
Generation of Pearl/Calcium Phosphate Composite Particles and Their Integration into Porous Chitosan Scaffolds for Bone Regeneration


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fabrication of Composite Particles
2.3. Fabrication of Scaffolds
2.4. Characterization
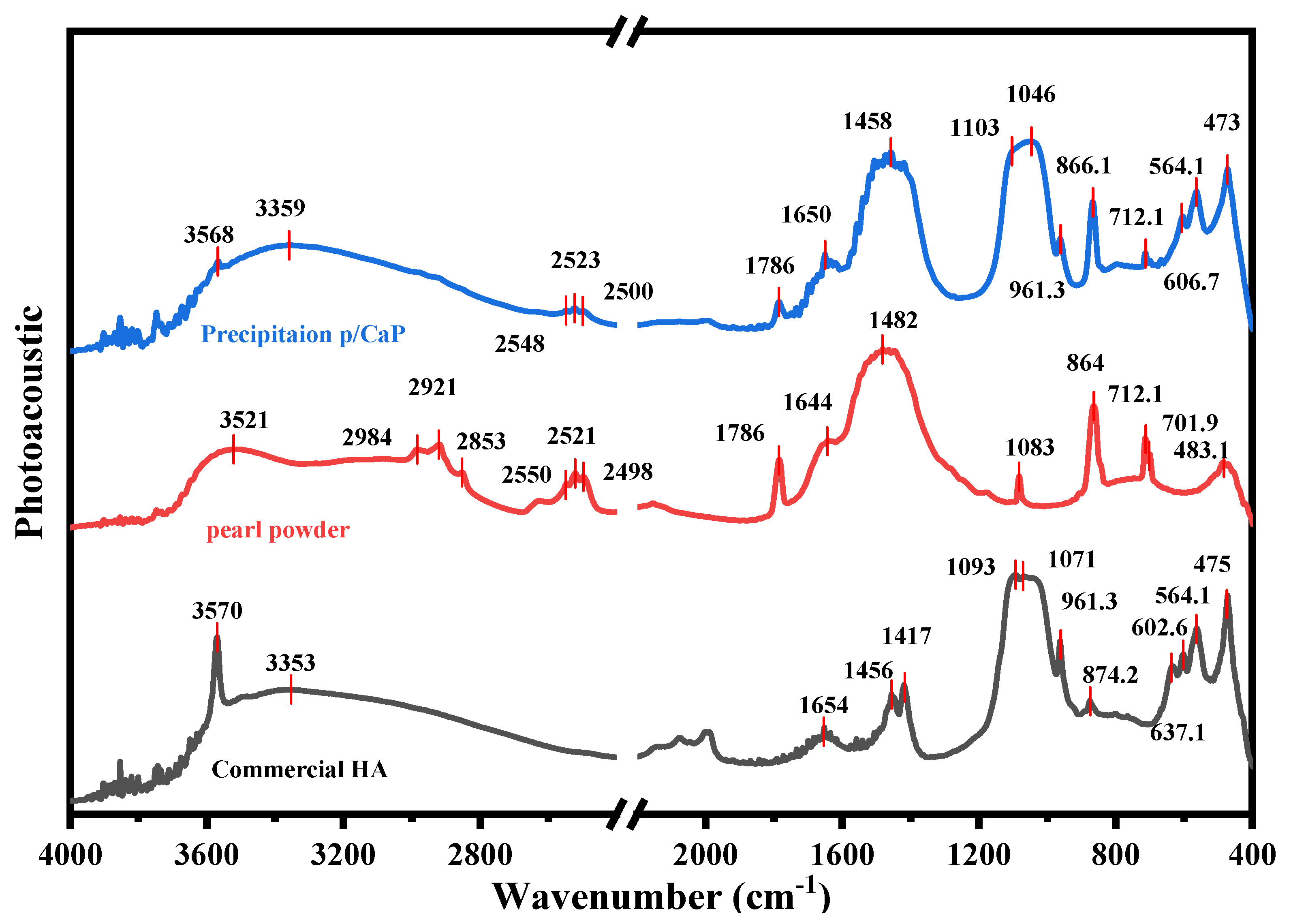
2.4.1. FTIR Analysis
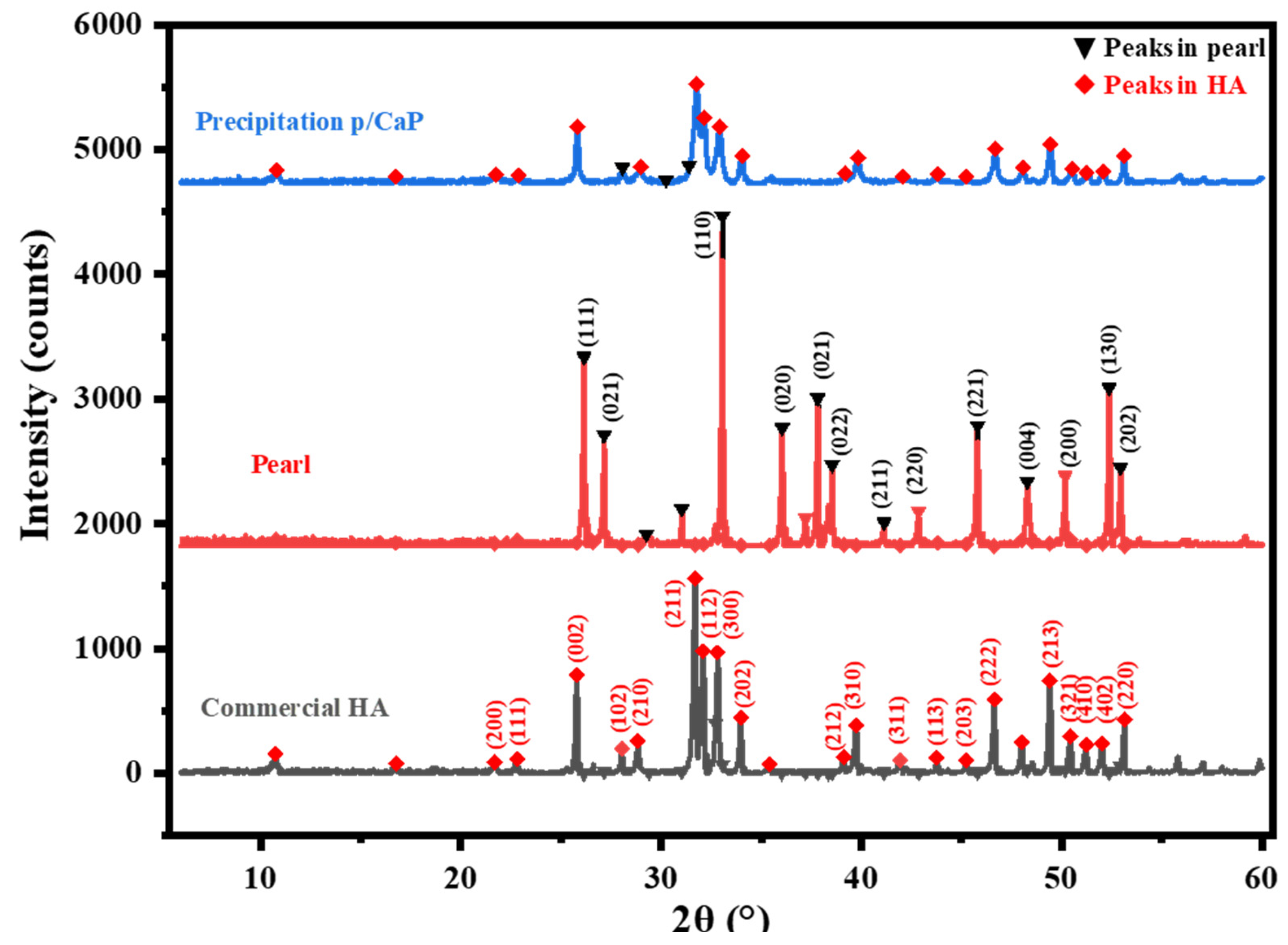
2.4.2. XRD Characterization
2.4.3. SEM Analysis
2.4.4. Porosity Analysis
2.4.5. Compressive Properties Analysis
2.5. In Vitro Cytotoxicity and Compatibility
2.5.1. Cell Morphology
2.5.2. Cell Cytotoxicity and Proliferation Analysis
2.5.3. Statistical Analysis
3. Results
3.1. Composite Particle Properties
3.2. Composite Scaffold Properties
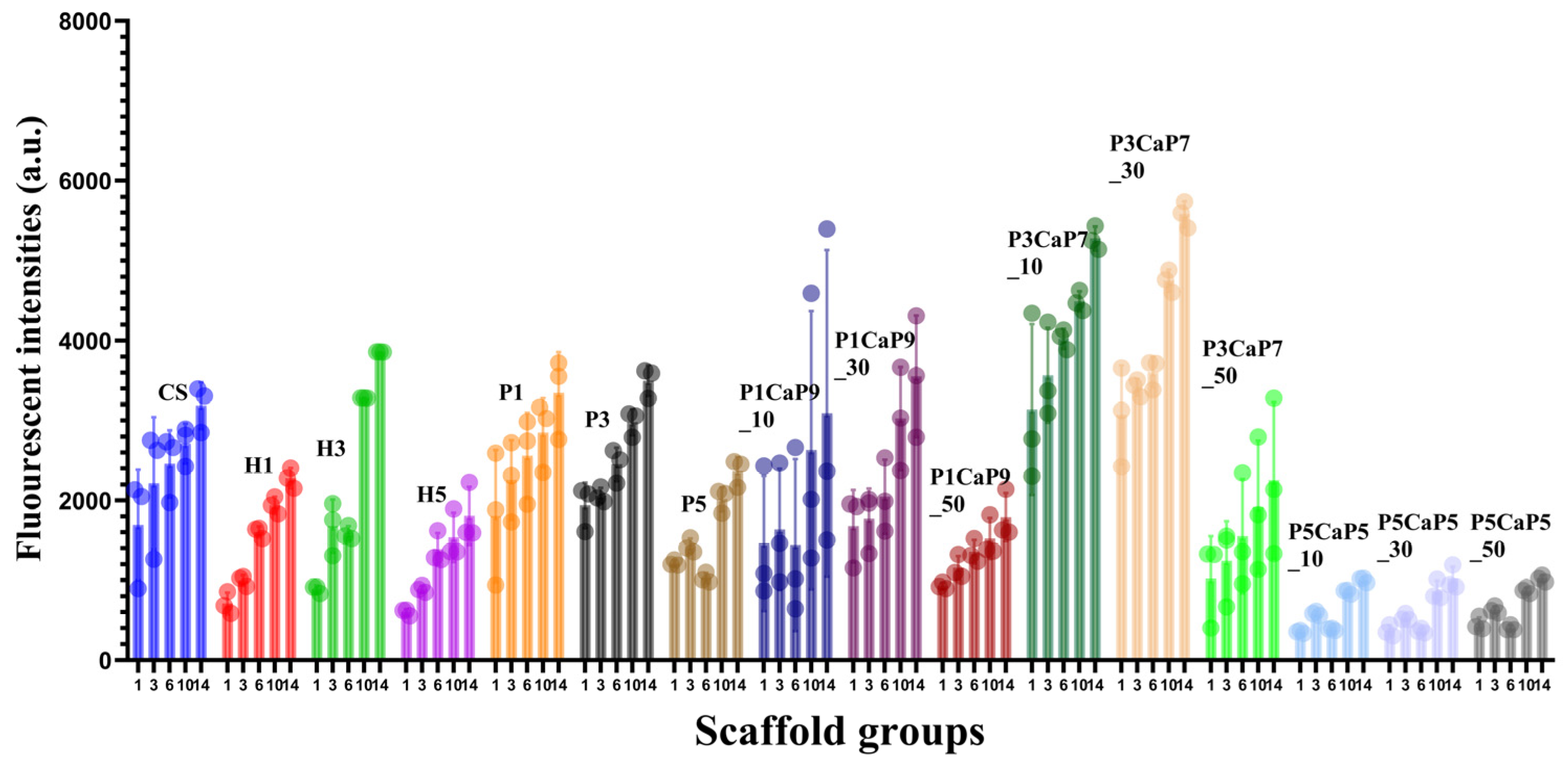
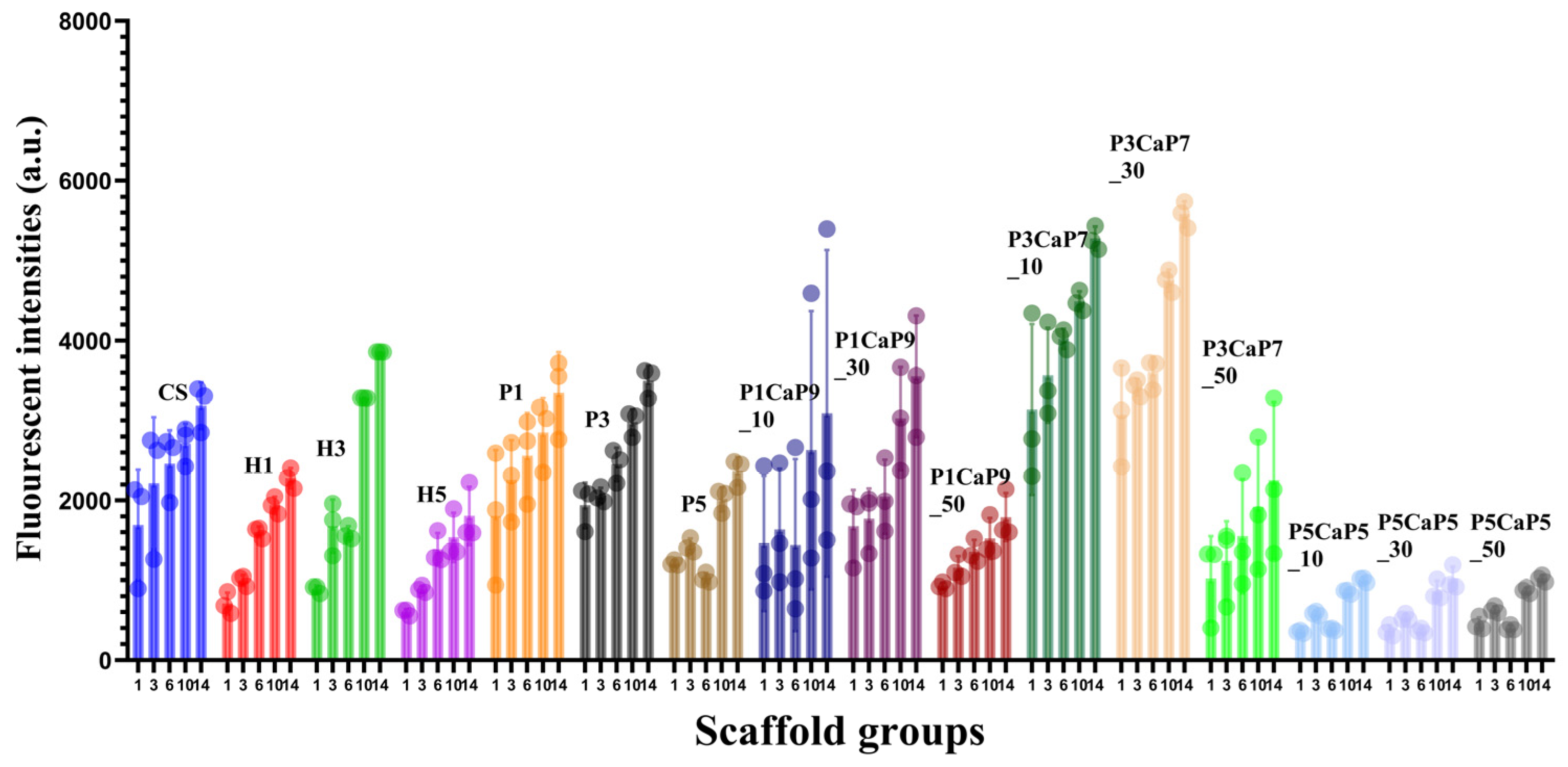
3.3. MG-63 Cell Growth on Scaffolds
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dimitriou, R.; Jones, E.; McGonagle, D.; Giannoudis, P.V. Bone regeneration: Current concepts and future directions. BMC Med. 2011, 9, 66. [Google Scholar] [CrossRef]
- Petite, H.; Viateau, V.; Bensaid, W.; Meunier, A.; de Pollak, C.; Bourguignon, M.; Oudina, K.; Sedel, L.; Guillemin, G. Tissue-engineered bone regeneration. Nat. Biotechnol. 2000, 18, 959–963. [Google Scholar] [CrossRef]
- Wang, W.; Yeung, K.W.K. Bone grafts and biomaterials substitutes for bone defect repair: A review. Bioact. Mater. 2017, 2, 224–247. [Google Scholar] [CrossRef]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–492. [Google Scholar] [CrossRef]
- Lanza, R.; Langer, R.; Vacanti, J.P.; Atala, A. Principles of Tissue Engineering; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Ager, J.W.; Balooch, G.; Ritchie, R.O. Fracture, aging, and disease in bone. J. Mater. Res. 2006, 21, 1878–1892. [Google Scholar] [CrossRef]
- Melton, L.J.B., III. Epidemiology of hip fractures: Implications of the exponential increase with age. Bone 1996, 18, S121–S125. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, F.C. Calcium carbonate in biomineralisation and biomimetic chemistry. Int. Mater. Rev. 2003, 48, 187–224. [Google Scholar] [CrossRef]
- Morse, J.W.; Arvidson, R.S.; Luttge, A. Calcium carbonate formation and dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef] [PubMed]
- Murr, L.E.; Ramirez, D.A. The Microstructure of the Cultured Freshwater Pearl. JOM 2012, 64, 469–474. [Google Scholar] [CrossRef]
- Sun, J.; Bhushan, B.J.R.A. Hierarchical structure and mechanical properties of nacre: A review. RSC Adv. 2012, 2, 7617–7632. [Google Scholar] [CrossRef]
- Pei, J.; Wang, Y.; Zou, X.; Ruan, H.; Tang, C.; Liao, J.; Si, G.; Sun, P. Extraction, Purification, Bioactivities and Application of Matrix Proteins From Pearl Powder and Nacre Powder: A Review. Front. Bioeng. Biotechnol. 2021, 9, 649665. [Google Scholar] [CrossRef]
- Lopez, E.; Vidal, B.; Berland, S.; Camprasse, S.; Camprasse, G.; Silve, C. Demonstration of the capacity of nacre to induce bone formation by human osteoblasts maintained in vitro. Tissue Cell 1992, 24, 667–679. [Google Scholar] [CrossRef]
- Li, X.; Xu, P.; Cheng, Y.; Zhang, W.; Zheng, B.; Wang, Q. Nano-pearl powder/chitosan-hyaluronic acid porous composite scaffold and preliminary study of its osteogenesis mechanism. Mater. Sci. Eng. C 2020, 111, 110749. [Google Scholar] [CrossRef]
- Liu, Y.S.; Huang, Q.L.; Kienzle, A.; Muller, W.E.; Feng, Q.L. In vitro degradation of porous PLLA/pearl powder composite scaffolds. Mater. Sci. Eng. C 2014, 38, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Bai, J.; Jin, J.; Yang, S.; Li, G.J.A.E.M. Stimulation by pearl of mineralization and biocompatibility of PLA. Adv. Eng. Mater. 2015, 17, 1691–1697. [Google Scholar] [CrossRef]
- Kattimani, V.S.; Kondaka, S.; Lingamaneni, K.P.J.B.; Insights, T.R. Hydroxyapatite–-Past, present, and future in bone regeneration. Bone Tissue Regen. Insights 2016, 7. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Maxian, S.H.; Zawadsky, J.P.; Dunn, M.G. In vitro evaluation of amorphous calcium phosphate and poorly crystallized hydroxyapatite coatings on titanium implants. J. Biomed. Mater. Res. 1993, 27, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Nagano, M.; Nakamura, T.; Kokubo, T.; Tanahashi, M.; Ogawa, M. Differences of bone bonding ability and degradation behaviour in vivo between amorphous calcium phosphate and highly crystalline hydroxyapatite coating. Biomaterials 1996, 17, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Balasundaram, G.; Webster, T.J. Increased osteoblast adhesion on nanoparticulate crystalline hydroxyapatite functionalized with KRSR. Int. J. Nanomed. 2006, 1, 339–349. [Google Scholar]
- Karampas, I.; Kontoyannis, C.J.V.S. Characterization of calcium phosphates mixtures. Vib. Spectrosc. 2013, 64, 126–133. [Google Scholar] [CrossRef]
- Denry, I.; Kuhn, L.T. Design and characterization of calcium phosphate ceramic scaffolds for bone tissue engineering. Dent. Mater. 2016, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Fada, R.; Shahgholi, M.; Azimi, R.; Babadi, N.F. Estimation of Porosity Effect on Mechanical Properties in Calcium Phosphate Cement Reinforced by Strontium Nitrate Nanoparticles: Fabrication and FEM Analysis. Arab. J. Sci. Eng. 2023, 49, 1815–1825. [Google Scholar] [CrossRef]
- Dillon, J.P.; Waring-Green, V.J.; Taylor, A.M.; Wilson, P.J.M.; Birch, M.; Gartland, A.; Gallagher, J.A. Primary Human Osteoblast Cultures. In Bone Research Protocols; Methods in Molecular Biology Series; Springer: Berlin/Heidelberg, Germany, 2012; Volume 816, pp. 3–18. [Google Scholar]
- Balmain, J.; Hannoyer, B.; Lopez, E. Fourier transform infrared spectroscopy (FTIR) and X-ray diffraction analyses of mineral and organic matrix during heating of mother of pearl (nacre) from the shell of the mollusc Pinctada maxima. J. Biomed. Mater. Res. 1999, 48, 749–754. [Google Scholar] [CrossRef]
- Wang, J.F.; Cheng, Q.F.; Tang, Z.Y. Layered nanocomposites inspired by the structure and mechanical properties of nacre. Chem. Soc. Rev. 2012, 41, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.Y.; Lee, I.S. Characterization of vaterite in low quality freshwater-cultured pearls. Mater. Sci. Eng. C 2006, 26, 721–723. [Google Scholar] [CrossRef]
- Rehman, I.; Bonfield, W. Characterization of hydroxyapatite and carbonated apatite by photo acoustic FTIR spectroscopy. J. Mater. Sci. Mater. Med. 1997, 8, 1–4. [Google Scholar] [CrossRef]
- Raynaud, S.; Champion, E.; Bernache-Assollant, D.; Thomas, P. Calcium phosphate apatites with variable Ca/P atomic ratio I. Synthesis, characterisation and thermal stability of powders. Biomaterials 2002, 23, 1065–1072. [Google Scholar] [CrossRef]
- Lotsari, A.; Rajasekharan, A.K.; Halvarsson, M.; Andersson, M. Transformation of amorphous calcium phosphate to bone-like apatite. Nat. Commun. 2018, 9, 4170. [Google Scholar] [CrossRef]
- Januariyasa, I.K.; Ana, I.D.; Yusuf, Y. Nanofibrous poly(vinyl alcohol)/chitosan contained carbonated hydroxyapatite nanoparticles scaffold for bone tissue engineering. Mater. Sci. Eng. C 2020, 107, 110347. [Google Scholar] [CrossRef]
- Maji, K.; Dasgupta, S.; Kundu, B.; Bissoyi, A. Development of gelatin-chitosan-hydroxyapatite based bioactive bone scaffold with controlled pore size and mechanical strength. J. Biomater. Sci. Polym. Ed. 2015, 26, 1190–1209. [Google Scholar] [CrossRef]
- Sari, M.; Hening, P.; Chotimah; Ana, I.D.; Yusuf, Y. Porous structure of bioceramics carbonated hydroxyapatite-based honeycomb scaffold for bone tissue engineering. Mater. Today Commun. 2021, 26, 102135. [Google Scholar] [CrossRef]
- Abbasi, N.; Hamlet, S.; Love, R.M.; Nguyen, N.T. Porous scaffolds for bone regeneration. J. Sci. Adv. Mater. Dev. 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Hannink, G.; Arts, J.C.J.I. Bioresorbability, porosity and mechanical strength of bone substitutes: What is optimal for bone regeneration? Injury 2011, 42, S22–S25. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Haugh, M.G.; O’brien, F.J.J.B. The effect of mean pore size on cell attachment, proliferation and migration in collagen–glycosaminoglycan scaffolds for bone tissue engineering. Biomaterials 2010, 31, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Park, I.K.; Kim, J.M.; Lee, J.H. In vitro and in vivo characteristics of PCL scaffolds with pore size gradient fabricated by a centrifugation method. Biomaterials 2007, 28, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Loh, Q.L.; Choong, C. Three-dimensional scaffolds for tissue engineering applications: Role of porosity and pore size. Tissue Eng. Part B Rev. 2013, 19, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.C.; Erkal, J.L.; Lockwood, S.Y.; Chen, C.; Spence, D.M. Evaluation of 3D printing and its potential impact on biotechnology and the chemical sciences. Anal. Chem. 2014, 86, 3240–3253. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Jones, W.R.; Ting-Beall, H.P.; Lee, G.M. The deformation behavior and mechanical properties of chondrocytes in articular cartilage. Osteoarthr. Cartil. 1999, 7, 59–70. [Google Scholar] [CrossRef]
- Mow, V.C.; Kuei, S.C.; Lai, W.M.; Armstrong, C.G. Biphasic creep and stress relaxation of articular cartilage in compression: Theory and experiments. J. Biomech. Eng. 1980, 102, 73–84. [Google Scholar] [CrossRef]
- Odgaard, A.; Linde, F. The underestimation of Young’s modulus in compressive testing of cancellous bone specimens. J. Biomech. 1991, 24, 691–698. [Google Scholar] [CrossRef]
- Rho, J.Y.; Ashman, R.B.; Turner, C.H. Young’s modulus of trabecular and cortical bone material: Ultrasonic and microtensile measurements. J. Biomech. 1993, 26, 111–119. [Google Scholar] [CrossRef]
- Kim, H.L.; Jung, G.Y.; Yoon, J.H.; Han, J.S.; Park, Y.J.; Kim, D.G.; Zhang, M.; Kim, D.J. Preparation and characterization of nano-sized hydroxyapatite/alginate/chitosan composite scaffolds for bone tissue engineering. Mater. Sci. Eng. C 2015, 54, 20–25. [Google Scholar] [CrossRef]
- Venkatesan, J.; Kim, S.-K. Chitosan composites for bone tissue engineering—An overview. Mar. Drugs 2010, 8, 2252–2266. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Z. Chitosan-calcium carbonate scaffold with high mineral content and hierarchical structure for bone regeneration. Smart Mater. Med. 2023, 4, 552–561. [Google Scholar] [CrossRef]
- Boonrungsiman, S.; Gentleman, E.; Carzaniga, R.; Evans, N.D.; McComb, D.W.; Porter, A.E.; Stevens, M.M. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14170–14175. [Google Scholar] [CrossRef] [PubMed]
- Omelon, S.; Georgiou, J.; Henneman, Z.J.; Wise, L.M.; Sukhu, B.; Hunt, T.; Wynnyckyj, C.; Holmyard, D.; Bielecki, R.; Grynpas, M.D. Control of vertebrate skeletal mineralization by polyphosphates. PLoS ONE 2009, 4, e5634. [Google Scholar] [CrossRef] [PubMed]
- Rustom, L.E.; Poellmann, M.J.; Wagoner Johnson, A.J. Mineralization in micropores of calcium phosphate scaffolds. Acta Biomater. 2019, 83, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Qi, C.; Zhang, X.M. Chitosan microspheres supported palladium heterogeneous catalysts modified with pearl shell powders. Int. J. Biol. Macromol. 2013, 55, 240–245. [Google Scholar] [CrossRef]
- Zhao, X.X.; Ng, S.; Heng, B.C.; Guo, J.; Ma, L.; Tan, T.T.Y.; Ng, K.W.; Loo, S.C.J. Cytotoxicity of hydroxyapatite nanoparticles is shape and cell dependent. Arch. Toxicol. 2013, 87, 1037–1052. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M. Calcium phosphate/chitosan composite scaffolds for controlled in vitro antibiotic drug release. J. Biomed. Mater. Res. 2002, 62, 378–386. [Google Scholar] [CrossRef]
- Wei, G.; Ma, P.X. Structure and properties of nano-hydroxyapatite/polymer composite scaffolds for bone tissue engineering. Biomaterials 2004, 25, 4749–4757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, X.; Li, D.; Ao, R.; Yu, B.; Yu, B. Three dimensionally printed pearl powder/poly-caprolactone composite scaffolds for bone regeneration. J. Biomater. Sci. Polym. Ed. 2018, 29, 1686–1700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M. Synthesis and characterization of macroporous chitosan/calcium phosphate composite scaffolds for tissue engineering. J. Biomed. Mater. Res. 2001, 55, 304–312. [Google Scholar] [CrossRef]
- Li, G.; Zhao, X.; Zhao, W.; Zhang, L.; Wang, C.; Jiang, M.; Gu, X.; Yang, Y. Porous chitosan scaffolds with surface micropatterning and inner porosity and their effects on Schwann cells. Biomaterials 2014, 35, 8503–8513. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ao, Q.; Gong, K.; Wang, A.; Zheng, L.; Gong, Y.; Zhang, X. The effect of topology of chitosan biomaterials on the differentiation and proliferation of neural stem cells. Acta Biomater. 2010, 6, 3630–3639. [Google Scholar] [CrossRef]
- Ragetly, G.R.; Griffon, D.J.; Lee, H.-B.; Fredericks, L.P.; Gordon-Evans, W.; Chung, Y.S. Effect of chitosan scaffold microstructure on mesenchymal stem cell chondrogenesis. Acta Biomater. 2010, 6, 1430–1436. [Google Scholar] [CrossRef]
- Ross, A.M.; Jiang, Z.; Bastmeyer, M.; Lahann, J. Physical aspects of cell culture substrates: Topography, roughness, and elasticity. Small 2012, 8, 336–355. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Scaffolds | Abbreviation | Particles Added to Chitosan Scaffold |
|---|---|---|
| Pure CS scaffolds | CS | - |
| Pearl/CS scaffolds | P1 | 10 wt.% pearl |
| P3 | 30 wt.% pearl | |
| P5 | 50 wt.% pearl | |
| HA/CS scaffolds | H1 | 10 wt.% HA |
| H3 | 30 wt.% HA | |
| H5 | 50 wt.% HA | |
| Pearl/CaP (1:9)(P1CaP9)/CS scaffolds | P1CaP9_10 | 10 wt.% P1CaP9 |
| P1CaP9_30 | 30 wt.% P1CaP9 | |
| P1CaP9_50 | 50 wt.% P1CaP9 | |
| P3CaP7/CS scaffolds | P3CaP7_10 | 10 wt.% P3CaP7 |
| P3CaP7_30 | 30 wt.% P3CaP7 | |
| P3CaP7_50 | 50 wt.% P3CaP7 | |
| P5CaP5/CS scaffolds | P5CaP5_10 | 10 wt.% P5CaP5 |
| P5CaP5_30 | 30 wt.% P5CaP5 | |
| P5CaP5_50 | 50 wt.% P5CaP5 |
| Names | N Total | Mean μm | Standard Deviation μm | Minimum μm | Median μm | Maximum μm | Median Absolute Deviation μm |
|---|---|---|---|---|---|---|---|
| CS | 351 | 110.30 | 104.35 | 31.86 | 98.58 | 589.97 | 11.72 |
| P1 | 358 | 121.40 | 115.40 | 32.18 | 99.27 | 595.87 | 22.14 |
| P3 | 363 | 134.78 | 129.14 | 30.27 | 95.15 | 560.47 | 39.63 |
| P5 | 373 | 114.50 | 108.88 | 30.19 | 82.98 | 558.97 | 21.52 |
| H1 | 349 | 109.03 | 103.70 | 28.61 | 87.57 | 529.70 | 21.57 |
| H3 | 388 | 118.86 | 113.54 | 28.56 | 91.47 | 528.81 | 27.38 |
| H5 | 353 | 127.04 | 121.82 | 28.03 | 88.34 | 519.07 | 38.69 |
| P1CaP9_10 | 347 | 121.88 | 116.40 | 29.43 | 83.35 | 544.97 | 28.53 |
| P1CaP9_30 | 355 | 128.20 | 122.92 | 28.37 | 91.06 | 525.30 | 37.13 |
| P1CaP9_50 | 358 | 128.93 | 123.07 | 31.49 | 107.73 | 582.67 | 20.23 |
| P3CaP7_10 | 356 | 126.24 | 120.52 | 30.69 | 96.06 | 568.29 | 30.55 |
| P3CaP7_30 | 362 | 138.07 | 132.25 | 31.22 | 107.20 | 578.07 | 20.88 |
| P3CaP7_50 | 370 | 107.58 | 121.79 | 31.08 | 96.89 | 575.43 | 30.69 |
| P5CaP5_10 | 349 | 110.85 | 115.96 | 26.24 | 100.49 | 485.95 | 34.36 |
| P5CaP5_30 | 352 | 102.86 | 105.97 | 26.25 | 92.50 | 486.01 | 32.35 |
| P5CaP5_50 | 355 | 99.52 | 104.17 | 28.75 | 88.88 | 532.34 | 27.71 |
| Composition | Density (g/cm3) | Porosity |
|---|---|---|
| CS | 0.061 | 80.33 ± 10.37% |
| P1 | 0.065 | 81.77 ± 3.76% |
| P3 | 0.071 | 79.00 ± 4.49% |
| P5 | 0.076 | 76.23 ± 5.42% |
| H1 | 0.079 | 81.35 ± 3.42% |
| H3 | 0.082 | 79.23 ± 4.17% |
| H5 | 0.089 | 75.10 ± 7.27% |
| P1CaP9_10 | 0.067 | 78.64 ± 1.08% |
| P1CaP9_30 | 0.072 | 75.83 ± 5.40% |
| P1CaP9_50 | 0.075 | 69.25 ± 5.42% |
| P3CaP7_10 | 0.065 | 78.17 ± 1.18% |
| P3CaP7_30 | 0.068 | 76.13 ± 1.70% |
| P3CaP7_50 | 0.074 | 70.90 ± 6.79% |
| P5CaP5_10 | 0.064 | 69.73 ± 8.62% |
| P5CaP5_30 | 0.068 | 67.73 ± 4.62% |
| P5CaP5_50 | 0.071 | 63.73 ± 3.92% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Ur Rehman, I.; Shepherd, R.; Douglas, T.E.L. Generation of Pearl/Calcium Phosphate Composite Particles and Their Integration into Porous Chitosan Scaffolds for Bone Regeneration. J. Funct. Biomater. 2024, 15, 55. https://doi.org/10.3390/jfb15030055
Li Z, Ur Rehman I, Shepherd R, Douglas TEL. Generation of Pearl/Calcium Phosphate Composite Particles and Their Integration into Porous Chitosan Scaffolds for Bone Regeneration. Journal of Functional Biomaterials. 2024; 15(3):55. https://doi.org/10.3390/jfb15030055
Chicago/Turabian StyleLi, Zhiyi, Ihtesham Ur Rehman, Rebecca Shepherd, and Timothy E. L. Douglas. 2024. "Generation of Pearl/Calcium Phosphate Composite Particles and Their Integration into Porous Chitosan Scaffolds for Bone Regeneration" Journal of Functional Biomaterials 15, no. 3: 55. https://doi.org/10.3390/jfb15030055