
Identification of an Individualized Prognostic Biomarker for Serous Ovarian Cancer: A Qualitative Model
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
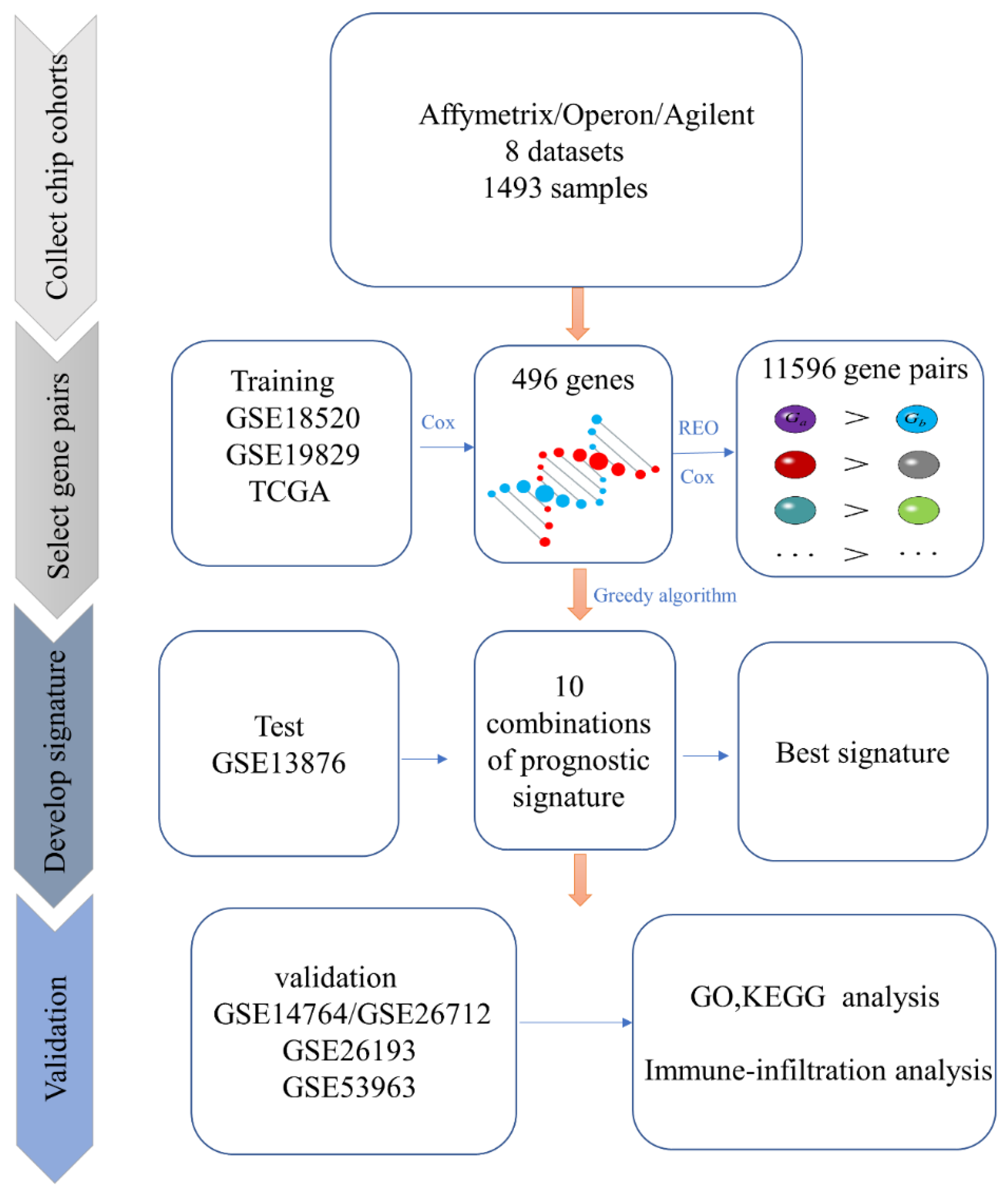
2.2. Identification of Candidate Prognosis-Related Gene Pairs
2.3. Identification of The Prognostic Biomarker
2.4. Performance Evaluation of The Prognostic Biomarker
2.5. Functional Enrichment Analysis
2.6. Immune Infiltration Analysis
2.7. Drug SensitivityAnalysis
2.8. Performance Comparison with Other Prognostic Biomarkers
2.9. Statistical Analysis
3. Results
3.1. Identification of the Prognosis-Related Biomarker
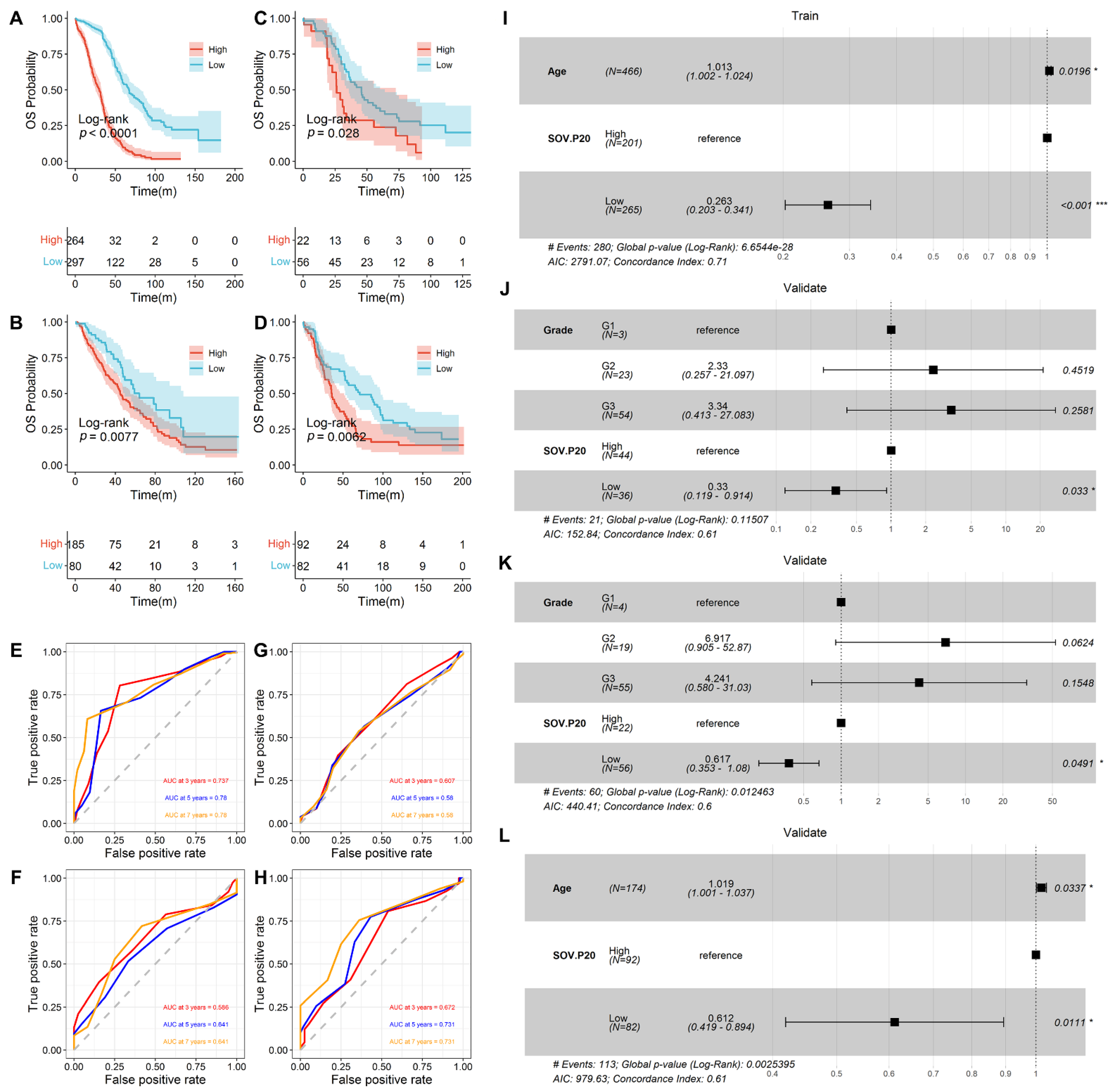
3.2. Prediction of Overall Survival by SOV-P20
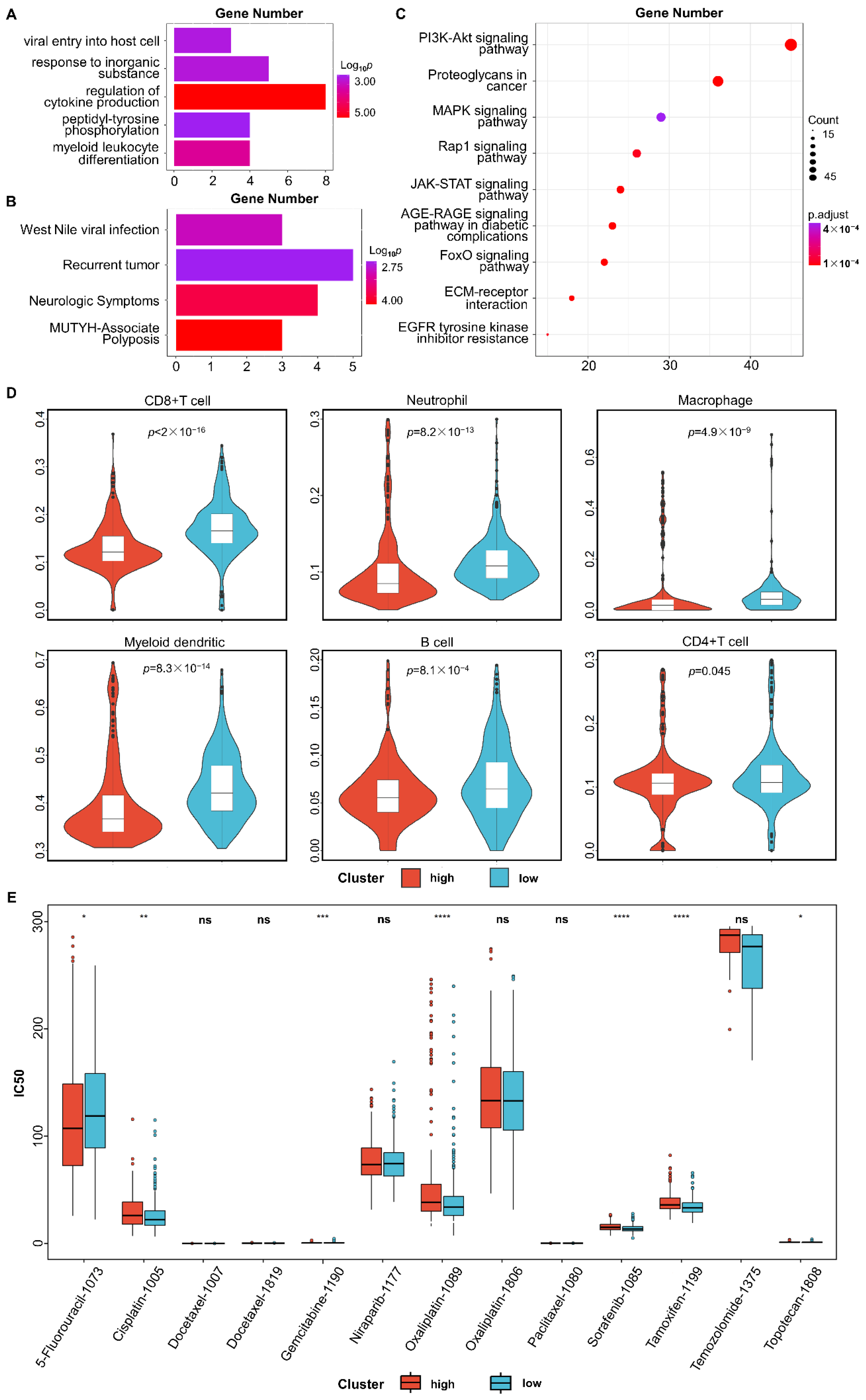
3.3. Functional Enrichment Analysis
3.4. Immune Infiltration Analysis
3.5. Tumor Response to Drug Treatment
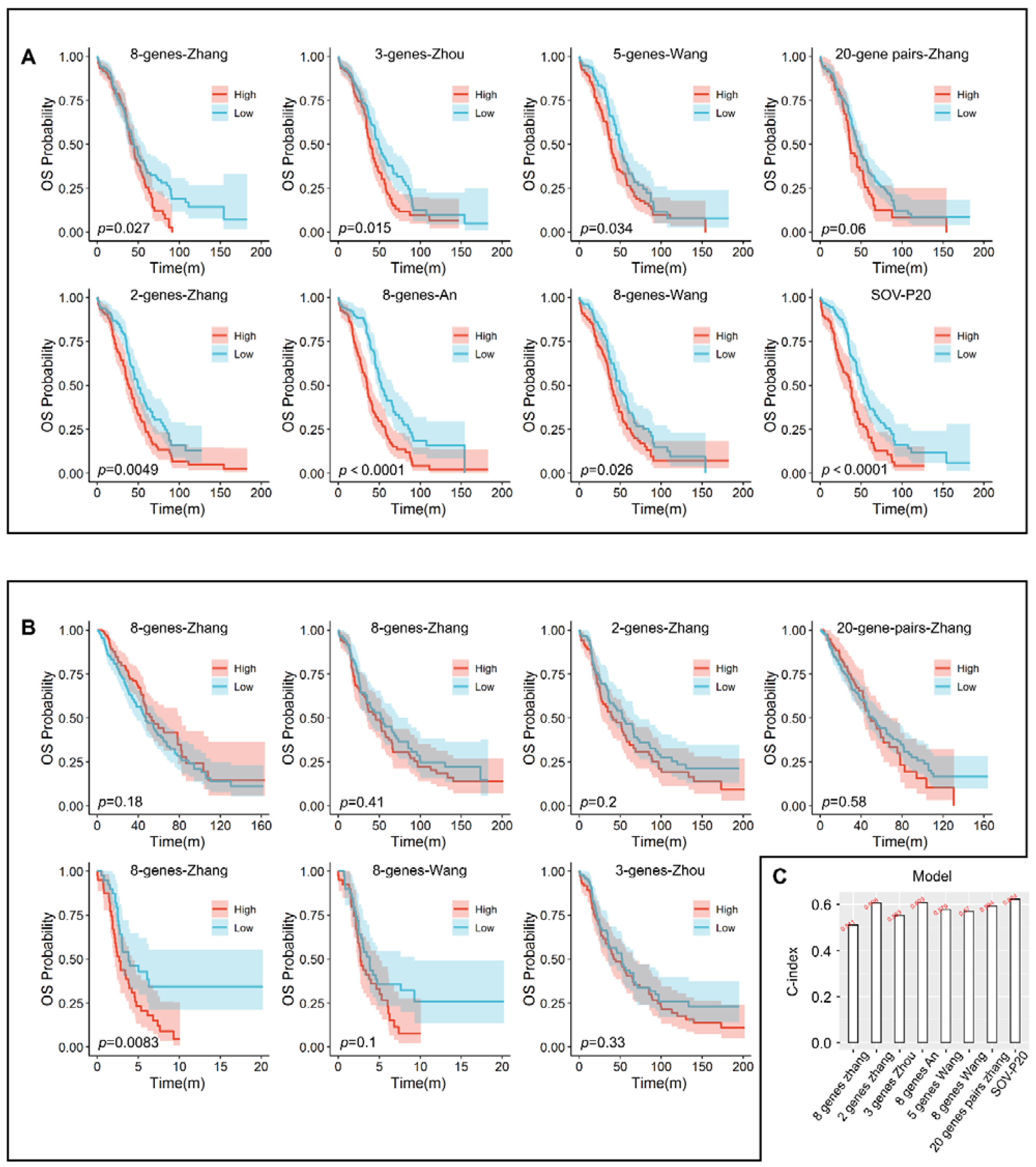
3.6. Comparison with Other Models
4. Discussion
4.1. Main Findings
4.2. Strengths and Limitations
4.3. Interpretation of Findings
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SOV-P20 | Serous ovarian cancer prognostic biomarker consisting of 20 gene pairs |
| AUC | Area under the curve |
| C-index | Index of the concordance |
| REO | Relative expression ordering |
| OV | Ovarian cancer |
| GO | Gene ontology |
| GEO | Gene Expression Omnibus |
| TCGA | The Cancer Genome Atlas |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| BH | Benjamini–Hochberg |
| PI3K-AKT | Phosphatidylinositol 3 kinase/protein kinase B |
| AGE-RAGE | Advanced glycation end products |
| TIMER | Tumor Immune Estimation Resource |
| UCSC | University of California Santa Cruz |
| RMA | Robust multi-array average |
| ROC | Receiver operating characteristic |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| CR | Complete response |
| PR | Partial response |
| SD | Stable disease |
| PD | Progressive disease |
| CSC | Cancer stem cell |
| RT-PCR | Real-time PCR |
References
- Chen, H.; Zhou, S.; Sun, Z. Research status of the prevalence and burden of disease of three malignant tumors in common gynecology Chin. J. Mod. Med. 2015, 25, 108–112. [Google Scholar]
- Zhang, S.; Xia, Q.; Zheng, R.; Chen, W. Analysis of ovarian cancer incidence and mortality in China in 2010. China Oncol. 2016, 25, 169–173. [Google Scholar]
- Colombo, N.; Sessa, C.; Du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO consensus conference recommendations on ovarian cancer: Pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Zhang, Y.; Deng, Q.; Liang, H. Expression and mutations of BRCA in breast cancer and ovarian cancer: Evidence from bioinformatics analyses. Int. J. Mol. Med. 2018, 42, 3542–3550. [Google Scholar] [CrossRef] [Green Version]
- Laganà, A.S.; Colonese, F.; Colonese, E.; Sofo, V.; Salmeri, F.M.; Granese, R.; Chiofalo, B.; Ciancimino, L.; Triolo, O. Cytogenetic analysis of epithelial ovarian cancer’s stem cells: An overview on new diagnostic and therapeutic perspectives. Eur. J. Gynaecol. Oncol. 2015, 36, 495–505. [Google Scholar]
- Buttarelli, M.; De Donato, M.; Raspaglio, G.; Babini, G.; Ciucci, A.; Martinelli, E.; Baccaro, P.; Pasciuto, T.; Fagotti, A.; Scambia, G.; et al. Clinical Value of lncRNA MEG3 in High-Grade Serous Ovarian Cancer. Cancers 2020, 12, 966. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Pan, W.; Sun, X.; Shen, H. Increased expression of TET3 predicts unfavorable prognosis in patients with ovarian cancer-a bioinformatics integrative analysis. J. Ovarian Res. 2019, 12, 101. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Gu, Z.-Y.; Li, Q.; Liu, Q.-H.; Yang, X.-Y.; Zhang, J.-J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Mullikin, H.; Hester, A.; Czogalla, B.; Heidegger, H.; Vilsmaier, T.; Vattai, A.; Chelariu-Raicu, A.; Jeschke, U.; Trillsch, F.; et al. Development and Validation of a Novel 11-Gene Prognostic Model for Serous Ovarian Carcinomas Based on Lipid Metabolism Expression Profile. Int. J. Mol. Sci. 2020, 21, 9169. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Chen, L.; Li, Y.; Qin, Y.; Pan, R.; Zhao, W.; Gu, Y.; Wang, H.; Wang, R.; Chen, X.; et al. Critical limitations of prognostic signatures based on risk scores summarized from gene expression levels: A case study for resected stage I non-small-cell lung cancer. Briefings Bioinform. 2016, 17, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Ray, S.; Zhu, Q.; A Kon, M. Top scoring pairs for feature selection in machine learning and applications to cancer outcome prediction. BMC Bioinform. 2011, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ding, K.; Strumpf, D.; Weir, B.A.; Meyerson, M.; Pennell, N.; Thomas, R.K.; Naoki, K.; Ladd-Acosta, C.; Liu, N.; et al. Prognostic and Predictive Gene Signature for Adjuvant Chemotherapy in Resected Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 4417–4424. [Google Scholar] [CrossRef]
- Geman, D.; D’Avignon, C.; Naiman, D.Q.; Winslow, R.L. Classifying Gene Expression Profiles from Pairwise mRNA Comparisons. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–19. [Google Scholar] [CrossRef]
- Kadara, H.; Behrens, C.; Yuan, P.; Solis, L.; Liu, D.; Gu, X.; Minna, J.D.; Lee, J.J.; Kim, E.; Hong, W.-K.; et al. A Five-Gene and Corresponding Protein Signature for Stage-I Lung Adenocarcinoma Prognosis. Clin. Cancer Res. 2011, 17, 1490–1501. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Jiang, W.; Ao, L.; Song, K.; Chen, H.; Guan, Q.; Gao, Q.; Cheng, J.; Liu, H.; Wang, X.; et al. A qualitative signature for predicting pathological response to neoadjuvant chemoradiation in locally advanced rectal cancers. Radiother. Oncol. 2018, 129, 149–153. [Google Scholar] [CrossRef]
- Peng, F.; Wang, R.; Zhang, Y.; Zhao, Z.; Zhou, W.; Chang, Z.; Liang, H.; Zhao, W.; Qi, L.; Guo, Z.; et al. Differential expression analysis at the individual level reveals a lncRNA prognostic signature for lung adenocarcinoma. Mol. Cancer 2017, 16, 98. [Google Scholar] [CrossRef]
- Zhang, B.; Nie, X.; Miao, X.; Wang, S.; Li, J.; Wang, S. Development and verification of an immune-related gene pairs prognostic signature in ovarian cancer. J. Cell. Mol. Med. 2021, 25, 2918–2930. [Google Scholar] [CrossRef]
- Guan, Q.; Zeng, Q.; Yan, H.; Xie, J.; Cheng, J.; Ao, L.; He, J.; Zhao, W.; Chen, K.; Guo, Y.; et al. A qualitative transcriptional signature for the early diagnosis of colorectal cancer. Cancer Sci. 2019, 110, 3225–3234. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Guo, Y.; Wang, X.; Cai, H.; Zheng, W.; Li, N.; Song, X.; Ao, L.; Guo, Z.; Zhao, W. Transcriptional signatures for coupled predictions of stage II and III colorectal cancer metastasis and fluorouracil-based adjuvant chemotherapy benefit. FASEB J. 2019, 33, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirenajwis, H.; Lauss, M.; Planck, M.; Vallon-Christersson, J.; Staaf, J. Performance of gene expression–based single sample predictors for assessment of clinicopathological subgroups and molecular subtypes in cancers: A case comparison study in non-small cell lung cancer. Brief. Bioinform. 2020, 21, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Thissen, D.; Steinberg, L.; Kuang, D. Quick and Easy Implementation of the Benjamini-Hochberg Procedure for Controlling the False Positive Rate in Multiple Comparisons. J. Educ. Behav. Stat. 2002, 27, 77–83. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, M.; Gao, H.; Guo, J.; Guo, Y.; Zou, M.; Wu, X. Two protein-coding genes act as a novel clinical signature to predict prognosis in patients with ovarian serous cystadenocarcinoma. Oncol. Lett. 2018, 15, 3669–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yi, Y.; Wang, C.; Su, C.; Luo, Y. Identification of a 3-mRNA signature as a novel potential prognostic biomarker in patients with ovarian serous cystadenocarcinoma in G2 and G3. Oncol. Lett. 2019, 18, 3545–3552. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Bi, F.; You, Y.; Liu, X.; Yang, Q. Development of a Novel Autophagy-related Prognostic Signature for Serous Ovarian Cancer. J. Cancer 2018, 9, 4058–4071. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ye, X.H.; Zhao, X.L.; Liu, J.L.; Zhang, C.Y. Development of a five-gene signature as a novel prognostic marker in ovarian cancer. Neoplasma 2019, 66, 343–349. [Google Scholar] [CrossRef]
- Wang, L.; Li, X. Identification of an energy metabolism-related gene signature in ovarian cancer prognosis. Oncol. Rep. 2020, 43, 1755–1770. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, P.; Tong, Y.; Wang, Y.; Ma, H.; Xia, X.; Zhou, Y.; Zhang, X.; Gao, F.; Shu, P. An immune-related gene pairs signature predicts overall survival in serous ovarian carcinoma. OncoTargets Ther. 2019, ume 12, 7005–7014. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, V.D.; Huang, Y.-W.; Delgado-Gonzalez, A.; Chen, S.-Y.; Donoso, K.; Sachs, K.; Gentles, A.J.; Allard, G.M.; Kolahi, K.S.; Howitt, B.E.; et al. High-grade serous ovarian tumor cells modulate NK cell function to create an immune-tolerant microenvironment. Cell Rep. 2021, 36, 109632. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Hofmann, J.; Lu, Y.; Mills, G.B.; Jaffe, R.B. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002, 62, 1087–1092. [Google Scholar] [PubMed]
- Biffo, S.; Manfrini, N.; Ricciardi, S. Crosstalks between translation and metabolism in cancer. Curr. Opin. Genet. Dev. 2018, 48, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Q.; Wu, Z.; Zheng, J.; Ji, L. Integrated Bioinformatics Analysis for Identification of the Hub Genes Linked with Prognosis of Ovarian Cancer Patients. Comput. Math. Methods Med. 2022, 2022, 5113447. [Google Scholar] [CrossRef]
- Ko, Y.S.; Pyo, J.-S. Clinicopathological significance and prognostic role of tumor-infiltrating lymphocytes in colorectal cancer. Int. J. Biol. Markers 2019, 34, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.-F.; Li, Y.-K.; Chen, C.-Y.; Zhang, X.-D.; Cao, L.; Quan, F.-F.; Zeng, X.; Wang, J.; Liu, J. Identification and validation of a prognostic index based on a metabolic-genomic landscape analysis of ovarian cancer. Biosci. Rep. 2020, 40, bsr20201937. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Wang, S.; Tao, L.; Pang, L.; Fu, R.; Fu, Y.; Liang, W.; Li, F.; Jia, W. MiR-212-3p suppresses high-grade serous ovarian cancer progression by directly targeting MAP3K3. Am. J. Transl. Res. 2020, 12, 875–888. [Google Scholar]
- Yang, J.; Hong, S.; Zhang, X.; Liu, J.; Wang, Y.; Wang, Z.; Gao, L.; Hong, L. Tumor Immune Microenvironment Related Gene-Based Model to Predict Prognosis and Response to Compounds in Ovarian Cancer. Front. Oncol. 2021, 11, 807410. [Google Scholar] [CrossRef]
- Laganà, A.S.; Sofo, V.; Vitale, S.G.; Triolo, O. Epithelial ovarian cancer inherent resistance: May the pleiotropic interaction between reduced immunosurveillance and drug-resistant cells play a key role? Gynecol. Oncol. Rep. 2016, 18, 57–58. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training | Test | Validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Dataset 1 | Dataset 2 | Dataset 3 | ||||||
| GEO accession | GSE18520 | GSE19829 | TCGA | GSE13876 | GSE14764 | GSE26712 | GSE26193 | GSE53963 |
| Microarray platform | GPL570 | GPL8300 | GPL96 | GPL7759 | GPL96 | GPL96 | GPL570 | GPL6480 |
| Sample No. | 53 | 42 | 466 | 415 | 80 | 185 | 78 | 174 |
| Stage | ||||||||
| I | - | 0 | 15 | - | 8 | - | 12 | 0 |
| II | - | 1 | 28 | - | 1 | - | 20 | 8 |
| III | - | 35 | 369 | - | 69 | - | 53 | 125 |
| IV | - | 6 | 51 | - | 2 | - | 14 | 41 |
| Late | 53 (III–IV) | - | - | 415 (III–IV) | - | - | - | |
| Unstaged | - | - | 3 | - | - | - | - | - |
| Age, median | - | 58.3 | 59.9 | 57.9 | - | - | - | 63 |
| (range), y | (39–80) | (26–89) | (21–84) | - | - | (24–89) | ||
| Grade | ||||||||
| G1 | 1 | 5 | - | 3 | - | 7 | 0 | |
| G2 | 9 | 60 | - | 23 | - | 33 | 4 | |
| G3 | - | 32 | 389 | - | 54 | - | 67 | 90 |
| G4 | - | 0 | 1 | - | - | - | - | 80 |
| High-grade (G2/3/4) | 53 | - | - | - | - | - | - | - |
| Borderline | - | - | 9 | - | - | - | - | - |
| Ungraded | - | - | 2 | - | - | - | - | - |
| Survival, median (range), m | 40.4 (5–150) | 38.5 (1–68) | 59.5 (0.27–182.70) | 45.6 (1–234) | 35 (7–73) | 38.3 (0.72–163.80) | 60.2 (0.1–133.71) | 55.7 (0.3–201.61) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, F.; Li, N.; Zhang, Q.; Ma, L.; Li, X.; Hu, T.; Zhong, H.; Li, H.; Hong, G. Identification of an Individualized Prognostic Biomarker for Serous Ovarian Cancer: A Qualitative Model. Diagnostics 2022, 12, 3128. https://doi.org/10.3390/diagnostics12123128
Luo F, Li N, Zhang Q, Ma L, Li X, Hu T, Zhong H, Li H, Hong G. Identification of an Individualized Prognostic Biomarker for Serous Ovarian Cancer: A Qualitative Model. Diagnostics. 2022; 12(12):3128. https://doi.org/10.3390/diagnostics12123128
Chicago/Turabian StyleLuo, Fengyuan, Na Li, Qi Zhang, Liyuan Ma, Xinqiao Li, Tao Hu, Haijian Zhong, Hongdong Li, and Guini Hong. 2022. "Identification of an Individualized Prognostic Biomarker for Serous Ovarian Cancer: A Qualitative Model" Diagnostics 12, no. 12: 3128. https://doi.org/10.3390/diagnostics12123128