—Scope and perspectives molecules, supermolecules, and molecular devices. Angew. Chem. Int. Ed. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Takagi, M.; Nakamura, H. Analytical application of functionalized crown ether-metal complexes. J. Coord. Chem. 1986, 15, 53–82. [Google Scholar] [CrossRef]
- Nakashima, K.; Nagaoka, Y.; Nakatsuji, S.I.; Kaneda, T.; Tanigawa, I.; Hirose, K.; Misumi, S.; Akiyama, S. Fluorescence reactions of “crowned” benzothiazolylphenols with alkali and alkaline earth metal ions and their analytical applications. Bull. Chem. Soc. Jpn. 1987, 60, 3219–3223. [Google Scholar] [CrossRef]
- Desilva, A.P.; Gunaratne, H.Q.N.; Gunnlaugsson, T.; Huxley, A.J.M.; Mccoy, C.P.; Rademacher, J.T.; Rice, T.E. Signaling recognition events with fluorescent sensors and switches. Chem. Rev. 1997, 97, 1515–1566. [Google Scholar] [CrossRef]
- Hisamoto, H. Ion-selective optodes: Current developments and future prospects. Trac-Trends Anal. Chem. 1999, 18, 513–524. [Google Scholar] [CrossRef]
- Stradiotto, N.R.; Yamanaka, H.; Zanoni, M.V.B. Electrochemical sensors: A powerful tool in analytical chemistry. J. Braz. Chem. Soc. 2003, 14, 159–173. [Google Scholar] [CrossRef]
- Faridbod, F.; Ganjali, M.R.; Dinarvand, R.; Norouzi, P.; Riahi, S. Schiff’s bases and crown ethers as supramolecular sensing materials in the construction of potentiometric membrane sensors. Sensors 2008, 8, 1645–1703. [Google Scholar] [CrossRef] [PubMed]
- Sogah, G.D.Y.; Cram, D.J. Total chromatographic optical resolutions of alpha-amino acid and ester salts through chiral recognition by a host covalently bound to polystyrene resin. J. Am. Chem. Soc. 1976, 98, 3038–3041. [Google Scholar] [PubMed]
- Machida, Y.; Nishi, H.; Nakamura, K.; Nakai, H.; Sato, T. Enantiomer separation of amino compounds by a novel chiral stationary phase derived from crown ether. J. Chromatogr. A 1998, 805, 85–92. [Google Scholar] [CrossRef]
- Hyun, M.H.; Jin, J.S.; Lee, W.J. Liquid chromatographic resolution of racemic amino acids and their derivatives on a new chiral stationary phase based on crown ether. J. Chromatogr. A 1998, 822, 155–161. [Google Scholar] [CrossRef]
- Maier, N.M.; Franco, P.; Lindner, W. Separation of enantiomers: Needs, challenges, perspectives. J. Chromatogr. A 2001, 906, 3–33. [Google Scholar] [CrossRef]
- Hirose, K.; Nakamura, T.; Nishioka, R.; Ueshige, T.; Tobe, Y. Preparation and evaluation of novel chiral stationary phases covalently bound with chiral pseudo-18-crown-6 ethers. Tetrahedron Lett. 2003, 44, 1549–1551. [Google Scholar] [CrossRef]
- Hirose, K.; Jin, Y.; Nakamura, T.; Nishioka, R.; Ueshige, T.; Tobe, Y. Chiral stationary phase covalently bound with a chiral pseudo-18-crown-6 ether for enantiomer separation of amino compounds using a normal mobile phase. Chirality 2005, 17, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Cram, D.J.; Cram, J.M. Design of complexes between synthetic hosts and organic guests. Acc. Chem. Res. 1978, 11, 8–14. [Google Scholar] [CrossRef]
- Cram, D.J.; Trueblood, K.N. Concept, structure, and binding in complexation. Top. Curr. Chem. 1981, 98, 43–106. [Google Scholar]
- Wenzel, T.J.; Freeman, B.E.; Sek, D.C.; Zopf, J.J.; Nakamura, T.; Jin, Y.; Hirose, K.; Tobe, Y. Chiral recognition in NMR spectroscopy using crown ethers and theirYtterbium(III) complexes. Anal. Bioanal. Chem. 2004, 378, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, T.; Hirose, K.; Misumi, S. Chiral azophenolic acerands: Color indicator to judge the absolute configuration of chiral amines. J. Am. Chem. Soc. 1989, 111, 742–743. [Google Scholar] [CrossRef]
- Hirose, K.; Goshima, Y.; Wakebe, T.; Tobe, Y.; Naemura, K. Supramolecular method for the determination of absolute configuration of chiral compounds: Theoretical derivatization and a demonstration for phenolic crown ether—2-amino-1-ethanol system. Anal. Chem. 2007, 79, 6295–6302. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Maeda, S.; Tokita, S.; Kubo, M. Colorimetric chiral recognition by a molecular sensor. Nature 1996, 382, 522–524. [Google Scholar] [CrossRef]
- Naemura, K.; Takeuchi, S.; Hirose, K.; Tobe, Y.; Kaneda, T.; Sakata, Y. Preparation and enantiomer recognition behaviour of azophenolic crown ethers containing cis-cyclohexane-1,2-diol as the chiral centre. J. Chem. Soc. Perkin Trans. 1995, 213–219. [Google Scholar] [CrossRef]
- Van Delden, R.A.; Feringa, B.L. Color indicators of molecular chirality based on doped liquid crystals. Angew. Chem. Int. Ed. 2001, 40, 3198–3200. [Google Scholar] [CrossRef]
- Kim, H.N.; Guo, Z.; Zhu, W.; Yoon, J.; Tian, H. Recent progress on polymer-based fluorescent and colorimetric chemosensors. Chem. Soc. Rev. 2011, 40, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Takai, Y.; Yamada, H.; Hirayama, S.; Kaneda, T.; Tanaka, T.; Kamada, K.; Mizooku, T.; Takeuchi, S.; Ueno, K.; et al. Chiral recognition in host-guest complexation determined by the enantiomer-labeled guest method using fast atom bombardment mass spectrometry. J. Am. Chem. Soc. 1995, 117, 7726–7736. [Google Scholar] [CrossRef]
- Sawada, M.; Takai, Y.; Yamada, H.; Nishida, J.; Kaneda, T.; Arakawa, R.; Okamoto, M.; Hirose, K.; Tanaka, T.; Naemura, K. Chiral amino acid recognition detected by electrospray ionization (ESI) and fast atom bombardment (FAB) mass spectrometry (MS) coupled with the enantiomer labelled (EL) guest method. J. Chem. Soc. Perkin Trans. 1998, 2, 701–710. [Google Scholar] [CrossRef]
- Finn, M.G. Emerging methods for the rapid determination of enantiomeric excess. Chirality 2002, 14, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.H.; Lin, C.-Y.; Anslyn, E.V. Rapid optical methods for enantiomeric excess analysis: From enantioselective indicator displacement assays to exciton-coupled circular dichroism. Acc. Chem. Res. 2014, 47, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, J.P. Interlacing molecular threads on transition-metals: Catenands, catenates, and knots. Acc. Chem. Res. 1990, 23, 319–327. [Google Scholar] [CrossRef]
- Blanco, M.J.; Jimenez, M.C.; Chambron, J.C.; Heitz, V.; Linke, M.; Sauvage, J.P. Rotaxanes as new architectures for photoinduced electron transfer and molecular motions. Chem. Soc. Rev. 1999, 28, 293–305. [Google Scholar] [CrossRef]
- Kawasaki, H.; Kihara, N.; Takata, T. High yielding and practical synthesis of rotaxanes by acylative end-capping catalyzed by tributylphosphine. Chem. Lett. 1999, 10, 1015–1016. [Google Scholar] [CrossRef]
- Schalley, C.A.; Weilandt, T.; Bruggemann, J.; Vögtle, F. Hydrogen-bond-mediated template synthesis of rotaxanes, catenanes, and knotanes. In Templates in Chemistry I; Springer: Berlin/Heidelberg, Germany, 2004; pp. 141–200. [Google Scholar] [CrossRef]
- Arico, F.; Badjic, J.D.; Cantrill, S.J.; Flood, A.H.; Leung, K.C.F.; Liu, Y.; Stoddart, J.F. Templated synthesis of interlocked molecules. In Templates in Chemistry II; Springer: Berlin/Heidelberg, Germany, 2005; pp. 203–259. [Google Scholar]
- Narita, M.; Yoon, I.; Aoyagi, M.; Goto, M.; Shimizu, T.; Asakawa, M. Transition metal(II)-salen and -salophen macrocyclic complexes for rotaxane formation: Syntheses and crystal structures. Eur. J. Inorg. Chem. 2007, 4229–4237. [Google Scholar] [CrossRef]
- Harrison, I.T.; Harrison, S. Synthesis of a stable complex of a macrocycle and a threaded chain. J. Am. Chem. Soc. 1967, 89, 5723–5724. [Google Scholar] [CrossRef]
- Schill, G.; Zollenko, H. Rotaxane compounds 1. Liebigs Ann. Chem. 1969, 721, 53–74. [Google Scholar] [CrossRef]
- Hiratani, K.; Suga, J.; Nagawa, Y.; Houjou, H.; Tokuhisa, H.; Numata, M.; Watanabe, K. A new synthetic method for rotaxanes via tandem claisen rearrangement, diesterification, and aminolysis. Tetrahedron Lett. 2002, 43, 5747–5750. [Google Scholar] [CrossRef]
- Hirose, K.; Nishihara, K.; Harada, N.; Nakamura, Y.; Masuda, D.; Araki, M.; Tobe, Y. Highly selective and high-yielding rotaxane synthesis via aminolysis of prerotaxanes consisting of a ring component and a stopper unit. Org. Lett. 2007, 9, 2969–2972. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, J.P. From chemical topology to molecular machines (nobel lecture). Angew. Chem. Int. Ed. 2017, 56, 11080–11093. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, J.F. Mechanically interlocked molecules (MIMS)-molecular shuttles, switches, and machines (nobel lecture). Angew. Chem. Int. Ed. 2017, 56, 11094–11125. [Google Scholar] [CrossRef] [PubMed]
- Feringa, B.L. The art of building small: From molecular switches to motors (nobel lecture). Angew. Chem. Int. Ed. 2017, 56, 11059–11078. [Google Scholar] [CrossRef] [PubMed]
- Bordoli, R.J.; Goldup, S.M. An efficient approach to mechanically planar chiral rotaxanes. J. Am. Chem. Soc. 2014, 136, 4817–4820. [Google Scholar] [CrossRef] [PubMed]
- Goldup, S.M. Mechanical chirality a chiral catalyst with a ring to it. Nat. Chem. 2016, 8, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, K.; Takata, T. Chiral interlocked molecule: Synthesis and function. J. Synth. Organ. Chem. Jpn. 2017, 75, 491–502. [Google Scholar] [CrossRef]
- Bruns, C.J.; Stoddart, J.F.; Sauvage, J.-P.; Fujita, M. The Nature of the Mechanical Bond: From Molecules to Machines; Wiley: Hoboken, NJ, USA, 2017; pp. 471–554. ISBN 9781119044000. [Google Scholar]
- Borsche, W. Ueber die beziehungen zwischen chinonhydrazonen und p-oxyazoverbindungen. (vierte abhandlung): Ueber die condensation von nitroderivaten des phenylhydrazins mit chinonen und chinonoximen der benzolreihe. Liebigs Ann. Chem. 1907, 357, 171–191. [Google Scholar] [CrossRef]
- Naemura, K.; Asada, M.; Hirose, K.; Tobe, Y. Preparation and enantiomer recognition of chiral azophenolic crown ethers having three chiral barriers on each of the homotopic faces. Tetrahedron Asymmetry 1995, 6, 1873–1876. [Google Scholar] [CrossRef]
- Naemura, K.; Takeuchi, S.; Sawada, M.; Ueno, K.; Hirose, K.; Tobe, Y.; Kaneda, T.; Sakata, Y. Synthesis of azophenolic crown ethers of cs symmetry incorporating cis-1-phenylcyclohexane-1,2-diol residues as a steric barrier and diastereotopic face selectivity in complexation of amines by their diastereotopic faces. J. Chem. Soc. Perkin Trans. 1995, 1, 1429–1435. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Stoncius, S.; Bagdziunas, G.; Malinauskiene, J.; Butkus, E. A study of planar chromophores in dichromophoric molecules by circular dichroism spectroscopy. Chirality 2008, 20, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K. A practical guide for the determination of binding constants. J. Incl. Phenom. 2001, 39, 193–209. [Google Scholar] [CrossRef]
- Hirose, K. Quantitative analysis of binding properties. In Analytical Methods in Supramolecular Chemistry; Schalley, C.A., Ed.; John Wiley & Sons: Weinheim, Germany, 2012; pp. 27–66. ISBN 9783527644131. [Google Scholar]
- Requirements for an application to a chromatography: Resolution factor (α) for baseline separation (Resolution (R) > 1.25, 99.4% separation) through normal size column (Number of theoretical plates (N) = 5000) is around 1.10 (Retention factor (k’2) = 10) where ratio of binding constant corresponds to resolution factor (α) of HPLC separation on chiral column.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
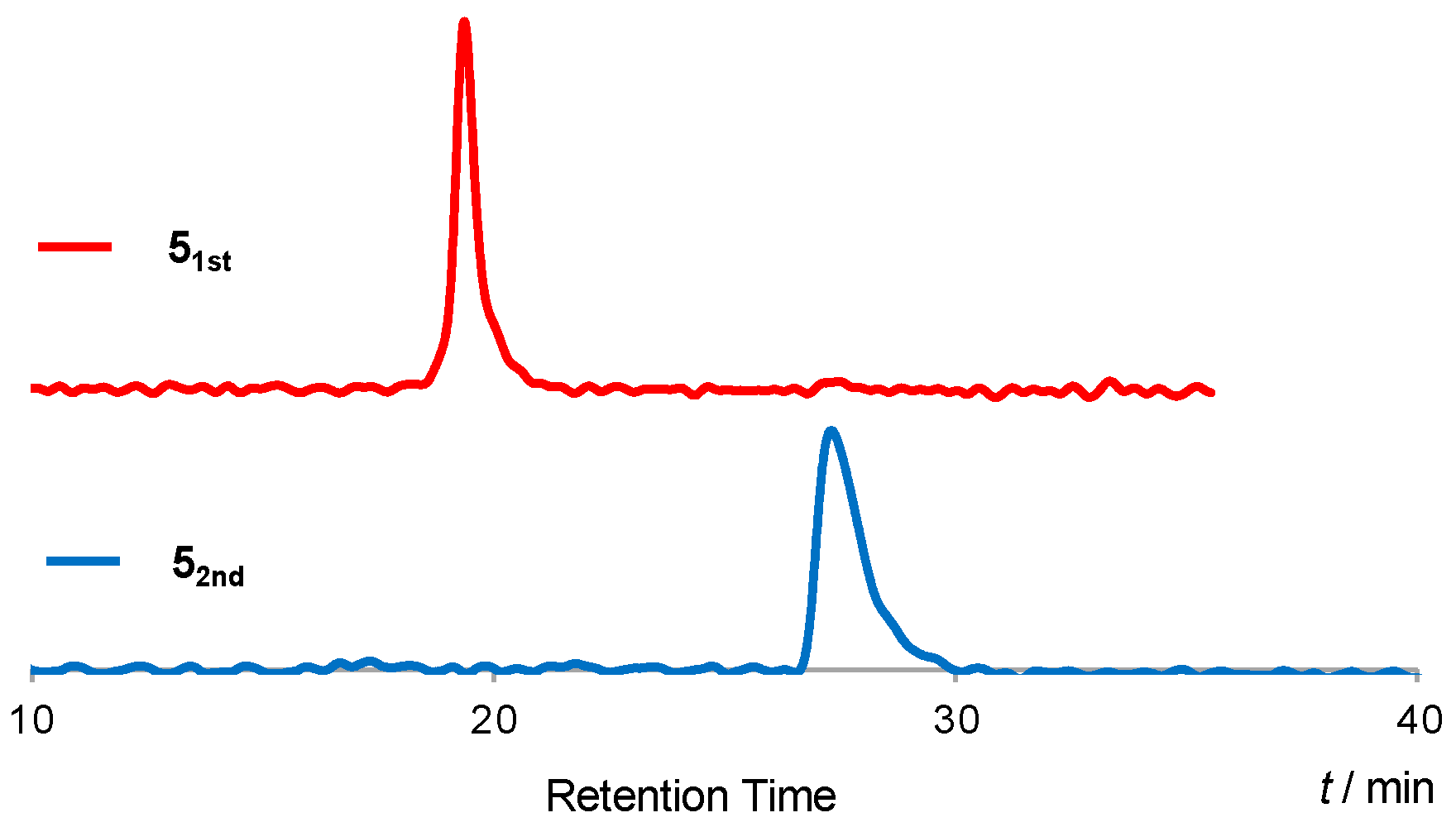{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guests | K/L mol−1 | KR/KS |
|---|---|---|
| (R)-PGO | (1.25 ± 0.03) × 102 | 1.48 |
| (S)-PGO | (8.47 ± 0.40) × 101 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirose, K.; Ukimi, M.; Ueda, S.; Onoda, C.; Kano, R.; Tsuda, K.; Hinohara, Y.; Tobe, Y. The Asymmetry is Derived from Mechanical Interlocking of Achiral Axle and Achiral Ring Components –Syntheses and Properties of Optically Pure [2]Rotaxanes–. Symmetry 2018, 10, 20. https://doi.org/10.3390/sym10010020
Hirose K, Ukimi M, Ueda S, Onoda C, Kano R, Tsuda K, Hinohara Y, Tobe Y. The Asymmetry is Derived from Mechanical Interlocking of Achiral Axle and Achiral Ring Components –Syntheses and Properties of Optically Pure [2]Rotaxanes–. Symmetry. 2018; 10(1):20. https://doi.org/10.3390/sym10010020
Chicago/Turabian StyleHirose, Keiji, Masaya Ukimi, Shota Ueda, Chie Onoda, Ryohei Kano, Kyosuke Tsuda, Yuko Hinohara, and Yoshito Tobe. 2018. "The Asymmetry is Derived from Mechanical Interlocking of Achiral Axle and Achiral Ring Components –Syntheses and Properties of Optically Pure [2]Rotaxanes–" Symmetry 10, no. 1: 20. https://doi.org/10.3390/sym10010020
APA StyleHirose, K., Ukimi, M., Ueda, S., Onoda, C., Kano, R., Tsuda, K., Hinohara, Y., & Tobe, Y. (2018). The Asymmetry is Derived from Mechanical Interlocking of Achiral Axle and Achiral Ring Components –Syntheses and Properties of Optically Pure [2]Rotaxanes–. Symmetry, 10(1), 20. https://doi.org/10.3390/sym10010020