
Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms
by
 , , , and
, , , and
Lucero A. Ramon-Luing
1 ,
,
Yadira Palacios
2,3,
Andy Ruiz
1,
Norma A. Téllez-Navarrete
4 and
Leslie Chavez-Galan
1,* 1
Laboratory of Integrative Immunology, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosío Villegas”, Mexico City 14080, Mexico
2
Escuela Militar de Graduados de Sanidad, Secretaría de la Defensa Nacional, Mexico City 11200, Mexico
3
Department of Biological Systems, Universidad Autónoma Metropolitana, Campus Xochimilco, Mexico City 04960, Mexico
4
Department of Healthcare Coordination, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosío Villegas”, Mexico City 14080, Mexico
*
Author to whom correspondence should be addressed.
Pathogens 2023, 12(6), 839; https://doi.org/10.3390/pathogens12060839
Submission received: 2 May 2023
/
Revised: 29 May 2023
/
Accepted: 16 June 2023
/
Published: 18 June 2023
(This article belongs to the Section Immunological Responses and Immune Defense Mechanisms)
Abstract
:Mycobacterium tuberculosis (Mtb) modulates diverse cell death pathways to escape the host immune responses and favor its dissemination, a complex process of interest in pathogenesis-related studies. The main virulence factors of Mtb that alter cell death pathways are classified according to their origin as either non-protein (for instance, lipomannan) or protein (such as the PE family and ESX secretion system). The 38 kDa lipoprotein, ESAT-6 (early antigen-secreted protein 6 kDa), and another secreted protein, tuberculosis necrotizing toxin (TNT), induces necroptosis, thereby allowing mycobacteria to survive inside the cell. The inhibition of pyroptosis by blocking inflammasome activation by Zmp1 and PknF is another pathway that aids the intracellular replication of Mtb. Autophagy inhibition is another mechanism that allows Mtb to escape the immune response. The enhanced intracellular survival (Eis) protein, other proteins, such as ESX-1, SecA2, SapM, PE6, and certain microRNAs, also facilitate Mtb host immune escape process. In summary, Mtb affects the microenvironment of cell death to avoid an effective immune response and facilitate its spread. A thorough study of these pathways would help identify therapeutic targets to prevent the survival of mycobacteria in the host.
1. Introduction
Mycobacterium tuberculosis (Mtb) is a member of the Mycobacterium tuberculosis complex (MtbC) and the primary causative agent of tuberculosis (TB) in humans. Mycobacterium africanum and Mycobacterium bovis also induce TB in humans under specific conditions, such as immunosuppression [1,2]. According to the World Health Organization (WHO), despite a reduction in the number of new TB cases in 2020 and 2021, the last WHO report (2022) showed an increase in TB-related deaths. This observation could possibly be attributed to the overestimation of the TB reduction, mainly due to undiagnosed TB due to disrupted access to TB diagnosis as a consequence of the COVID-19 pandemic [3].
Mtb is an intracellular pathogen, mainly infecting the respiratory tract; initially, it specifically infects resident alveolar macrophages (AMs) and replicates to survive intracellularly by avoiding the host immune response [4]. The innate immune cells in the lungs are the earliest defenders; they engulf the pathogen, degrade it, and regulate inflammation, whereas adaptive immune cells, such as CD4 and CD8 T cells, engage in the defense through an antigen-specific T cell response [5].
Cell death is an active process that critically regulates embryonic development, maintaining tissue homeostasis and eliminating damaged tissue and potentially harmful cells [6]. Although cell death is considered generally destructive, it is also a defensive process to preserve homeostasis. However, simultaneously activating several types of cell death could be detrimental to the organism that can be attributed to the induction of a hyper-inflammatory process [7,8].
Some Mtb antigens block apoptosis and activate other cell death pathways, such as necrosis, which allow its spread, thereby modulating cell death as a survival mechanism [9]. Currently, how Mtb leverages cell death to evade the host immune response is not entirely understood. However, a growing body of evidence indicates that virulence is a critical determining factor in the type of cell death triggered or inhibited by Mtb [10,11,12].
This review analyzes Mtb virulence factors that directly induce and modulate cell death pathways. This knowledge will be crucial for identifying critical cell death regulators that Mtb may inhibit to help develop therapeutic strategies.
2. Mtb Virulence Factors: Who Are Involved in Disturbing Cell Death?
Virulence is the ability of a pathogen to cause disease [2]. To define Mtb virulence, the following factors should be considered: (1) the ability of a bacterium to avoid the host’s immune response; (2) its capacity to cause lung damage; and (3) its successful transmission to infect a new host [2,13].
Unlike other pathogens, such as Vibrio cholerae or Corynebacterium diphtheriae, Mtb does not use toxins and enzymes, which are the typical virulence factors; instead, several virulence-associated genes compensate for them [2].
Mycobacterial virulence factors can be divided based on their nature into (a) non-protein molecules, such as lipids, sugars, and (b) proteins. The following Mtb molecules can modify cell death pathways:
2.1. Non-Protein Virulence Factors
Lipids, glycolipids, glycans, nucleic acids, and metabolites are included in this group; many are vital cell surface components involved in host–pathogen interaction, recognition, intracellular survival, and virulence [14].
Major Non-Protein Virulence Factors Inside the Cell Wall
Mtb has a lipid-rich envelope essential for its survival and virulence [14]. The mycobacterial cell wall contains up to 60% lipids [15], and many cell wall components that are secreted, shed, or localized on the bacterial surface, interact with the host cells [16]. Before describing how virulence factors modulate the cell death pathway, a brief description of the more abundant non-protein ones is provided below:
- Phosphatidyl-myo-inositol mannosides (PIMs) are the most abundant glycolipids in the mycobacterial cell envelope and are precursors of lipomannan (LM) and lipoarabinomannan (LAM) [17]. The PIMs comprise variable numbers of mannose units and levels of acylation. Virulent species possess PIMs with five or six mannoses that bind to the mannose receptor (MR), contributing to macrophage uptake. A few of the mannoses also interact with the dendritic cell (DC)-specific intercellular adhesion molecule-3-grabbing non-integrin DC-SIGN from DC [2,18]. Acyl phosphatidyl-myo-inositol dimannoside (AcPIM2) is a part of the inner membrane in most mycobacterial species, and AcPIM6 is involved in maintaining cell envelope integrity [16];
- LM is a multi-glycosylated lipid or polymannosylated PIMs, which is the basic structure of LAM. LM efficiently activates the innate immune response via a tetra-acylated form that activates macrophages through toll-like receptor-2 and 4 (TLR2 and TLR4, respectively). In contrast, its di-acylated form regulates and inhibits nitric oxide (NO) production and cytokine secretion in activated macrophages [19];
- LAM is a glycolipoconjugate composed of LM bound to multiple arabinose residues. When LAM acquires extra and random formation of mannose-capped LM, it is called ManLAM. An addition of phosphoinositol-capped LM makes it known as PILAM, and uncapped or arabinofuranosyl-terminated LM makes it AraLAM. PIMs, LM, LAM, and ManLAM, are recognized by specific receptors, expressed on the cell surface of antigen-presenting cells, such as MR, DC-SIGN, and dendritic cell activating receptor (DCAR), facilitating Mtb uptake into host cells [20,21]. Indeed, the diversity in Mtb mannosylated cell walls alters its virulence, affecting pathogeny and host adaptation [21];
- Phthiocerol dimycocerosates (PDIM) are lipids present on the outer leaflet of the outer membrane that may be secreted or shed from mycobacteria. PDIM promotes Mtb’s evasion of TLR-mediated pathogen detection, delaying the recruitment of immune cells and adaptive responses to the infection site [22]. In addition, PDIM has been suggested to be crucial for phagocytosis and in the rupture of phagosomes and mitochondrial membranes [22,23].
2.2. Protein Virulence Factors
Along with abundant lipids, the Mtb cell wall also contains proteins that directly affect the host’s immune response. Various reports have shown that protein families, such as PE, PPE, and lipoproteins, alter the host immune response. The effects include modulating cytokine production and arresting phagosome maturation, phagosome escape, autophagy, and cell death [2,16].
2.2.1. Major Proteins Virulence Factors from Mtb
Mtb possesses an early secretory antigenic target (ESAT-6) secretion (ESX) system, also known as the type VII secretion system. Mtb has five secretion systems, or ESX, ranging from ESX-1 to ESX-5. The ESX-1, -3, and -5 are essential for mycobacterial virulence and regulate protein secretion and transport across the cytoplasmic membrane and complex mycobacterial cell wall [24].
ESX genes encode the ESX-type proteins EspA, EspB, EspC, and EspG, the secreted proteins ESAT-6 and CFP-10, PE-PPE family proteins, and the conserved components EccB, EccC, EccD, and MycP [25,26].
ESAT-6, delivered through the ESX system, is also known as EsxA or Rv3875. It is either secreted alone or in a complex with the chaperone CFP-10 (EsxB) [27]. It regulates Mtb’s colonization, survival, pathogenesis, and granuloma formation. Additionally, it has been implicated in inhibiting phagosome maturation and modulation of host cell death [28,29].
PE and PPE proteins are transported across the bacterial inner membranes by the ESX-1, ESX-3, and ESX-5 secretion systems through the PE and PPE domains [30]. The PE and PPE protein families have conserved Pro-Glu and Pro-Pro-Glu motifs in their N-terminal regions and are named after these conserved amino acid residues. It has been estimated that 10% of the Mtb genome encodes PE/PPE proteins, which participate in infection, antigenic variation, and host–pathogen interactions [2]. In addition, some PE-PPE members, such as PPE34, PE-PGRS11, PE-PGRS17, PE-PGRS33, PPE26, PPE57, and PPE60, interact with TLR2 to modulate the host innate immune response by inducing cytokine secretion, necrosis, and apoptosis, and enhancing mycobacterial survival [31,32].
2.2.2. Major Lipoproteins Virulence Factors from Mtb
Lipoarabinomannan carrier protein (LprG) is a 27 kDa triacylated lipoprotein of the Mtb cell wall, which is involved in cell wall biosynthesis, and is essential for the expression of surface LAM [33]. LprG displays TLR2 agonist activity; it binds to LAM, PIMs, and LM, enhancing their recognition by TLRs. It also inhibits antigen processing in human macrophage MHC II [34] through TLR-2 signaling and inhibits and controls phagosome–lysosome fusion [35]. LpqH is a 19 kDa O-glycosylated and acylated glycolipoprotein and a primary cell wall antigen recognized by T cells. It induces T cell proliferation in vitro, stimulates DC maturation and autophagy, activates TLR-2 [36], and can affect the expression and antigen presentation of MHCII [37].
PstS1 is a 38-kDa glycolipoprotein that acts as a phosphate transporter recognized by the MR, promotes phagocytosis, reduces the production of reactive oxygen species (ROS), and induces tumor necrosis factor (TNF) and interleukin (IL)-6 production in human monocytes [38].
2.2.3. Major Phosphatases and Kinases Virulence Factors from Mtb
SapM and PtpA are phosphatases that participate in phagosomal arrest. SapM is a 28-kDa Mtb acid phosphatase found in the culture filtrate, indicating that it is secreted. SapM dephosphorylates phosphatidylinositol 3-phosphate (PI3P), an essential molecule for phagosomes to acquire lysosomal constituents; thus, its presence in the phagosome inhibits phagosome–lysosome fusion in macrophage cell lines from both murine and human origin (RAW 264.7 and THP-1, respectively) [39,40]. Moreover, SapM is essential for the pathogenesis of Mtb, and its inhibition leads to impaired mycobacterial growth in THP-1 cells and guinea pig tissues [40].
PtpA is a low-molecular-weight secreted protein tyrosine phosphatase (PTP) activator that dephosphorylates several proteins to suppress the immune response and, hence, is essential for Mtb survival and pathogenicity [41].
PtpA binds to ubiquitin (via a ubiquitin-interacting motif-like region) and dephosphorylates molecules, such as JNK, p38, and VPS33B. Specifically, dephosphorylated VPS33B excludes V-ATPase from phagosomes during Mtb infection and inhibits phagosome acidification [42,43]. Nuclear PtpA binds to host DNA and regulates the transcription of genes involved in host innate immunity, cell proliferation, and migration [42].
Mtb secretes two serine/threonine kinases with different effector functions, protein kinase E (PknE), which regulates Mtb survival response by inhibiting apoptosis [44], and protein kinase G (PknG), which modulates autophagy [45]. Both kinases aid in the intracellular survival of the pathogen, conferring the ability to suppress the host’s innate immune response. The serine/threonine kinase PknF is another bacterial transmembrane protein that inhibits the NLR family pyrin domain, containing three (NLRP3) inflammasomes [46].
Figure 1 shows the locations of some of the more critical virulence factors that alter cell death mechanisms in bacilli.
3. Cell Death Mechanisms Activated by Mtb Virulence Factors: The Good and the Bad
Cell death was initially classified into three main types based on morphological, biochemical, and immunological criteria: type I (apoptosis); type II (autophagy); and type III (necrosis) [47]. Currently, novel types of cell death are characterized by a more integrative profile, including trigger stimuli, molecular mechanisms, and morphological, enzymological, and immunological characteristics.
In 2018, the Nomenclature Committee on Cell Death (NCCD) proposed a new classification based on the mechanistic aspects of regulated cell death (RCD) and a definition based on the molecular and essential aspects. Some of these cell death types include intrinsic apoptosis, extrinsic apoptosis, mitochondrial permeability transition (MPT)-driven necrosis, necroptosis, ferroptosis, pyroptosis, autophagy-dependent cell death, parthanatos, endothelial cell death, NETotic cell death, lysosome-dependent cell death, immunogenic cell death, cellular senescence, and mitotic catastrophe [6].
Intriguingly, the molecular mechanisms involved in each RCD type have a considerable degree of interconnectivity, sharing key mediators. The novel signaling pathways involved in RCD are still being characterized.
Apoptosis and necrosis can be considered as light and shadows in the immune response. Apoptosis is a “double-edged sword“ for the host because Mtb can inhibit or activate apoptosis for its benefit. Grover et al. showed that apoptosis disseminates Mtb infection in lung granulomas during later stages [48]. Necrosis enhances bacterial replication and facilitates reinfection [49].
Recently, it was reported that Mtb virulence factors block molecules of the BCL-2 family to inhibit conventional apoptosis. Moreover, it also simultaneously alters the levels of molecules involved in inducing apoptosis (related to RE stress), necroptosis, and pyroptosis; thus, this phenomenon might possibly be a strategy for Mtb to survive [10].
3.1. Apoptosis
Apoptosis is a highly regulated cell death process, wherein a broad definition of molecular mechanisms allows it to be classified as type I cell death. It is characterized by cytoplasmic contraction, confinement of cytoplasmic contents, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), and blebs in the plasma membrane, forming tiny, apparently intact vesicles (known as apoptotic bodies) that can be removed by phagocytosis [6]. Furthermore, the nature of the stimuli may trigger two different apoptotic pathways, the extrinsic and intrinsic (or mitochondrial) pathways, with convergence, overlap, and synergism in different steps, mainly in the effector caspases [6].
The factors initiating the intrinsic pathway include cytotoxic stresses, such as radiation, toxins, hypoxia, reactive oxygen species, virus infection, and cytokine deprivation [48]. Extracellular alterations induce the extrinsic pathway, where cell-surface death receptors, such as FAS (also CD95 or APO-1), members of the TNF receptor superfamily [6,50], and dependence receptors (such as netrin receptors DCC and UNC5B) trigger apoptosis in the absence of ligands [51].
Extrinsic apoptosis mediated by the oligomerization of death receptor signaling may occur in two ways: (a) caspase-8/-10 activation, leading to caspase-3 activation to cleave critical proteins for cell survival and DNA fragmentation [52]; and (b) caspase-8 activation leading to BID cleavage and mitochondrial outer membrane permeabilization (MOMP) induction. It has also been suggested that extrinsic apoptosis mediated by dependent receptors leads to the activation of caspase-9 and caspase-3 [51].
After ligand-death receptor interactions, monomeric caspase-8 is recruited through its death-effector domain (DED) to the death-inducing signal complex (DISC) in the cytoplasmic domain of the death receptor [53]. In addition, a conformational change allows the association of the adapter protein FAS-associated death domain (FADD) or TNF receptor-associated death domain (TRADD), leading to caspase-8 dimerization and activation of effector caspases, thereby impacting the methods previously described to induce cell death [6,53]. Caspase-8 autocleavage is essential for apoptosis; however, this process also inhibits necroptosis and affects lymphocyte homeostasis via the RIPK3/ mixed lineage kinase domain-like protein (MLKL) signaling axis [54].
In the intrinsic apoptosis, the BH3-only proteins activate the Bcl-2 family members BAX and BAK, leading to MOMP, impact on loss of mitochondrial transmembrane potential (ΔΨm), and induce release to cytosol apoptotic mitochondrial factors, such as cytochrome c, which interacts with Apaf-1 (Apoptotic protease activating factor-1) via their WD domain. This interaction induces conformational changes to induce APAF-1 oligomerization and interaction with dATP. Apaf-1-CARD domains are exposed in the apoptosome complex to recruit and activate initiator caspase-9. The assembly of CARD domains occurs in two kinds of complexes depending on apoptosome localization, which stimulates caspase-9 catalytic activity and suppresses inhibitory CARD activity in free caspase-9 [50,55]. Some pro-apoptotic proteins, such as SMAC and OMI, may be released by MOMP, inhibiting the caspase inhibitor XIAP (X-linked inhibitor of apoptosis protein). Generating tBID (a BH3-only protein) by caspase-8 may link the intrinsic and extrinsic apoptotic pathways. However, tBID may promote MOMP, and proapoptotic factors are released independently of BAX and BAK but positively impact SMAC [56,57].
3.1.1. Modulation of Apoptosis in the Mtb Infection: Proteins Involved
Apoptosis is an RCD that helps eliminate the pathogen during mycobacterial infection. However, apoptosis can be a process with dual impact: on one side, it is a defense mechanism to control the pathogen, but on another side, it can be a strategy for its multiplication and dissemination [9].
Several virulence factors affecting apoptosis have been described in mycobacteria. For example, virulence factors of Mtb may lead to the capacity of infected cells to undergo or inhibit apoptosis. Macrophages may eliminate non-pathogenic mycobacteria, such as M. fortuitum and M. smegmatis, and induce an innate immune response [58]. In this regard, particular variations in mycobacterial strains influence their ability to affect the balance between cell death and proliferation [59].
It is well established that TNF signaling is essential to promote cell recruitment and granuloma formation. Inhibiting TNF by anti-TNF monoclonal antibodies, evidenced via in vitro models that change granuloma formation and structure, increases the risk of activating latent tuberculosis [60]. Furthermore, Mtb reactivation is dependent on TGF-β1 in particular in combination with Adalimumab, a fully humanized IgG1 TNF inhibitor [61].
Mtb-infected macrophages can up-regulate apoptotic signals through the differential expression of 42 genes belonging to the TNFR family [44]. All these pathways may be influenced by MAPSKs/NF-kB activity. Moreover, dual-specificity phosphatase 1 (DUSP1) has a relevant proapoptotic role in Bacillus Calmette–Guérin (BCG) infection [62]. Furthermore, the virulent strains of Mtb interrupt TNF death signals to affect apoptosis evolution, specifically, the DISC conformation or initiator caspase activation, even with low-dose infection [63]. It has been reported that soluble TNF Receptor-2 (TNFR2) blocks the axis TNF/TNFR1, reducing the Fas ligand expression [64].
TNF expression may be associated with the clearance of non-pathogenic strains in human alveolar macrophages and is induced during the primary immune response [65]. TNF also increases caspase-3, leading to a convergence of the intrinsic and extrinsic apoptotic pathways and the final consequences of apoptosis [66]. Soluble TNFR2 may inhibit induced apoptosis during the Mtb infection. However, this phenomenon was not observed during the BCG infection, suggesting that the impact of TNFR2 may be modulated by mycobacterial virulence [67,68]. In macrophages, TNF-induced ROS production increases the associated kinases, c-Abl, ASK1, and p38, leading to FLIP (FLICE-inhibitory protein) phosphorylation, promoting its interaction with the ubiquitin E3 ligase c-Cbl. FLIPS is ubiquitinated and degraded by the proteasome, allowing procaspase-8 and FADD interaction to trigger apoptosis [69].
The Mtb H37Rv strain was associated with the inhibition of apoptosis in U937 and THP-1 cells and human monocyte-derived macrophages, interfering with the extrinsic but not the intrinsic pathway. Furthermore, the putative proteins Rv3654c and Rv3655c have been implicated in inhibiting apoptosis via the extrinsic pathway [70]. Rv3654c interacts with PFS (polypyrimidine tract-binding Protein-associated Splicing Factor), a transcriptional regulator of caspase activity, whereas Rv3655c interacts with ALO17 associated with ALK (anaplastic lymphoma kinase). However, these proteins have been associated with anti-apoptotic activity [70]. The mitochondrial apoptosis pathway is not blocked; additionally, it has been suggested that it may induce necrosis in virulent mycobacteria when the cell death signal is from internally infected macrophages [71].
PE-PGRS33, the most extensively characterized PE family protein, is involved in different types of cell death; PE-PGRS33 co-localization in the host-cell mitochondria induces necrosis and apoptosis. PE_PGRS5 (Rv0297) leads to apoptosis through TLR-4-dependent endoplasmic reticulum stress via its PE domain [48,72].
In addition, ESAT-6, together with PDIMs, induced apoptosis [73]. ESAT-6 increases the gene expression of caspases 1, 3, 5, 7, and 8. Caspases -1 and -5 are associated with the inflammatory process, and the rest participate in the apoptotic pathways [74,75]. Moreover, ESAT-6 increases the miR-155 expression via the TLR2/NF-kB/ SOCS1 signaling axis for promoting apoptosis and impacts the proinflammatory response mediated by IL-6 and TNFs [76].
Among the anti-apoptotic Mtb genes, nuoG, a subunit of type I NADH dehydrogenase (type NADH-1), affects TNF apoptotic signaling by neutralizing NOX2-derived ROS. Thus, there is a close connection between NOX2, phagosomal ROS, and TNF-signaling in apoptosis in macrophages [77]. In a model to improve Mycobacterium bovis BCG vaccine efficacy, the nuoG gene was deleted, and the mutant was evaluated as an immunogen in C57BL/6 mice. After Mtb infection, the up-regulation of apoptosis, autophagy, and inflammasomes was identified, suggesting diverse roles for the antiapoptotic virulence gene nuoG in host–pathogen interactions that impact the induction of the immune response [78].
Mycobacterial factors that participate in redox balance, such as katG, sodA, PknE, and secA2, have been identified as anti-apoptotic agents [79]. The secA-2 system, an ATPase in the Sec secretion system, exports proteins required for bacterial cell growth [80]. Several studies have documented the antiapoptotic properties of secA2 [80,81].
Serine–threonine protein kinases (STPKs) are molecules involved in signal transduction, which play a role in Mtb survival. The serine/threonine kinase PknE has defined roles during NO exposure linked to infected cell apoptosis; PknE contributes to apoptosis resistance during nitrate stress [82]. In an infected macrophage model, the deletion of PknE induced high levels of apoptosis mediated by Bax, Bid, TP53, and caspases, suggesting the involvement of the intrinsic apoptotic pathway. Importantly, in this model, the authors identified the down-regulation of Mcl-1 and decreased TNF- and IL-6 levels [44]. Similarly, PstS1 (38 kDa lipoprotein) induces apoptosis via TNF and FasL [83]. This efficient signal affects the activation of intrinsic and extrinsic pathways, activating initiator procaspases -8, -9, and effector procaspase -3 [84].
3.1.2. Modulation of Apoptosis in the Mtb Infection: Lipid Molecules Involved
Mycobacterial lipoglycans also play a significant role in the intrinsic apoptotic pathways [14,85]. Evidence suggests that TLR-2 senses LM/LAM, which affects apoptosis to varying degrees because non-pathogenic strains trigger apoptosis in contrast to pathogenic strains [86]. LM from Mtb and other Mycobacterium species, such as M. kansasii and M. chelonae, induce apoptosis in THP1 macrophages [86].
Regarding ManLAM, it has been shown that the expression of proapoptotic Bcl-2-associated X protein (Bax) is down-regulated in murine macrophages infected with Mycobacterium bovis, and the antiapoptotic B-cell CLL/lymphoma 2 (Bcl2) family member A1 (Bcl-2) expression is stabilized in M. tuberculosis H37Ra [85]. In addition, ManLAM inhibits apoptosis in one way because it can up-regulate Bcl-2; however, it also up-regulates FasL expression and down-regulates IL-12, which protects against apoptosis [85,87].
LpqH interacts with TLR2 and up-regulates death expression [88] in a dose-dependent model using LpqH, isolated from M. smegmatis, incubated with human macrophages isolated from peripheral blood from healthy individuals, induced loss of ΔΨm, the release of cytochrome c, and formation of apoptosome with the subsequent activation of procaspases-9 and -3 [89].
Figure 2 presents a general overview of apoptotic events and how Mtb virulence factors modulate them.
In summary, the Mtb’s protein- and lipid-based molecules significantly affect apoptosis. Therefore, various efforts to activate or inhibit apoptosis have been implemented as therapeutic strategies to fight Mtb infections [90,91]. For example, in TB, the induction of apoptosis might be therapeutic, not only through the deregulation of the expression of virulence factors that inhibit the process but also through the direct activation of components, such as initiator or effector caspases, similar to other pathologies or health conditions [92].
3.2. Necrosis
Necrosis is considered to be a form of accidental cell death induced by pathological or physiological conditions, such as heat shock, mechanical stress, oxidative stress, inhibition of caspase activity, reduced levels of ATP, and radiation. This process encompasses several cell death modalities, commonly including the loss of plasma membrane integrity and the release of intracellular components that favor the inflammatory response, unlike that in apoptosis [7,93].
NCCD includes MPT-driven necrosis, a form of RCD induced by specific perturbations of the intracellular microenvironment triggered by severe oxidative stress and cytosolic Ca2+ overload [6]. An abrupt loss of the inner mitochondrial membrane’s impermeability to small solutes leads to a rapid Δψm dissipation and osmotic breakdown of both mitochondrial membranes and, consequently, RCD [94].
The presence of a necrotic morphology (i.e., rupture of the plasma membrane) can also be observed in the late stages of apoptotic or autophagic cell death by the absence of phagocytosis; apoptotic bodies may lose their integrity, and this is denominated as secondary necrosis caused by apoptosis or autophagy [95]. Secondary necrosis is not accidental but is controlled by a specific biochemical pathway regulated by caspase-3 and mediated by the GSDMD-related protein DFNA5 (deafness-associated tumor suppressor (GSDMD-related protein). Thus, this is a form of programmed necrosis similar to pyroptosis; caspase-3 cleaves DFNA5 to produce a necrosis-promoting DFNA5-N fragment that forms a large pore in the plasma membrane to release inflammatory molecules [96]. Other forms of regulated necrosis, including necroptosis, pyroptosis, and ferroptosis, have also been described.
Necrosis Modulation in Mycobacterial Infection
Macrophage necrosis is a strategy by which Mtb evades innate defense and could be a critical factor in forming granulomas, inflammatory tissue damage, and Mtb transmission [12]. Necrotic infected cells contribute to bacterial dissemination. Mtb inhibits plasma membrane repair, leading to necrosis and the release of mycobacteria into the extracellular environment [97,98]. Reports suggest that macrophage and neutrophil necrosis induced by Mtb contribute to its growth and sustained infection [99,100].
Among the virulence factors that activate necrosis, ESAT-6 induces neutrophil necrosis and NETs production by triggering intracellular Ca2+ influx [75]. The ESX-1 secretion system is essential in many virulence-related mechanisms to mediate macrophage necrosis, including host cell necrosis, extracellular trap formation, and mycobacterial aggregation [101]. PE17 (Rv1646) inhibits IL-6, IL-12, and TNF production, leading to macrophage necrosis [102]; as well, PPE11 (Rv0453) causes necrosis promoting mycobacterial survival [103]. Rv2626c interacts with PPE68 for secretion and is associated with host cell necrosis [104].
In a murine model, the accumulation of mycobacterial antigens in foamy macrophages from TB lesions was observed, which favors necrosis, tissue destruction, and mortality [105]. Figure 3 shows a diagram that summarizes how Mtb infection induces necrotic-like cell death, including pyroptosis, necroptosis, and ferroptosis, to promote disease progression [106,107].
3.3. Pyroptosis
Pyroptosis is a type of RCD that depends on the formation of pores in the plasma membrane by members of the gasdermin protein family and is often induced by the inflammatory activation of caspases [6]. Thus, this inflammatory form of programmed necrosis occurs mainly in myeloid cells after activating the pattern recognition receptor (PRR) [108]. In humans, it is triggered by caspase-1 and/or caspase-4/-5, inflammasome activation, and the maturation and release of I-β and IL-18 [109,110].
The inflammasome is a multiprotein complex containing PRRs, such as TLRs, nucleotide-binding domains, and leucine-rich repeat-containing receptors (NLRs), and is Absent in Melanoma (AIM)-like receptors (ALRs). It is activated by pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) when host cells are exposed to microbial infections, stress, or tissue damage [110]. NLRP1, NLRP3, AIM2, and pyrin require an adaptor protein for inflammasome assembly, an apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), which facilitates the recruitment of pro-caspase-1 to the inflammasome complex [111].
NLRP3 is critical in bacterial, fungal, and viral infections and contributes to the host immune response [112]. The canonical activation of NRLP3 could be induced by two pathways: 1) by the detection of PAMPs, leading to up-regulated transcription of NLRP3, ASC, pro-IL-1β, pro-IL-18, and pro-caspase-11 by activation of NF-κB; or 2) by detection of DAMPS, such as abnormal levels of potassium (K+) efflux, chloride (Cl−) efflux, calcium (Ca2+) influx, oxidized mitochondrial DNA (ox-mtDNA), lysosomal rupture and intracellular ROS production. After recognizing PAMP or DAMP, the NLRP3 complex is oligomerized and assembled with ASC to recruit pro-caspase-1 for cleavage into active caspase-1. Finally, caspase-1 activates pro-IL-1β and pro-IL-18 via proteolytic cleavage into their active forms, IL-1β and IL-18, respectively [110].
As a result of inflammasome activation, caspase-1 cleaves Gasdermin D (GSDMD) into its pore-forming N-terminal fragment, GSDMD-N. Indeed, GSDMD is the primary executor of pyroptosis, forming pores in the plasma membrane resulting in pyroptosis [113,114]. Pyroptosis is another cell death mechanism that eliminates infected or compromised cells and plays a protective role in the host’s immune response [115].
Modulation of Pyroptosis by Mtb
Mtb can manipulate pyroptosis and take advantage of it for survival, and mycobacteria modulate the host cell inflammasome [110].
NLRP3/AIM2 is activated by sensing Mtb effector molecules, such as ESX-1, LpqH, PPE13, EST12, and double-stranded RNA, as well as Mtb lipids, such as TDM and trehalose dimycolate (TDB) [110]. The role of the inflammasome is to limit Mtb growth; however, ESAT-6 has been reported to stimulate the activation of caspase-1 and the secretion of IL-1β to promote inflammasome activation and facilitate the spread of bacteria to neighboring cells [116,117,118]. Other Mtb proteins, such as PPE60 and PE_PGRS19, also induce pyroptosis by cleaving caspase-11, a non-classical pathway that probably benefits the spread of bacteria [119,120].
In contrast, Zmp1 and Rv3364c Mtb proteins inhibit pyroptosis. Zmp1 inhibits inflammasome activation by interacting with the mitochondrial respiratory chain complex 1 protein GRIM-19, an essential regulator of the NLRP3 inflammasome. Rv3364c translocates into the host cytosol to inhibit cathepsin G activity, affecting caspase-1 activation [121,122].
Recently, the phosphokinase PknF was implicated in inhibiting the NLRP3 inflammasome and inhibiting pyroptosis activation independently of the ESX-1 secretion system [46].
These findings suggest that Mtb may temporally regulate host cell death by inhibiting pyroptosis (and other cell death types) to establish an intracellular niche for replication. Once established, Mtb undergoes pyroptosis to escape and spread.
3.4. Necroptosis
Necroptosis is a highly regulated type of necrosis; this modality of RCD is triggered by perturbations of extracellular or intracellular homeostasis and is modulated by the kinase activity of the receptor-interacting protein 1 (RIPK1), RIPK3, and MLKL [6,123]. RIP Kinases play an essential role not only in inflammatory cell death even also in inflammatory signaling [124].
RIPK1 participates in TNF-induced NF-κB activation to regulate the switch between TNF-induced apoptosis and necroptosis, and when it is associated with RIPK3, necroptosis is induced [125,126].
Necroptosis can be triggered by impaired apoptosis, that is, the formation of necrosomes [127], by RIPK1-3; RIPK2 and RIPK3 are activated by TRADD, FAS, TNFR1, or PRRs. PPRs include TLR3, TLR4, and Z-DNA-binding protein 1 (ZBP1, also known as DAI) [125,128].
Additionally, RIPK3 can be activated following a domain-dependent interaction of the RIP-homotypic interaction motif (RHIM) due to the activation of TLR3 by double-stranded RNA (dsRNA) within endosomes and TLR4 activation by lipopolysaccharide (LPS) or DAMPs at the plasma membrane. RIPK3 catalyzes the phosphorylation of MLKL, leading to the formation of MLKL oligomers (trimers or tetramers) and their translocation to the plasma membrane, where they bind to specific phosphatidylinositol phosphate species [129,130].
MLKL promotes Ca2+ influx, probably by its target, the transient receptor potential cation channel, receptor subfamily M member 7 (TRPM7). When MLK is present on the membrane, the ADAM family proteases promote the shedding of membrane-associated proteins and form Mg2+ channels [131,132].
Some of the factors that regulate necrosome formation include stress-induced phosphoprotein 1 (STIP1) protein, homology, U-box containing protein 1 (STUB1 or CHIP), A20 protein, Mg2+/Mn2+-dependent protein phosphatase 1 B (PPM1B), aurora kinase A (AURKA), and RIPK3 [133,134].
STIP1 and STUB1/CHIP degrade RIPK1 and RIPK3 via ubiquitination and lysosomal degradation. A20 inhibits necrosome formation by eliminating RIPK3 ubiquitination. PPM1B prevents MLKL from binding to the necrosome by reducing RIPK3 phosphorylation. Finally, AURKA physically interacts with RIPK1 and RIPK3 to inhibit the necrosomal function [134,135].
These negative necrosome regulators play an essential role in regulating necroptosis and may modulate the pathogenesis of various diseases.
Modulation of Necroptosis by Mtb
Mtb employs virulence factors, such as the 38 kDa lipoprotein and ESAT-6, to induce TNF production, which induces necroptosis. High soluble levels of TNF and a high bacterial load indicate necroptosis instead of apoptosis [9,136].
Mtb triggers necrosis through tuberculosis necrotizing toxin (TNT), another secreted protein with NAD+-glycohydrolase activity that degrades all intracellular NAD+, activating necroptotic cell death via the RIPK3/MLKL pathway, independent of TNF signaling or RIPK1 [137,138]. These findings suggest that Mtb can induce different necroptotic pathways by switching the TNF/TNFR1/RIPK1 cascade to avoid immune responses.
ESX-1 also induces necroptosis. The ESAT-6 protein, together with the lipid PDIM, forms pores in the host membranes, causing destabilization and loss of integrity of the plasma membrane, which finally triggers necroptosis [139].
Mtb virulence also plays an essential role in the networking of cell death pathways displayed by the pathogen during infection. Mtb inhibits FADD expression from decreasing apoptosis, inducing necroptosis [140]. In addition, macrophages from patients with pulmonary TB infected with the Beijing strain developed severe pulmonary damage and inhibited FADD expression, exhibiting reduced apoptosis [141].
Afriyie-Asante et al. showed that host cells overexpress focal adhesion kinase (FAK) in response to mycobacterial infection. This cytoplasmic non-receptor protein tyrosine kinase promotes bacterial survival by inhibiting necroptosis and increasing ROS production [107].
Additionally, prevention of necrosome assembly and necroptosis are induced by caspase 8-mediated degradation of necrosome components, including the deubiquitinase CYLD, an essential component for necroptosis, which may be induced by cFLIP of cellular FLICE-like inhibitory protein (cFLIP). Although necroptosis is primed in Mtb-infected macrophages, necroptotic signaling is interrupted, inhibiting the cell death pathway [128].
The molecular mechanisms underlying necroptosis induced by mycobacterial infections remain poorly understood. More studies are necessary to elucidate how Mtb modulates necroptosis through its virulence factors and how the host immune response can abrogate it.
3.5. Autophagy-Dependent Cell Death
Autophagy-dependent cell death is a form of RCD that mechanistically depends on the autophagic machinery [6], an evolutionarily conserved catabolic process of eukaryotes, to maintain homeostasis in response to different stress conditions, such as starvation, hypoxia, absence of growth factors, and infection [142]. During autophagy, double-membrane vesicles, called autophagosomes, fuse with acidic compartments, or lysosomes, to give rise to autolysosomes that remove unwanted components from the cell, such as long half-life proteins, damaged organelles, and intracellular pathogens, such as Mtb. This phenomenon allows the cells to economize on macromolecule synthesis and increases cell survival [142,143].
Three types of autophagy have been described: chaperone-mediated autophagy (CMA); microautophagy; and macroautophagy. During CMA, cytosolic chaperones interact with soluble proteins and bind to the lysosomal receptor LAMP-2A (lysosome-associated membrane protein type 2a) for transportation to the lysosome and degradation of proteins [142,144]. Microautophagy involves the direct immersion of the material to be degraded by the lysosome through the lysosomal membrane’s extension, protrusion, and septation [145]. Macroautophagy, also known as the classical autophagy pathway, sequesters cytoplasmic portions and is named according to the content or organelle to be eliminated; for instance, when it contains intracellular pathogens, it is called xenophagy; when it contains mitochondria, it is called mitophagy and lysophagy [143,146,147].
Modulation of Autophagy by Mtb
Several Mtb virulence factors primarily prevent autophagy by arresting the fusion of phagosomes with lysosomes. For example, Eis, ESAT-6, PtpA, and PKnG are among the factors secreted by Mtb into phagosomes. These factors inhibit autophagosome formation [9,148].
PE6, another virulence factor, suppresses autophagy by inhibiting ULK1 or activating the phosphorylation of the autophagy master regulator MtorC1. This observation was reflected in the reduced conversion of the autophagy marker Microtubule-associated protein 1A/1B-light chain 3 (LC3) BI to LC3BII and the increased accumulation of the autophagy substrate p6T2. Furthermore, this suppression is also TLR4-dependent [149,150].
Mtb also inhibits the recruitment of Rab7, a critical component of mycobacteria-containing autophagosomes that mature into autolysosomes. Rab7 recruitment is notably affected by ESAT-6 and SapM proteins [145,151]. Moreover, the secreted SapM and PknG proteins enter the cytoplasm through EsxA-mediated phagosome disruption, further interfering with autophagosome maturation [152].
In addition, lipids from Mtb can also inhibit autophagy; for example, glycosylated PI, an analog of LAM, inhibits PI(3)P production by the Vps34 protein, whereas the SapM protein dephosphorylates PI(3)P to arrest phagosome maturation and increase Mtb survival [146,153].
Mtb uses several strategies to evade the host immune system and promote its survival. miR-889-5p expression inhibits autophagy and promotes bacterial survival. Similarly, miR-125a and miR-30a target UVRAG and Beclin-1, respectively, to inhibit autophagy [143].
Although different drugs, such as isoniazid, carbamazepine, loperamide, verapamil, valproic acid, and metformin, can activate the autophagy mechanism, the challenge remains to eliminate mycobacteria effectively. Further research is needed to understand the complexity of autophagy regulation in Mtb infection and to develop effective therapeutic interventions.
3.6. Ferroptosis
Ferroptosis is another form of RCD triggered by oxidative perturbations of the intracellular microenvironment under the constitutive control of glutathione peroxidase 4 (GPX4). Severe lipid peroxidation mainly depends on ROS generation and iron availability. Iron chelators and lipophilic antioxidants can inhibit ferroptosis [6].
Ferroptosis is regulated by molecules of the metabolic pathways that regulate cysteine exploitation, glutathione status, and nicotinamide adenine dinucleotide phosphate (NADP) function. Unlike other forms of RCD, such as apoptosis and necroptosis, ferroptosis is independent of caspases, necrosome components, and autophagy. Instead, ferroptotic cells display necrotic morphology with mitochondrial alterations, shrinkage, and electrodense ultrastructure. Additionally, they may release damage-associated molecular patterns (DAMPs) [154,155].
Modulation of Ferroptosis by Mtb
During Mtb infection, ferroptosis is triggered by various stimuli, including oxidative stress, lipid peroxide accumulation, and GPX4 inactivation or depletion [154,156]. Furthermore, ferroptosis contributes to tissue necrosis during TB infection.
The ferroptosis signaling pathway in Mtb infection involves several mechanisms. Heme oxygenase-1 (HO-1) regulates the expression of proteins, such as ferritin and ferroportin, and also mediates the immune response to TB infection. Nutritional immunity, which seeks to limit iron availability for invading microorganisms, plays an essential role in innate immunity [155,157].
The accumulation of ROS and lipid peroxides promotes tissue damage and bacterial dissemination, whereas HO-1 increases the levels of ferrous metals, contributing to Mtb survival [154]. Additionally, regulating ferritin and ferroportin by HO-1 can reduce intracellular oxidative stress and promote iron exit from the intracellular environment through the ferroportin protein [155,158]. Qiang et al. showed that PtpA triggers ferroptosis to promote Mtb pathogenicity and dissemination. PtpA inhibits GPX4 expression by inducing the formation of histone H3 arginine 2 (H3R2me2a), which, in turn, recruits methyltransferase 6 (PRMT6) to specifically down-regulate GPX4 expression, a key inhibitor of ferroptosis [159].
HO-1 and peptidyl-prolyl isomerase A (PPiA) are involved in Mtb survival. HO-1 can increase ferrous metal levels, whereas PPiA favors intracellular bacterial survival. Deleting PPiA in a murine infection model formed granuloma-like lesions and induced host cell death through ferroptosis [160].
Ferroptosis, which can be suppressed by inhibitors, such as ferrostatins and liproxstatins, is also involved in the oxidation of specific polyunsaturated fatty acids (PUFAs) containing phosphatidylethanolamines, such as arachidonic acid and adrenic acid. These oxidized PUFAs accumulate after GPX4 inactivation and may be involved in the inflammatory processes. Recent studies showed that specific inhibitors, including sorafenib, trigger ferroptosis [156,161,162,163,164].
The suppressor of cytokine signaling 1 (SOCS1) is the only gene related to ferroptosis in patients with TB and may regulate the microenvironment and bone destruction during TB infection. In addition, the release of damage-associated molecules and alarmins by the ferroptotic cells regulates immunity and pro-inflammatory activities during TB infection [165].
The PI3K signaling pathway may also regulate cell proliferation and growth by activating protein kinase B (Akt kinase, also known as PKB). Moreover, the p38 MAPK, ERK regulators, and JNK may participate in the intracellular signals that can induce HO-1 expression [155]. In contrast, the HO-1 enzyme has a dual role in TB, and its expression and activity can be therapeutically regulated. Finally, analysis of SOCS1_TB (profile GSE83456) identified 105 differentially expressed genes (DEGs), of which 97 were up-regulated, and six were down-regulated [165].
Therefore, ferroptosis plays an essential role in the pathogenesis of Mtb infection. Understanding the complex and multifaceted processes of ferroptosis regulation in TB is crucial for developing new therapies against TB and other infectious diseases. Identifying HO-1 and SOCS1 as potential therapeutic targets offers promising opportunities for the modulation of ferroptosis and for developing novel treatment strategies.
4. Conclusions
It is well-established that Mtb exploits its varied virulence factors to orchestrate the infection process, facilitating its growth, dissemination, and latency. Host cell death is a critical mechanism that determines the outcome of infection. It is one of the main processes manipulated by mycobacterium to favor itself, thereby having various consequences on the host cell. It has been proposed that apoptosis and pyroptosis, which are the cell death mechanisms helpful for Mtb, restrict intracellular bacterial growth and facilitate anti-Mtb immune responses. Meanwhile, necroptosis and ferroptosis benefit Mtb replication and transmission.
As shown in Figure 4 and Table 1, the activation and inhibition of host cell death are driven by Mtb through its virulence factors; in fact, one virulence factor can be critical to more than one cell death modality, allowing the persistence of Mtb in the host. Among the virulence factors, ESAT-6 is essential in Mtb-modulated host cell death. It can trigger apoptosis, necrosis, necroptosis, and pyroptosis, mainly favoring the spread of mycobacteria. It can also inhibit phagosome maturation, thereby affecting autophagy, an essential mechanism for eliminating Mtb. Thus, ESAT-6 is one of the main tools used by Mtb to disseminate and survive in host cells.
A complete understanding of how Mtb interacts with its host to manipulate cell death mechanisms is still in progress. Therefore, new insights and findings will help understand why mycobacterium uses a specific mode of cell death and how it confers its virulence to escape the host immune response if simultaneously activated by diverse cell death mechanisms.
Author Contributions
Conceptualization, L.A.R.-L. and L.C.-G.; investigation, L.A.R.-L., Y.P., A.R. and N.A.T.-N.; writing—original draft preparation, L.A.R.-L., Y.P., A.R. and N.A.T.-N.; writing—review and editing, L.C.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This study received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 16076. [Google Scholar] [CrossRef]
- Echeverria-Valencia, G.; Flores-Villalva, S.; Espitia, C.I. Virulence Factors and Pathogenicity of Mycobacterium; Ribón, W., Ed.; IntechOpen: Rijeka, Croatia, 2017; p. Ch. 12. ISBN 978-1-78923-211-0. [Google Scholar]
- Global Tuberculosis Report 2022. Available online: https://www.who.int/publications/i/item/9789240061729 (accessed on 2 February 2023).
- Lee, J.; Boyce, S.; Powers, J.; Baer, C.; Sassetti, C.M.; Behar, S.M. CD11cHi Monocyte-Derived Macrophages Are a Major Cellular Compartment Infected by Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008621. [Google Scholar] [CrossRef] [PubMed]
- Nadolinskaia, N.I.; Kotliarova, M.S.; Goncharenko, A.V. Fighting Tuberculosis: In Search of a BCG Replacement. Microorganisms 2022, 11, 51. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohareer, K.; Asalla, S.; Banerjee, S. Cell Death at the cross Roads of Host-Pathogen Interaction in Mycobacterium tuberculosis Infection. Tuberculosis 2018, 113, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Luing, L.A.; Olvera, Y.; Flores-Gonzalez, J.; Palacios, Y.; Carranza, C.; Aguilar-Duran, Y.; Vargas, M.A.; Gutierrez, N.; Medina-Quero, K.; Chavez-Galan, L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens 2022, 11, 492. [Google Scholar] [CrossRef]
- Feng, Z.; Bai, X.; Wang, T.; Garcia, C.; Bai, A.; Li, L.; Honda, J.R.; Nie, X.; Chan, E.D. Differential Responses by Human Macrophages to Infection With Mycobacterium tuberculosis and Non-Tuberculous Mycobacteria. Front. Microbiol. 2020, 11, 116. [Google Scholar] [CrossRef] [Green Version]
- Butler, R.E.; Brodin, P.; Jang, J.; Jang, M.S.; Robertson, B.D.; Gicquel, B.; Stewart, G.R. The Balance of Apoptotic and Necrotic Cell Death in Mycobacterium tuberculosis Infected Macrophages Is Not Dependent on Bacterial Virulence. PLoS ONE 2012, 7, e47573. [Google Scholar] [CrossRef] [Green Version]
- Coscolla, M.; Gagneux, S. Consequences of Genomic Diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzheimer, M.; Buter, J.; Minnaard, A.J. Chemical Synthesis of Cell Wall Constituents of Mycobacterium tuberculosis. Chem. Rev. 2021, 121, 9554–9643. [Google Scholar] [CrossRef]
- Chiaradia, L.; Lefebvre, C.; Parra, J.; Marcoux, J.; Burlet-Schiltz, O.; Etienne, G.; Tropis, M.; Daffé, M. Dissecting the Mycobacterial Cell Envelope and Defining the Composition of the Native Mycomembrane. Sci. Rep. 2017, 7, 12807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and Virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef] [PubMed]
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial Outer Membrane Is a Lipid Bilayer and the Inner Membrane Is Unusually Rich in Diacyl Phosphatidylinositol Dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963. [Google Scholar] [CrossRef] [Green Version]
- Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Fine Discrimination in the Recognition of Individual Species of Phosphatidyl-Myo-Inositol Mannosides from Mycobacterium tuberculosis by C-Type Lectin Pattern Recognition Receptors. J. Immunol. 2006, 177, 1805–1816. [Google Scholar] [CrossRef] [Green Version]
- Doz, E.; Rose, S.; Nigou, J.; Gilleron, M.; Puzo, G.; Erard, F.; Ryffel, B.; Quesniaux, V.F.J. Acylation Determines the Toll-like Receptor (TLR)-Dependent Positive versus TLR2-, Mannose Receptor-, and SIGNR1-Independent Negative Regulation of pro-Inflammatory Cytokines by Mycobacterial Lipomannan. J. Biol. Chem. 2007, 282, 26014–26025. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.; Torrelles, J.B. Mannose-Capped Lipoarabinomannan in Mycobacterium tuberculosis Pathogenesis. Pathog. Dis. 2018, 76, fty026. [Google Scholar] [CrossRef] [Green Version]
- Torrelles, J.B.; Schlesinger, L.S. Diversity in Mycobacterium tuberculosis Mannosylated Cell Wall Determinants Impacts Adaptation to the Host. Tuberculosis 2010, 90, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Rens, C.; Chao, J.D.; Sexton, D.L.; Tocheva, E.I.; Av-Gay, Y. Roles for Phthiocerol Dimycocerosate Lipids in Mycobacterium tuberculosis Pathogenesis. Microbiology 2021, 167, 001042. [Google Scholar] [CrossRef]
- Day, T.A.; Mittler, J.E.; Nixon, M.R.; Thompson, C.; Miner, M.D.; Hickey, M.J.; Liao, R.P.; Pang, J.M.; Shayakhmetov, D.M.; Sherman, D.R. Mycobacterium tuberculosis Strains Lacking Surface Lipid Phthiocerol Dimycocerosate Are Susceptible to Killing by an Early Innate Host Response. Infect. Immun. 2014, 82, 5214–5222. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Ghatak, D.; Das, P.; BoseDasgupta, S. ESX Secretion System: The Gatekeepers of Mycobacterial Survivability and Pathogenesis. Eur. J. Microbiol. Immunol. 2020, 10, 202–209. [Google Scholar] [CrossRef]
- Dumas, E.; Boritsch, E.C.; Vandenbogaert, M.; De La Vega, R.C.R.; Thiberge, J.M.; Caro, V.; Gaillard, J.L.; Heym, B.; Girard-Misguich, F.; Brosch, R.; et al. Mycobacterial Pan-Genome Analysis Suggests Important Role of Plasmids in the Radiation of Type VII Secretion Systems. Genome Biol. Evol. 2016, 8, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Briken, V. Modular Organization of the ESX-5 Secretion System in Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 2016, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augenstreich, J.; Briken, V. Host Cell Targets of Released Lipid and Secreted Protein Effectors of Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 2020, 10, 595029. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-W. The Role of ESX-1 in Mycobacterium tuberculosis Pathogenesis. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Peng, X.; Sun, J. Mechanism of ESAT-6 Membrane Interaction and Its Roles in Pathogenesis of Mycobacterium tuberculosis. Toxicon 2016, 116, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cheng, H.F.; Zhou, J.; Chan, C.Y.; Lau, K.F.; Tsui, S.K.W.; Au, S.W. ngor Structural Basis of the PE–PPE Protein Interaction in Mycobacterium tuberculosis. J. Biol. Chem. 2017, 292, 16880. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Pathak, S.K.; Banerjee, A.; Pathak, S.; Bhattacharyya, A.; Yang, Z.; Talarico, S.; Kundu, M.; Basu, J. Execution of Macrophage Apoptosis by PE_PGRS33 of Mycobacterium tuberculosis Is Mediated by Toll-like Receptor 2-Dependent Release of Tumor Necrosis Factor-Alpha. J. Biol. Chem. 2007, 282, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucci, I.; Camassa, S.; Cascioferro, A.; Sali, M.; Anoosheh, S.; Zumbo, A.; Minerva, M.; Iantomasi, R.; De Maio, F.; Di Sante, G.; et al. PE-PGRS33 Contributes to Mycobacterium tuberculosis Entry in Macrophages through Interaction with TLR2. PLoS ONE 2016, 11, e0150800. [Google Scholar] [CrossRef]
- Shukla, S.; Richardson, E.T.; Drage, M.G.; Boom, W.H.; Harding, C.V. Mycobacterium tuberculosis Lipoprotein and Lipoglycan Binding to Toll-like Receptor 2 Correlates with Agonist Activity and Functional Outcomes. Infect. Immunol. 2018, 86, e00450-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, A.J.; Dobos, K.M.; Belisle, J.T.; Harding, C.V.; Boom, W.H. Mycobacterium tuberculosis LprG (Rv1411c): A Novel TLR-2 Ligand That Inhibits Human Macrophage Class II MHC Antigen Processing. J. Immunol. 2004, 173, 2660–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Richardson, E.T.; Athman, J.J.; Shi, L.; Wearsch, P.A.; McDonald, D.; Banaei, N.; Boom, W.H.; Jackson, M.; Harding, C.V. Mycobacterium tuberculosis Lipoprotein LprG Binds Lipoarabinomannan and Determines Its Cell Envelope Localization to Control Phagolysosomal Fusion. PLoS Pathog. 2014, 10, e1004471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, K.; Sinha, A.Y.; Ghorpade, D.S.; Togarsimalemath, S.K.; Patil, S.A.; Kaveri, S.V.; Balaji, K.N.; Bayry, J. Src Homology 3-Interacting Domain of Rv1917c of Mycobacterium tuberculosis Induces Selective Maturation of Human Dendritic Cells by Regulating PI3K-MAPK-NF-KappaB Signaling and Drives Th2 Immune Responses. J. Biol. Chem. 2010, 285, 36511–36522. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Zhu, S.; Zhu, L.; Huang, W.; Wang, H.; Zhang, Z.; Xu, Y. Recombinant Lipoprotein Rv1016c Derived from Mycobacterium tuberculosis Is a TLR-2 Ligand That Induces Macrophages Apoptosis and Inhibits MHC II Antigen Processing. Front. Cell Infect. Microbiol. 2016, 6, 147. [Google Scholar] [CrossRef] [Green Version]
- Esparza, M.; Palomares, B.; García, T.; Espinosa, P.; Zenteno, E.; Mancilla, R. PstS-1, the 38-KDa Mycobacterium tuberculosis Glycoprotein, Is an Adhesin, Which Binds the Macrophage Mannose Receptor and Promotes Phagocytosis. Scand. J. Immunol. 2015, 81, 46–55. [Google Scholar] [CrossRef]
- Saleh, M.T.; Belisle, J.T. Secretion of an Acid Phosphatase (SapM) by Mycobacterium tuberculosis That Is Similar to Eukaryotic Acid Phosphatases. J. Bacteriol. 2000, 182, 6850–6853. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.V.; Reddy, P.V.; Tyagi, A.K. Secreted Acid Phosphatase (SapM) of Mycobacterium tuberculosis Is Indispensable for Arresting Phagosomal Maturation and Growth of the Pathogen in Guinea Pig Tissues. PLoS ONE 2013, 8, e70514. [Google Scholar] [CrossRef] [Green Version]
- Bach, H.; Papavinasasundaram, K.G.; Wong, D.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis Virulence Is Mediated by PtpA Dephosphorylation of Human Vacuolar Protein Sorting 33B. Cell Host Microbe 2008, 3, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ge, P.; Qiang, L.; Tian, F.; Zhao, D.; Chai, Q.; Zhu, M.; Zhou, R.; Meng, G.; Iwakura, Y.; et al. The Mycobacterial Phosphatase PtpA Regulates the Expression of Host Genes and Promotes Cell Proliferation. Nat. Commun. 2017 8:1 2017, 8, 244. [Google Scholar] [CrossRef]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis Protein Tyrosine Phosphatase (PtpA) Excludes Host Vacuolar-H+-ATPase to Inhibit Phagosome Acidification. Proc. Natl. Acad. Sci. USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Narayanan, S. PknE, a Serine/Threonine Kinase of Mycobacterium tuberculosis Modulates Multiple Apoptotic Paradigms. Infect. Genet. Evol. 2012, 12, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Lei, Z.; Yu, Y.; Lu, Z.; Qiang, L.; Chai, Q.; Zhang, Y.; Zhao, D.; Li, B.; Pang, Y.; et al. M. Tuberculosis PknG Manipulates Host Autophagy Flux to Promote Pathogen Intracellular Survival. Autophagy 2022, 18, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Ellinwood, S.; Augenstreich, J.; Mayer-Barber, K.D.; Briken, V. Mycobacterium tuberculosis Inhibits the NLRP3 Inflammasome Activation via Its Phosphokinase PknF. PLoS Pathog. 2021, 17, e1009712. [Google Scholar] [CrossRef]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple Cell Death Modalities and Their Key Features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.; Sharma, T.; Singh, Y.; Kohli, S.; Manjunath, P.; Singh, A.; Wieler, L.H.; Tedin, K.; Ehtesham, N.Z.; Hasnain, S.E.; et al. The PGRS Domain of Mycobacterium tuberculosis PE_PGRS Protein Rv0297 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis through Toll-like Receptor 4. mBio 2018, 9, e01017-18. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New Insights into the Evasion of Host Innate Immunity by Mycobacterium tuberculosis. Cell. Mol. Immunol. 2020, 17, 901–913. [Google Scholar] [CrossRef]
- Lossi, L. The Concept of Intrinsic versus Extrinsic Apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef]
- Negulescu, A.M.; Mehlen, P. Dependence Receptors—The Dark Side Awakens. FEBS J. 2018, 285, 3909–3924. [Google Scholar] [CrossRef] [Green Version]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, Function, and Biotechnological Aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Li, X.; Li, F.; Zhang, X.; Zhang, H.; Zhao, Q.; Li, M.; Wu, X.; Wang, L.; Liu, J.; Wu, X.; et al. Caspase-8 Auto-Cleavage Regulates Programmed Cell Death and Collaborates with RIPK3/MLKL to Prevent Lymphopenia. Cell Death Differ. 2022, 29, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.W.; Shi, Y. Mechanistic Insights into Caspase-9 Activation by the Structure of the Apoptosome Holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-Family Protein TBID Can Act as a BAX-like Effector of Apoptosis. EMBO J. 2022, 41, e108690. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Wallace, R.J. Clinical and Taxonomic Status of Pathogenic Nonpigmented or Late-Pigmenting Rapidly Growing Mycobacteria. Clin. Microbiol. Rev. 2002, 15, 716–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadifar, S.; Mostafaei, S.; Behrouzi, A.; Fateh, A.; Riahi, P.; Siadat, S.D.; Vaziri, F. Strain-Specific Behavior of Mycobacterium tuberculosis in A549 Lung Cancer Cell Line. BMC Bioinform. 2021, 22, 154. [Google Scholar] [CrossRef]
- Alves da Silva, D.A.; Da Silva, M.V.; Oliveira Barros, C.C.; Dias Alexandre, P.B.; Timóteo, R.P.; Catarino, J.S.; Sales-Campos, H.; Machado, J.R.; Rodrigues, D.B.R.; Oliveira, C.J.; et al. TNF-α Blockade Impairs in Vitro Tuberculous Granuloma Formation and down Modulate Th1, Th17 and Treg Cytokines. PLoS ONE 2018, 13, e0194430. [Google Scholar] [CrossRef] [Green Version]
- Arbués, A.; Brees, D.; Chibout, S.D.; Fox, T.; Kammüller, M.; Portevin, D. TNF-α Antagonists Differentially Induce TGF-Β1-Dependent Resuscitation of Dormant-like Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008312. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, J.; Dai, F.; Zhang, D.; Li, W. DUSP1 Mediates BCG Induced Apoptosis and Inflammatory Response in THP-1 Cells via MAPKs/NF-ΚB Signaling Pathway. Sci. Rep. 2023, 13, 2606. [Google Scholar] [CrossRef]
- Riendeau, C.J.; Kornfeld, H. THP-1 Cell Apoptosis in Response to Mycobacterial Infection. Infect. Immunol. 2003, 71, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.F.; Alves, C.C.S.; Figueiredo, B.B.M.; Rezende, A.B.; Wohlres-Viana, S.; da Silva, V.L.; Machado, M.A.; Teixeira, H.C. Tumour Necrosis Factor Receptors and Apoptosis of Alveolar Macrophages during Early Infection with Attenuated and Virulent Mycobacterium Bovis. Immunology 2013, 139, 503–512. [Google Scholar] [CrossRef]
- Woo, M.; Wood, C.; Kwon, D.; Park, K.H.P.; Fejer, G.; Delorme, V. Mycobacterium tuberculosis Infection and Innate Responses in a New Model of Lung Alveolar Macrophages. Front. Immunol. 2018, 9, 438. [Google Scholar] [CrossRef]
- Bohsali, A.; Abdalla, H.; Velmurugan, K.; Briken, V. The Non-Pathogenic Mycobacteria M. Smegmatis and M. Fortuitum Induce Rapid Host Cell Apoptosis via a Caspase-3 and TNF Dependent Pathway. BMC Microbiol. 2010, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Walters, A.; Keeton, R.; Labuschagné, A.; Hsu, N.J.; Jacobs, M. TNFRp75-Dependent Immune Regulation of Alveolar Macrophages and Neutrophils during Early Mycobacterium tuberculosis and Mycobacterium Bovis BCG Infection. Immunology 2021, 162, 220–234. [Google Scholar] [CrossRef]
- Balcewicz-Sablinska, M.K.; Keane, J.; Kornfeld, H.; Remold, H.G. Pathogenic Mycobacterium tuberculosis Evades Apoptosis of Host Macrophages by Release of TNF-R2, Resulting in Inactivation of TNF-α. J. Immunol. 1998, 161, 2636–2641. [Google Scholar] [CrossRef]
- Kundu, M.; Pathak, S.K.; Kumawat, K.; Basu, S.; Chatterjee, G.; Pathak, S.; Noguchi, T.; Takeda, K.; Ichijo, H.; Thien, C.B.F.; et al. A TNF- and c-Cbl-Dependent FLIP(S)-Degradation Pathway and Its Function in Mycobacterium tuberculosis-Induced Macrophage Apoptosis. Nat. Immunol. 2009, 10, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Danelishvili, L.; Yamazaki, Y.; Selker, J.; Bermudez, L.E. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c Proteins Participate in the Suppression of Macrophage Apoptosis. PLoS ONE 2010, 5, e10474. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between Apoptosis, Necrosis and Autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramarska, E.; Squeglia, F.; De Maio, F.; Delogu, G.; Berisio, R. PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response. Cells 2021, 10, 161. [Google Scholar] [CrossRef]
- Augenstreich, J.; Arbues, A.; Simeone, R.; Haanappel, E.; Wegener, A.; Sayes, F.; Le Chevalier, F.; Chalut, C.; Malaga, W.; Guilhot, C.; et al. ESX-1 and Phthiocerol Dimycocerosates of Mycobacterium tuberculosis Act in Concert to Cause Phagosomal Rupture and Host Cell Apoptosis. Cell Microbiol. 2017, 19, e12726. [Google Scholar] [CrossRef] [Green Version]
- Derrick, S.C.; Morris, S.L. The ESAT6 Protein of Mycobacterium tuberculosis Induces Apoptosis of Macrophages by Activating Caspase Expression. Cell Microbiol. 2007, 9, 1547–1555. [Google Scholar] [CrossRef]
- Francis, R.J.; Butler, R.E.; Stewart, G.R. Mycobacterium tuberculosis ESAT-6 Is a Leukocidin Causing Ca2+ Influx, Necrosis and Neutrophil Extracellular Trap Formation. Cell Death Dis. 2014, 5, e1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Li, F.; Jia, S.; Zhang, K.; Jiang, W.; Shang, Y.; Chang, K.; Deng, S.; Chen, M. Early Secreted Antigen ESAT-6 of Mycobacterium tuberculosis Promotes Apoptosis of Macrophages via Targeting the MicroRNA155-SOCS1 Interaction. Cell Physiol. Biochem. 2015, 35, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Velmurugan, K.; Cowan, M.J.; Briken, V. The Type I NADH Dehydrogenase of Mycobacterium tuberculosis Counters Phagosomal NOX2 Activity to Inhibit TNF-Alpha-Mediated Host Cell Apoptosis. PLoS Pathog. 2010, 6, e1000864. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Nieuwenhuizen, N.; Vogelzang, A.; Liu, H.; Kaiser, P.; Schuerer, S.; Lazar, D.; Wagner, I.; Mollenkopf, H.J.; Kaufmann, S.H.E. Deletion of NuoG from the Vaccine Candidate Mycobacterium Bovis BCG ΔureC::Hly Improves Protection against Tuberculosis. mBio 2016, 7, e00679-16. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, M.; Bensing, B.A.; Sullam, P.M. The Two Distinct Types of SecA2-Dependent Export Systems. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Sullivan, J.T.; Young, E.F.; Mccann, J.R.; Braunstein, M. The Mycobacterium tuberculosis SecA2 System Subverts Phagosome Maturation to Promote Growth in Macrophages. Infect. Immunol. 2012, 80, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Hinchey, J.; Lee, S.; Jeon, B.Y.; Basaraba, R.J.; Venkataswamy, M.M.; Chen, B.; Chan, J.; Braunstein, M.; Orme, I.M.; Derrick, S.C.; et al. Enhanced Priming of Adaptive Immunity by a Proapoptotic Mutant of Mycobacterium tuberculosis. J. Clin. Investig. 2007, 117, 2279–2288. [Google Scholar] [CrossRef]
- Jayakumar, D.; Jacobs, W.R.; Narayanan, S. Protein Kinase E of Mycobacterium tuberculosis Has a Role in the Nitric Oxide Stress Response and Apoptosis in a Human Macrophage Model of Infection. Cell Microbiol. 2008, 10, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chen, Z.; Bai, N.; Yan, R.; Xu, M.; Wu, W.; Liang, W.; Li, H.; Mao, Y. Construction of a Eukaryotic Expression System with Stable and Secretory Expression of Mycobacterium tuberculosis 38 KDa Protein. World J. Microbiol. Biotechnol. 2021, 37, 175. [Google Scholar] [CrossRef]
- Sanchez, A.; Espinosa, P.; Esparza, M.A.; Colon, M.; Bernal, G.; Mancilla, R. Mycobacterium tuberculosis 38-KDa Lipoprotein Is Apoptogenic for Human Monocyte-Derived Macrophages. Scand. J. Immunol. 2009, 69, 20–28. [Google Scholar] [CrossRef]
- Wojtas, B.; Fijalkowska, B.; Wlodarczyk, A.; Schollenberger, A.; Niemialtowski, M.; Hamasur, B.; Pawlowski, A.; Krzyzowska, M. Mannosylated Lipoarabinomannan Balances Apoptosis and Inflammatory State in Mycobacteria-Infected and Uninfected Bystander Macrophages. Microb. Pathog. 2011, 51, 9–21. [Google Scholar] [CrossRef]
- Dao, D.N.; Kremer, L.; Guérardel, Y.; Molano, A.; Jacobs, W.R.; Porcelli, S.A.; Briken, V. Mycobacterium tuberculosis Lipomannan Induces Apoptosis and Interleukin-12 Production in Macrophages. Infect. Immun. 2004, 72, 2067–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, P.; Kumar, R.; Jana, K.; Chakraborty, S.; Ghosh, Z.; Kundu, M.; Basu, J. Gene Expression Profiling of Mycobacterium tuberculosis Lipoarabinomannan-Treated Macrophages: A Role of the Bcl-2 Family Member A1 in Inhibition of Apoptosis in Mycobacteria-Infected Macrophages. IUBMB Life 2015, 67, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Kundapura, S.V.; Basak, A.J.; Mukherjee, D.; Dash, S.; Ganguli, N.; Das, A.K.; Mukherjee, G.; Samanta, D.; Ramagopal, U.A. High-Resolution Crystal Structure of LpqH, an Immunomodulatory Surface Lipoprotein of Mycobacterium tuberculosis Reveals a Distinct Fold and a Conserved Cleft on Its Surface. Int. J. Biol. Macromol. 2022, 210, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Espinosa, P.; García, T.; Mancilla, R. The 19 KDa Mycobacterium tuberculosis Lipoprotein (LpqH) Induces Macrophage Apoptosis through Extrinsic and Intrinsic Pathways: A Role for the Mitochondrial Apoptosis-Inducing Factor. Clin. Dev. Immunol. 2012, 2012, 950503. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Payá, E.; Orzáez, M.; Mondragón, L.; Wolan, D.; Wells, J.A.; Messeguer, A.; Vicent, M.J. Molecules That Modulate Apaf-1 Activity. Med. Res. Rev. 2011, 31, 649–675. [Google Scholar] [CrossRef]
- Orzáez, M.; Gortat, A.; Mondragón, L.; Pérez-Payá, E. Peptides and Peptide Mimics as Modulators of Apoptotic Pathways. ChemMedChem 2009, 4, 146–160. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as Anticancer Mechanism: Function and Dysfunction of Its Modulators and Targeted Therapeutic Strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Aging 2012, 7, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis Evades Macrophage Defenses by Inhibiting Plasma Membrane Repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.G.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis Replicates within Necrotic Human Macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Mahamed, D.; Boulle, M.; Ganga, Y.; Mc Arthur, C.; Skroch, S.; Oom, L.; Catinas, O.; Pillay, K.; Naicker, M.; Rampersad, S.; et al. Intracellular Growth of Mycobacterium tuberculosis after Macrophage Cell Death Leads to Serial Killing of Host Cells. Elife 2017, 6, e22028. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E.M. Tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.W.; Jacobs, W.R. Mycobacterium tuberculosis Exploits Human Interferon γ to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J. Infect. Dis. 2013, 208, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, H.; Li, H.; Dang, G.; Cui, Z.; Song, N.; Wang, Q.; Liu, S.; Chen, L. PE17 Protein from Mycobacterium tuberculosis Enhances Mycobacterium Smegmatis Survival in Macrophages and Pathogenicity in Mice. Microb. Pathog. 2019, 126, 63–73. [Google Scholar] [CrossRef]
- Peng, X.; Luo, T.; Zhai, X.; Zhang, C.; Suo, J.; Ma, P.; Wang, C.; Bao, L. PPE11 of Mycobacterium tuberculosis Can Alter Host Inflammatory Response and Trigger Cell Death. Microb. Pathog. 2019, 126, 45–55. [Google Scholar] [CrossRef]
- Danelishvili, L.; Everman, J.; Bermudez, L.E. Mycobacterium tuberculosis PPE68 and Rv2626c Genes Contribute to the Host Cell Necrosis and Bacterial Escape from Macrophages. Virulence 2016, 7, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Riaz, S.M.; Bjune, G.A.; Wiker, H.G.; Sviland, L.; Mustafa, T. Mycobacterial Antigens Accumulation in Foamy Macrophages in Murine Pulmonary Tuberculosis Lesions: Association with Necrosis and Making of Cavities. Scand. J. Immunol. 2020, 91, e12866. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Repasy, T.; Papavinasasundaram, K.; Sassetti, C.; Kornfeld, H. Mycobacterium tuberculosis Induces an Atypical Cell Death Mode to Escape from Infected Macrophages. PLoS ONE 2011, 6, e18367. [Google Scholar] [CrossRef] [PubMed]
- Afriyie-Asante, A.; Dabla, A.; Dagenais, A.; Berton, S.; Smyth, R.; Sun, J. Mycobacterium tuberculosis Exploits Focal Adhesion Kinase to Induce Necrotic Cell Death and Inhibit Reactive Oxygen Species Production. Front. Immunol. 2021, 12, 4401. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, S.; Briken, V. Interaction of Mycobacteria with Host Cell Inflammasomes. Front. Immunol. 2022, 13, 791136. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, J.; Zhao, Z.; Xin, T.; Fan, X.; Shen, Q.; Raheem, A.; Lee, C.R.; Jiang, H.; Ding, J. Regulated Necrosis, a Proinflammatory Cell Death, Potentially Counteracts Pathogenic Infections. Cell Death Dis. 2022, 13, 637. [Google Scholar] [CrossRef]
- Eklund, D.; Welin, A.; Andersson, H.; Verma, D.; Söderkvist, P.; Stendahl, O.; Särndahl, E.; Lerm, M. Human Gene Variants Linked to Enhanced NLRP3 Activity Limit Intramacrophage Growth of Mycobacterium tuberculosis. J. Infect. Dis. 2014, 209, 749. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis during Mycobacterium tuberculosis Infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Moura-Alves, P.; Sonawane, A.; Hacohen, N.; Griffiths, G.; Moita, L.F.; Anes, E. Mycobacterium tuberculosis Protein ESAT-6 Is a Potent Activator of the NLRP3/ASC Inflammasome. Cell Microbiol. 2010, 12, 1046–1063. [Google Scholar] [CrossRef]
- Gong, Z.; Kuang, Z.; Li, H.; Li, C.; Ali, M.K.; Huang, F.; Li, P.; Li, Q.; Huang, X.; Ren, S.; et al. Regulation of Host Cell Pyroptosis and Cytokines Production by Mycobacterium tuberculosis Effector PPE60 Requires LUBAC Mediated NF-ΚB Signaling. Cell Immunol. 2019, 335, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Hu, Y.; Zhang, X.; Chi, M.; Xu, S.; Wang, H.; Zhang, X. Mycobacterium tuberculosis PE_PGRS19 Induces Pyroptosis through a Non-Classical Caspase-11/GSDMD Pathway in Macrophages. Microorganisms 2022, 10, 2473. [Google Scholar] [CrossRef]
- Kurane, T.; Matsunaga, T.; Ida, T.; Sawada, K.; Nishimura, A.; Fukui, M.; Umemura, M.; Nakayama, M.; Ohara, N.; Matsumoto, S.; et al. GRIM-19 Is a Target of Mycobacterial Zn2+ Metalloprotease 1 and Indispensable for NLRP3 Inflammasome Activation. FASEB J. 2022, 36, e22096. [Google Scholar] [CrossRef]
- Danelishvili, L.; Everman, J.L.; McNamara, M.J.; Bermudez, L.E. Inhibition of the Plasma-Membrane-Associated Serine Protease Cathepsin G by Mycobacterium tuberculosis Rv3364c Suppresses Caspase-1 and Pyroptosis in Macrophages. Front. Microbiol. 2012, 2, 281. [Google Scholar] [CrossRef] [Green Version]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in Development and Diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef]
- Chirieleison, S.M.; Kertesy, S.B.; Abbott, D.W. Synthetic Biology Reveals the Uniqueness of the RIP Kinase Domain. J. Immunol. 2016, 196, 4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, E.; Park, K.A.; Shen, H.M.; Hur, G.M. The Resurrection of RIP Kinase 1 as an Early Cell Death Checkpoint Regulator-a Potential Target for Therapy in the Necroptosis Era. Exp. Mol. Med. 2022, 54, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 Activation by TAK1-Mediated Phosphorylation Dictates Apoptosis and Necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, L.; Jia, J.; Tian, W.; Lai, R.; Zhang, Z.; Li, J.; Ju, J.; Xu, H. Knowledge Mapping of Necroptosis From 2012 to 2021: A Bibliometric Analysis. Front. Immunol. 2022, 13, 2930. [Google Scholar] [CrossRef] [PubMed]
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic Signaling Is Primed in Mycobacterium tuberculosis-Infected Macrophages, but Its Pathophysiological Consequence in Disease Is Restricted. Cell Death Differ. 2018, 25, 951–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; He, Y.; Jiang, Y.; Wu, Q.; Liu, Y.; Xie, Q.; Zou, Y.; Wu, J.; Zhang, C.; Zhou, Z.; et al. PRMT1 Reverts the Immune Escape of Necroptotic Colon Cancer through RIP3 Methylation. Cell Death Dis. 2023, 14, 233. [Google Scholar] [CrossRef]
- Chen, D.; Tong, J.; Yang, L.; Wei, L.; Stolz, D.B.; Yu, J.; Zhang, J.; Zhang, L. PUMA Amplifies Necroptosis Signaling by Activating Cytosolic DNA Sensors. Proc. Natl. Acad. Sci. USA 2018, 115, 3930–3935. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL Forms Cation Channels. Cell Res. 2016, 26, 517. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of Cell-Surface Proteases Promotes Necroptosis, Inflammation and Cell Migration. Cell Res. 2016, 26, 886–900. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP Controls Necroptosis through Ubiquitylation- and Lysosome-Dependent Degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Molnár, T.; Pallagi, P.; Tél, B.; Király, R.; Csoma, E.; Jenei, V.; Varga, Z.; Gogolák, P.; Odile Hueber, A.; Máté, Z.; et al. Caspase-9 Acts as a Regulator of Necroptotic Cell Death. FEBS J. 2021, 288, 6476–6491. [Google Scholar] [CrossRef] [PubMed]
- Roca, F.J.; Ramakrishnan, L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria via Mitochondrial Reactive Oxygen Species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD+ Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429. [Google Scholar] [CrossRef] [Green Version]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Niederweis, M. NAD Hydrolysis by the Tuberculosis Necrotizing Toxin Induces Lethal Oxidative Stress in Macrophages. Cell Microbiol. 2020, 22, e13115. [Google Scholar] [CrossRef]
- Quigley, J.; Hughitt, V.K.; Velikovsky, C.A.; Mariuzza, R.A.; El-Sayed, N.M.; Briken, V. The Cell Wall Lipid PDIM Contributes to Phagosomal Escape and Host Cell Exit of Mycobacterium tuberculosis. mBio 2017, 8, e00148-17. [Google Scholar] [CrossRef] [Green Version]
- Dannappel, M.; Vlantis, K.; Kumari, S.; Polykratis, A.; Kim, C.; Wachsmuth, L.; Eftychi, C.; Lin, J.; Corona, T.; Hermance, N.; et al. RIPK1 Maintains Epithelial Homeostasis by Inhibiting Apoptosis and Necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Yanti, B.; Mulyadi, M.; Amin, M.; Harapan, H.; Mertaniasih, N.M.; Soetjipto, S. The Role of Mycobacterium tuberculosis Complex Species on Apoptosis and Necroptosis State of Macrophages Derived from Active Pulmonary Tuberculosis Patients. BMC Res. Notes 2020, 13, 415. [Google Scholar] [CrossRef]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in Inflammation, Infection, and Immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am. J. Physiol.-Cell Physiol. 2022, 323, C1444–C1474. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Sanicas, M.; Nigou, J.; Guilhot, C.; Astarie-Dequeker, C.; Vergne, I. The Lipid Virulence Factors of Mycobacterium tuberculosis Exert Multilayered Control over Autophagy-Related Pathways in Infected Human Macrophages. Cells 2020, 9, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Kim, J.K.; Jung, C.H.; Kim, Y.J.; Jung, E.J.; Lee, S.H.; Choi, H.R.; Son, Y.S.; Shim, S.M.; Jeon, S.M.; et al. Chemical Modulation of SQSTM1/P62-Mediated Xenophagy That Targets a Broad Range of Pathogenic Bacteria. Autophagy 2022, 18, 2926–2945. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Jeon, B.-Y.; Lee, H.-M.; Jin, H.S.; Yuk, J.-M.; Song, C.-H.; Lee, S.-H.; Lee, Z.-W.; Cho, S.-N.; Kim, J.-M.; et al. Mycobacterium tuberculosis Eis Regulates Autophagy, Inflammation, and Cell Death through Redox-Dependent Signaling. PLoS Pathog. 2010, 6, e1001230. [Google Scholar] [CrossRef] [Green Version]
- Pahari, S.; Negi, S.; Aqdas, M.; Arnett, E.; Schlesinger, L.S.; Agrewala, J.N. Induction of Autophagy through CLEC4E in Combination with TLR4: An Innovative Strategy to Restrict the Survival of Mycobacterium tuberculosis. Autophagy 2020, 16, 1021–1043. [Google Scholar] [CrossRef]
- Sharma, N.; Shariq, M.; Quadir, N.; Singh, J.; Sheikh, J.A.; Hasnain, S.E.; Ehtesham, N.Z. Mycobacterium tuberculosis Protein PE6 (Rv0335c), a Novel TLR4 Agonist, Evokes an Inflammatory Response and Modulates the Cell Death Pathways in Macrophages to Enhance Intracellular Survival. Front. Immunol. 2021, 12, 696491. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, R.; Zhang, R.; Wu, J.; Hu, D.; Xing, Y.; Ni, S.; Tie, B. [ESAT-6 inhibits autophagy in macrophages and promotes the growth of BCG]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2017, 33, 310–314. [Google Scholar]
- Zulauf, K.E.; Sullivan, J.T.; Braunstein, M. The SecA2 Pathway of Mycobacterium tuberculosis Exports Effectors That Work in Concert to Arrest Phagosome and Autophagosome Maturation. PLoS Pathog. 2018, 14, e1007011. [Google Scholar] [CrossRef]
- Vergne, I.; Gilleron, M.; Nigou, J. Manipulation of the Endocytic Pathway and Phagocyte Functions by Mycobacterium tuberculosis Lipoarabinomannan. Front. Cell Infect. Microbiol. 2014, 4, 187. [Google Scholar] [CrossRef] [Green Version]
- Amaral, E.P.; Foreman, T.W.; Namasivayam, S.; Hilligan, K.L.; Kauffman, K.D.; Bomfim, C.C.B.; Costa, D.L.; Barreto-Duarte, B.; Gurgel-Rocha, C.; Santana, M.F.; et al. GPX4 Regulates Cellular Necrosis and Host Resistance in Mycobacterium tuberculosis Infection. J. Exp. Med. 2022, 219, e20220504. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ouyang, J.; Lu, Y.; Harypursat, V.; Chen, Y. A Dual Role of Heme Oxygenase-1 in Tuberculosis. Front. Immunol. 2022, 13, 842858. [Google Scholar] [CrossRef]
- Amaral, E.P.; Namasivayam, S.; Costa, D.L.; Fisher, L.; Bomfim, C.C.B.; Andrade, B.B.; Sher, A. The Transcription Factor BACH1 Promotes Tissue Damage and Host Susceptibility in Mycobacterium tuberculosis Infection by Reducing Expression of Gpx4, a Major Negative Regulator of Ferroptosis. J. Immunol. 2020, 204, 227.16. [Google Scholar] [CrossRef]
- Ma, C.; Wu, X.; Zhang, X.; Liu, X.; Deng, G. Corrigendum: Heme Oxygenase-1 Modulates Ferroptosis by Fine-Tuning Levels of Intracellular Iron and Reactive Oxygen Species of Macrophages in Response to Bacillus Calmette-Guerin Infection. Front. Cell Infect. Microbiol. 2022, 12, 1105336. [Google Scholar] [CrossRef]
- Fan, J.; Jiang, T.; He, D. Emerging Insights into the Role of Ferroptosis in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2023, 14, 1579. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhang, Y.; Lei, Z.; Lu, Z.; Tan, S.; Ge, P.; Chai, Q.; Zhao, M.; Zhang, X.; Li, B.; et al. A Mycobacterial Effector Promotes Ferroptosis-Dependent Pathogenicity and Dissemination. Nat. Commun. 2023, 14, 1430. [Google Scholar] [CrossRef]
- Dubey, N.; Khan, M.Z.; Kumar, S.; Sharma, A.; Das, L.; Bhaduri, A.; Singh, Y.; Nandicoori, V.K. Mycobacterium tuberculosis Peptidyl Prolyl Isomerase A Interacts With Host Integrin Receptor to Exacerbate Disease Progression. J. Infect. Dis. 2021, 224, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Neyrolles, O. Die Another Way: Ferroptosis Drives Tuberculosis Pathology. J Exp Med 2019, 216, 471–473. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tang, P.; Cao, J.; Song, Q.; Zhu, L.; Ma, S.; Zhang, J. Lipid Peroxidation Aggravates Anti-Tuberculosis Drug-Induced Liver Injury: Evidence of Ferroptosis Induction. Biochem. Biophys. Res. Commun. 2020, 533, 1512–1518. [Google Scholar] [CrossRef]
- Xiao, L.; Huang, H.; Fan, S.; Zheng, B.; Wu, J.; Zhang, J.; Pi, J.; Xu, J.F. Ferroptosis: A Mixed Blessing for Infectious Diseases. Front. Pharmacol. 2022, 13, 992734. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, Y.; Hu, L.; Fu, J.; Li, D.; Chen, J.; Long, Y. Inhibition of HSPA8 by Rifampicin Contributes to Ferroptosis via Enhancing Autophagy. Liver Int. 2022, 42, 2889–2899. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Chen, J.; Xu, G.Y.; Zhang, Z.; Xue, J.; Zeng, H.; Jiang, J.; Chen, T.; Qin, Z.; Li, H.; et al. Ferroptosis-Related Gene SOCS1, a Marker for Tuberculosis Diagnosis and Treatment, Involves in Macrophage Polarization and Facilitates Bone Destruction in Tuberculosis. Tuberculosis 2022, 132, 102140. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Major virulent factors of Mtb involved in cell death mechanisms. (a) Mycobacterial virulence proteins are mainly secreted through one of the essential secretion systems, ESX. ESX1, ESX3, and ESX5 are localized across the membrane, where proteins, such as ESAT-6, CFP-10, PE_PGRS, PtpA, SapM, PknG, and PknE, are delivered (left). (b) Non-proteins are glycolipids and phospholipids forming mycobacterial membrane structures. LAM (lipoarabinomannan), ManLAM (mannose-capped LAM), PDIM (phthiocerol dimycocerosate), and PIMs (Phosphatidyl-myo-inositol mannosides) are inserted in the cell wall (right). TDM, trehalose mono, and di mycolate; PGL, phenolic glycolipid.
Figure 1.
Major virulent factors of Mtb involved in cell death mechanisms. (a) Mycobacterial virulence proteins are mainly secreted through one of the essential secretion systems, ESX. ESX1, ESX3, and ESX5 are localized across the membrane, where proteins, such as ESAT-6, CFP-10, PE_PGRS, PtpA, SapM, PknG, and PknE, are delivered (left). (b) Non-proteins are glycolipids and phospholipids forming mycobacterial membrane structures. LAM (lipoarabinomannan), ManLAM (mannose-capped LAM), PDIM (phthiocerol dimycocerosate), and PIMs (Phosphatidyl-myo-inositol mannosides) are inserted in the cell wall (right). TDM, trehalose mono, and di mycolate; PGL, phenolic glycolipid.

Figure 2.
Mycobacterium tuberculosis and apoptosis modulation. The mycobacteria or its virulence factors may modulate apoptosis, both intrinsic and extrinsic pathways at different levels, as inductors (red arrows) or inhibitors (black inhibitors). (a) Apoptosis may be detonated via TNF and FasL impacting initiator caspases-8 and -9 and effector caspases-3 and -7. (b) The intrinsic pathway also may control mycobacteria infection at the mitochondria level, cytochrome c released, and apoptosome formation by activating caspases-9 and -3. (c) TNF death signal may be blocked at DISC formation and caspase-8 activation. (d) The proapoptotic protein BAX may be down-regulated with the H37Ra strain. (e) Mycobacteria may stabilize the anti-apoptotic Bcl-2. (f) Up-regulation at the gene level of FasL and caspases may be done during mycobacteria infection; (g) down-regulation of IL-12 expression and TNF has also been reported.
Figure 2.
Mycobacterium tuberculosis and apoptosis modulation. The mycobacteria or its virulence factors may modulate apoptosis, both intrinsic and extrinsic pathways at different levels, as inductors (red arrows) or inhibitors (black inhibitors). (a) Apoptosis may be detonated via TNF and FasL impacting initiator caspases-8 and -9 and effector caspases-3 and -7. (b) The intrinsic pathway also may control mycobacteria infection at the mitochondria level, cytochrome c released, and apoptosome formation by activating caspases-9 and -3. (c) TNF death signal may be blocked at DISC formation and caspase-8 activation. (d) The proapoptotic protein BAX may be down-regulated with the H37Ra strain. (e) Mycobacteria may stabilize the anti-apoptotic Bcl-2. (f) Up-regulation at the gene level of FasL and caspases may be done during mycobacteria infection; (g) down-regulation of IL-12 expression and TNF has also been reported.

Figure 3.
Mycobacterium tuberculosis and the necrotic-like cell death modulation. The mycobacteria or its virulence factors may modulate pyroptosis, necroptosis, and ferroptosis pathways at different levels as inducers (red arrows) or inhibitors (black inhibitors). For example, Mtb through ESAT-6, LpqH activates inflammasome, favoring pyroptosis, while Zmp1 and PknF inhibit it. The Mtb ESX-1 secretion system, secreted protein ESAT-6, TNT, and PDIM lipid induce necroptosis. Meanwhile, multiple factors inhibit or activate ferroptosis, including the HO-1 enzyme, the SOCS1 protein, and the PI3K/Akt/mTORC1 signaling pathway, which act at different levels and points regulating the cell death pathway. PtpA favors ferroptosis by inhibiting GPX4 expression, a ferroptosis regulator.
Figure 3.
Mycobacterium tuberculosis and the necrotic-like cell death modulation. The mycobacteria or its virulence factors may modulate pyroptosis, necroptosis, and ferroptosis pathways at different levels as inducers (red arrows) or inhibitors (black inhibitors). For example, Mtb through ESAT-6, LpqH activates inflammasome, favoring pyroptosis, while Zmp1 and PknF inhibit it. The Mtb ESX-1 secretion system, secreted protein ESAT-6, TNT, and PDIM lipid induce necroptosis. Meanwhile, multiple factors inhibit or activate ferroptosis, including the HO-1 enzyme, the SOCS1 protein, and the PI3K/Akt/mTORC1 signaling pathway, which act at different levels and points regulating the cell death pathway. PtpA favors ferroptosis by inhibiting GPX4 expression, a ferroptosis regulator.

Figure 4.
Overview of cell death pathways modulated by Mtb through their virulence factors. Mtb can use its various virulence factors as inductors (red arrows) or inhibitors (black inhibitors) of diverse cell death mechanisms to orchestrate the infection process; as well, it allows it to disseminate and replicate for its survival.
Figure 4.
Overview of cell death pathways modulated by Mtb through their virulence factors. Mtb can use its various virulence factors as inductors (red arrows) or inhibitors (black inhibitors) of diverse cell death mechanisms to orchestrate the infection process; as well, it allows it to disseminate and replicate for its survival.


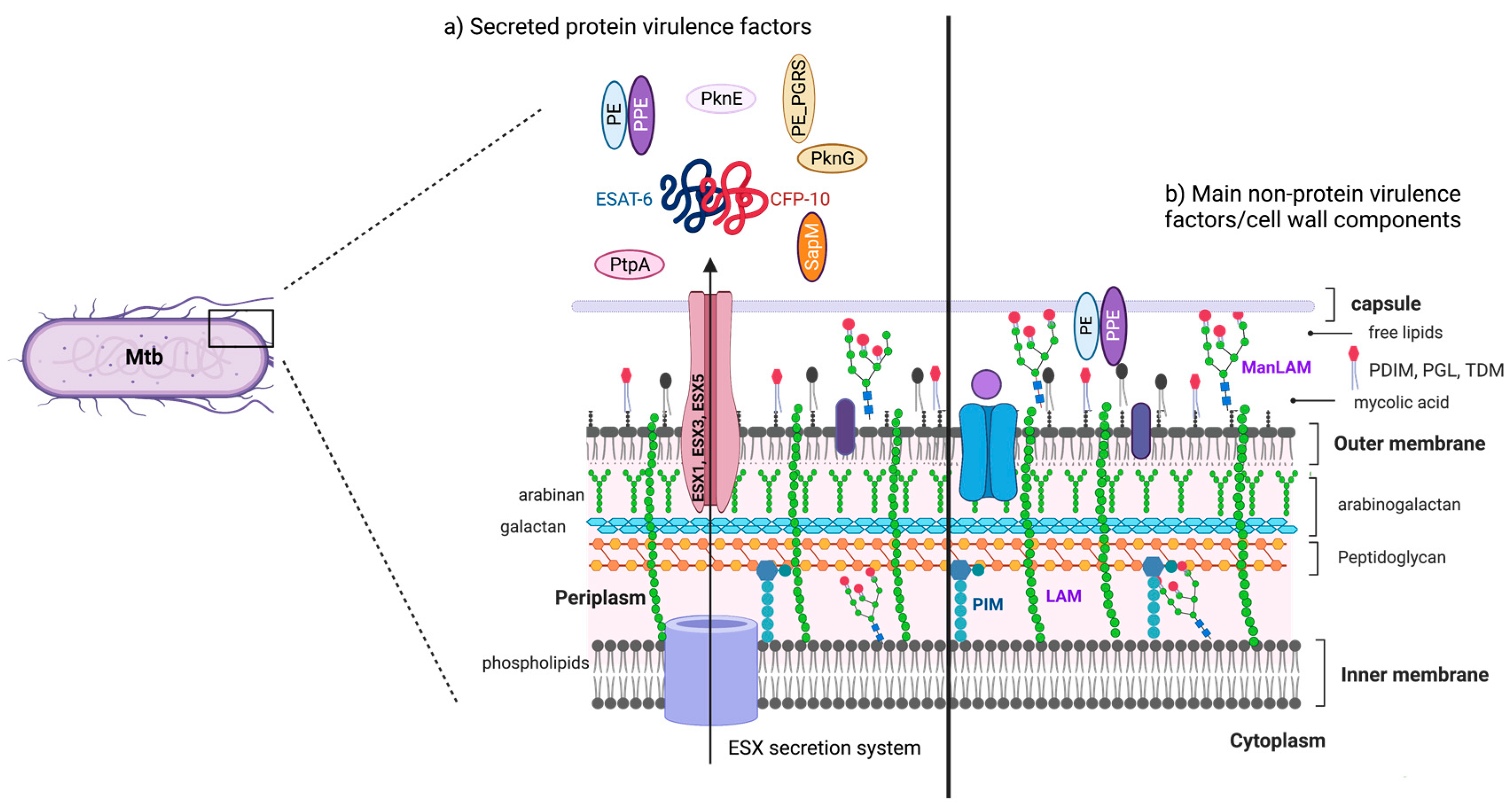{kind=link}
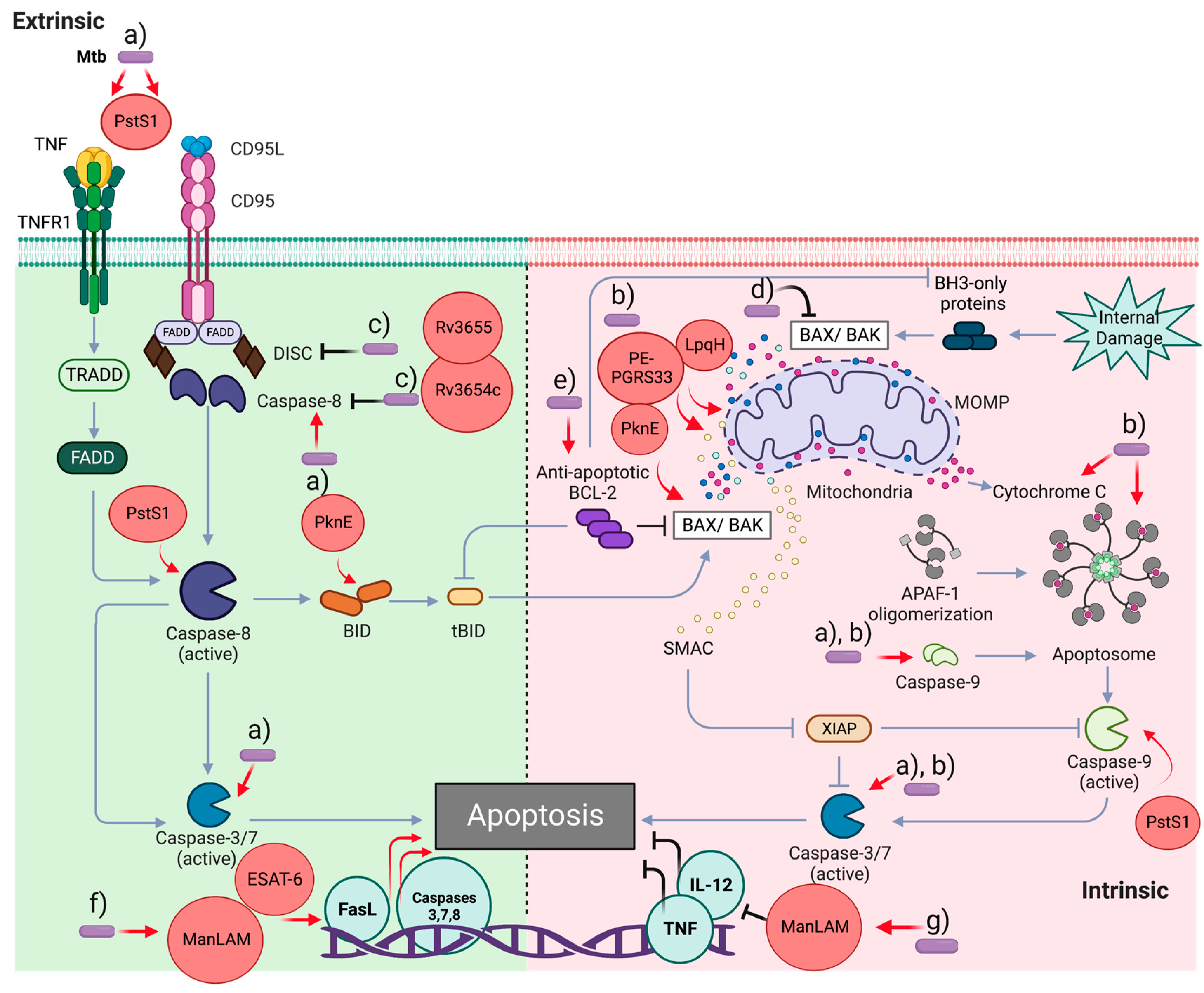{kind=link}
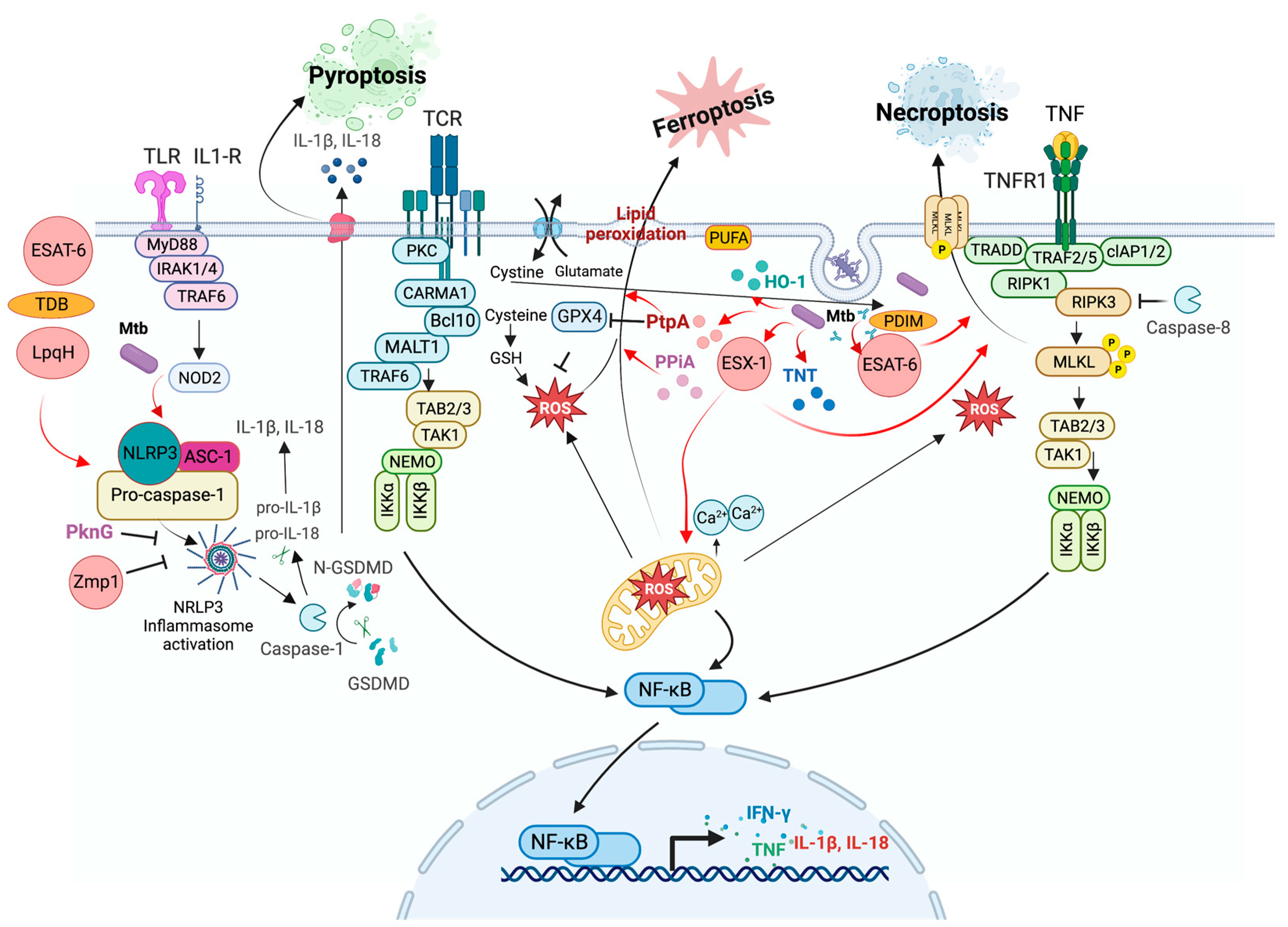{kind=link}
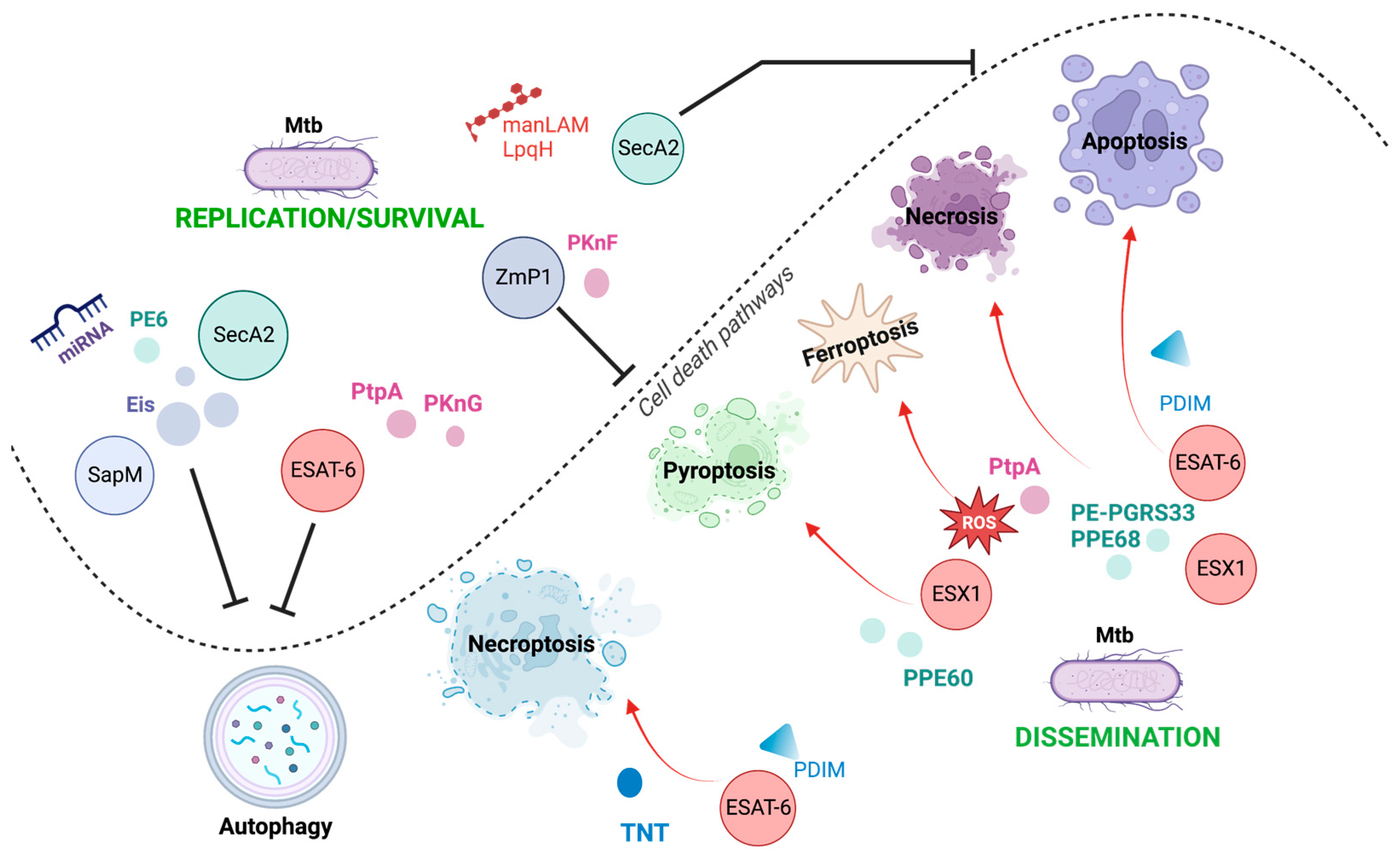{kind=link}
Table 1.
Mtb virulence factors involved in cell death modulation and its survival impact.
| Mtb Virulence Factor/Protein or Lipid Origin | Abbreviation | Function/Impact on Cell Death | Cell Type | Dissemination/ Intracellular Survival | References |
|---|---|---|---|---|---|
| Dual-Specificity Phosphatase 1 | DUSP1 | Proapoptotic, a regulator of MAPKs/NF-κB signaling pathway, induces apoptosis | THP-1 macrophages | Undefined | [62] |
| Rv3654c and Rv3655c putative proteins | Antiapoptotic interfering with the extrinsic pathway | Human monocytic cell line U937 | Undefined | [70] | |
| PE family proteins | PE-PGRS33, PE_PGRS5 (Rv0297) | Proapoptotic and induces necrosis | RAW 264.7 murine macrophages | Undefined | [72] |
| PE family proteins | PE17 (Rv1646) | Inhibits IL-6, IL-12, and TNF production and induces macrophage necrosis. | Mouse peritoneal macrophages, THP-1 macrophages, J774A.1 mouse cells, and BMDM C57BL/6 | ||
| PPE11 (Rv0453) | Induces macrophage necrosis | Intracellular survival | [102,103,104] | ||
| PPE68-Rv2626c | Induces necrosis | [119,120] | |||
| PPE60, and PE_PGRS19 | Induces pyroptosis by a non-classical-pathway | Dissemination | [149,150] | ||
| PE6 | Suppresses Autophagy | ||||
| Early secretory antigenic target 6 kD | ESAT-6 (EsxA) | Proapoptotic, increases gene expression of caspases-3, -5, -7, and -8. Promotes inflammasome activation-pyroptosis induction. Induces neutrophil necrosis and NETs production, and loss of integrity of the plasma membrane and triggers necroptosis Inhibits phagosome maturation | hMDM and THP-1 macrophages | [73,74,75] | |
| [116,117,118] | |||||
| Dissemination | [28,29] | ||||
| Zmp1, and Rv3364c | Inhibit inflammasome activation and pyroptosis | [121,122] | |||
| Subunit of type I NADH dehydrogenase | nuoG | Antiapoptotic, inhibits apoptosis in the infected cells | THP-1 macrophages, BMDM BALB/c mice | Undefined | [77,78] |
| ATPase of the Sec secretion system | SecA2 | Antiapoptotic, secretion of several proteins involved in blocking apoptosis | Undefined | [79] | |
| Serine threonine protein kinases | PknE | Proapoptotic | THP-1 macrophages | Intracellular survival | [44,82] |
| PknF | NLRP3 inflammasome inhibition | BMDM | [46] | ||
| Protein kinase G | PknG | Phagosome arresting, inhibition of phagosome–lysosome fusion | J774 macrophage cells | Intracellular survival | [45] |
| Phosphatase | SapM | Phagosome arresting, inhibition of phagosome–lysosome fusion | RAW 264.7 murine macrophages, THP-1 macrophages, and guinea pig tissues | Intracellular survival | [39,40] |
| Protein tyrosine phosphatase | PtpA | Phagosome arresting, inhibition phagosome acidification | THP-1 macrophages | Intracellular survival | [41,42,43] |
| Triggers ferroptosis | Dissemination | [159] | |||
| 38 kDa lipoprotein | PstS1 | Proapoptotic, TLR-2 and caspases -8, -9, and -3 activation, and promotes phagocytosis | hMDM | [84] | |
| Tuberculosis necrotizing toxin | TNT | Degrades all intracellular NAD+ and triggers necroptosis | THP-1 macrophages | Undefined | [137,138] |
| Enhanced intracellular survival protein | Eis | Abrogates ROS and proinflammatory cytokines production. Inhibits autophagosome formation | THP-1 macrophages | Undefined | [9,148] |
| Phthiocerol dimycocerosates | PDIMs | Proapoptotic, increases gene expression of caspases-3, -5, -7, and -8 | hMDM | Undefined | [73] |
| Induces necroptosis | THP-1 macrophages | [139] | |||
| Lipomannan (Glycolipid) | LM | Proapoptotic | THP-1 macrophages | Undefined | [86] |
| Mannosylated lipoarabinomannan (glycolipid) | ManLAM | Antiapoptotic, up-regulates Bcl-2 depending on the virulence | THP-1 macrophages, BMDM BALB/c mice | Undefined | [85,87] |
| Lipoprotein | LpqH | Proapoptotic, promotes apoptosome formation and activation of procaspases -9 and -3 | hMDM | Undefined | [89] |
hMDM, human monocyte-derived macrophages; BMDM, bone-marrow-derived macrophages; THP-1, human monocyte cell line.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ramon-Luing, L.A.; Palacios, Y.; Ruiz, A.; Téllez-Navarrete, N.A.; Chavez-Galan, L. Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms. Pathogens 2023, 12, 839. https://doi.org/10.3390/pathogens12060839
AMA Style
Ramon-Luing LA, Palacios Y, Ruiz A, Téllez-Navarrete NA, Chavez-Galan L. Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms. Pathogens. 2023; 12(6):839. https://doi.org/10.3390/pathogens12060839
Chicago/Turabian StyleRamon-Luing, Lucero A., Yadira Palacios, Andy Ruiz, Norma A. Téllez-Navarrete, and Leslie Chavez-Galan. 2023. "Virulence Factors of Mycobacterium tuberculosis as Modulators of Cell Death Mechanisms" Pathogens 12, no. 6: 839. https://doi.org/10.3390/pathogens12060839
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.