Metal Ion-Loaded Nanofibre Matrices for Calcification Inhibition in Polyurethane Implants

Abstract
:1. Introduction
2. Results and Discussion
2.1. Morphological Analysis
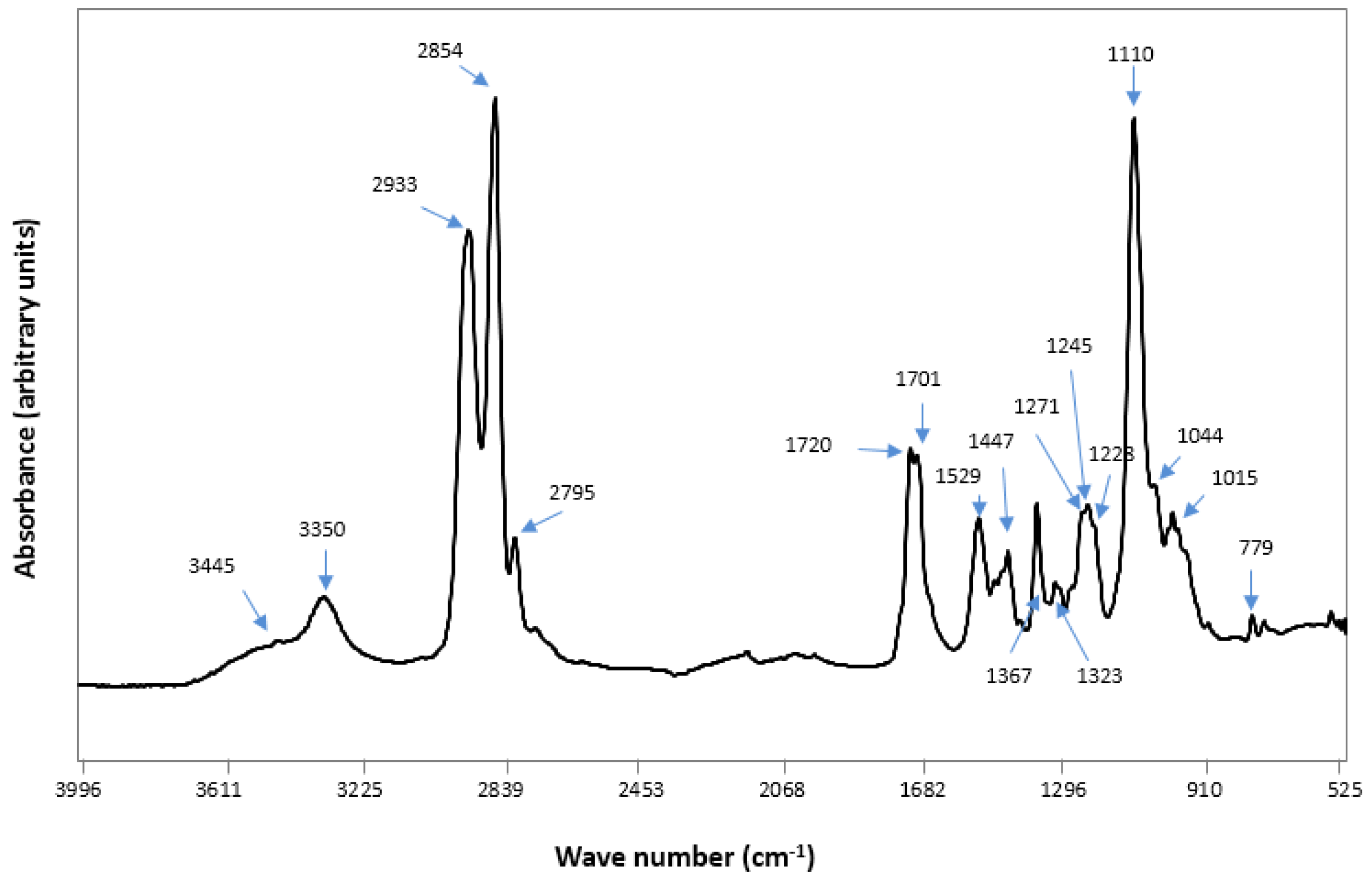
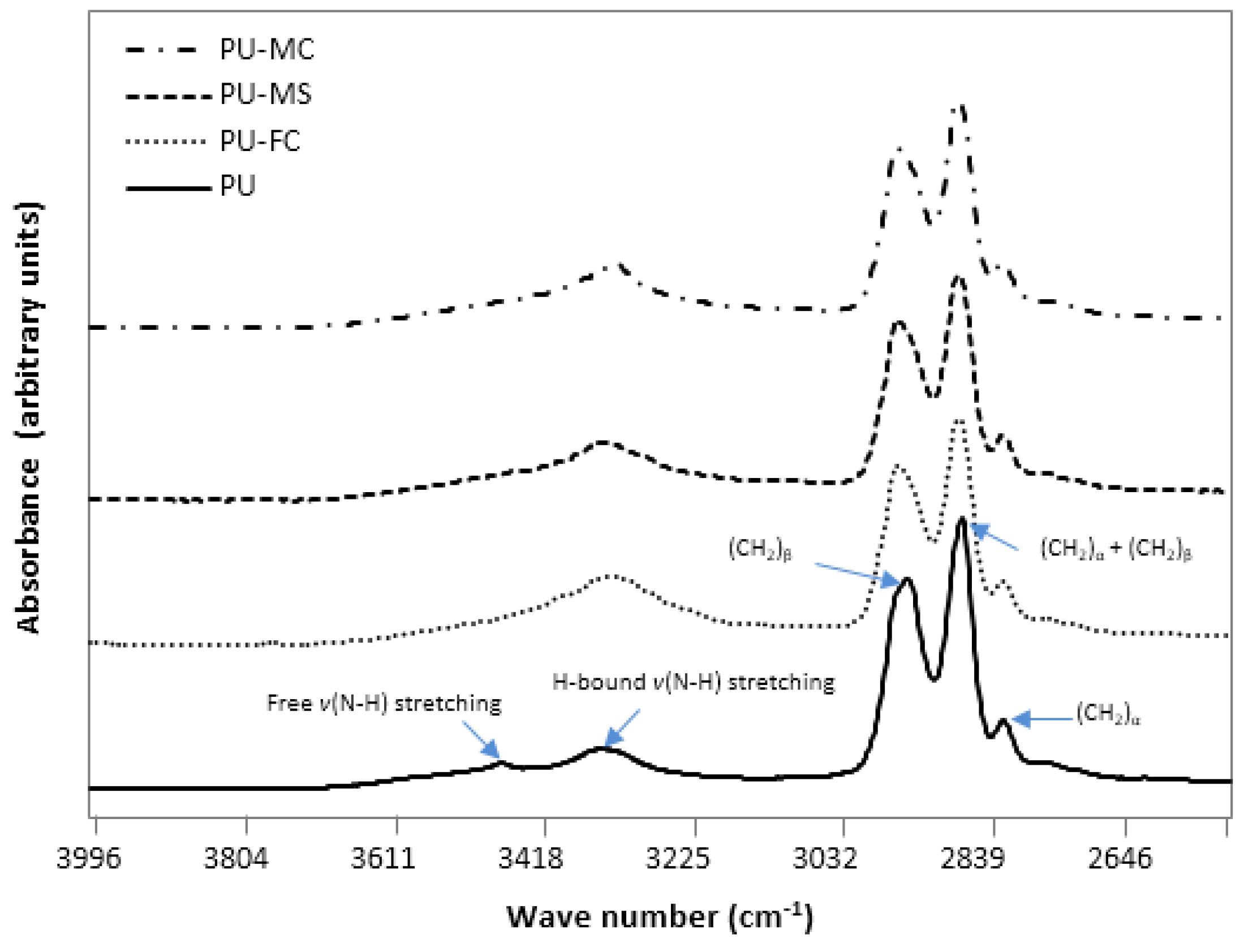
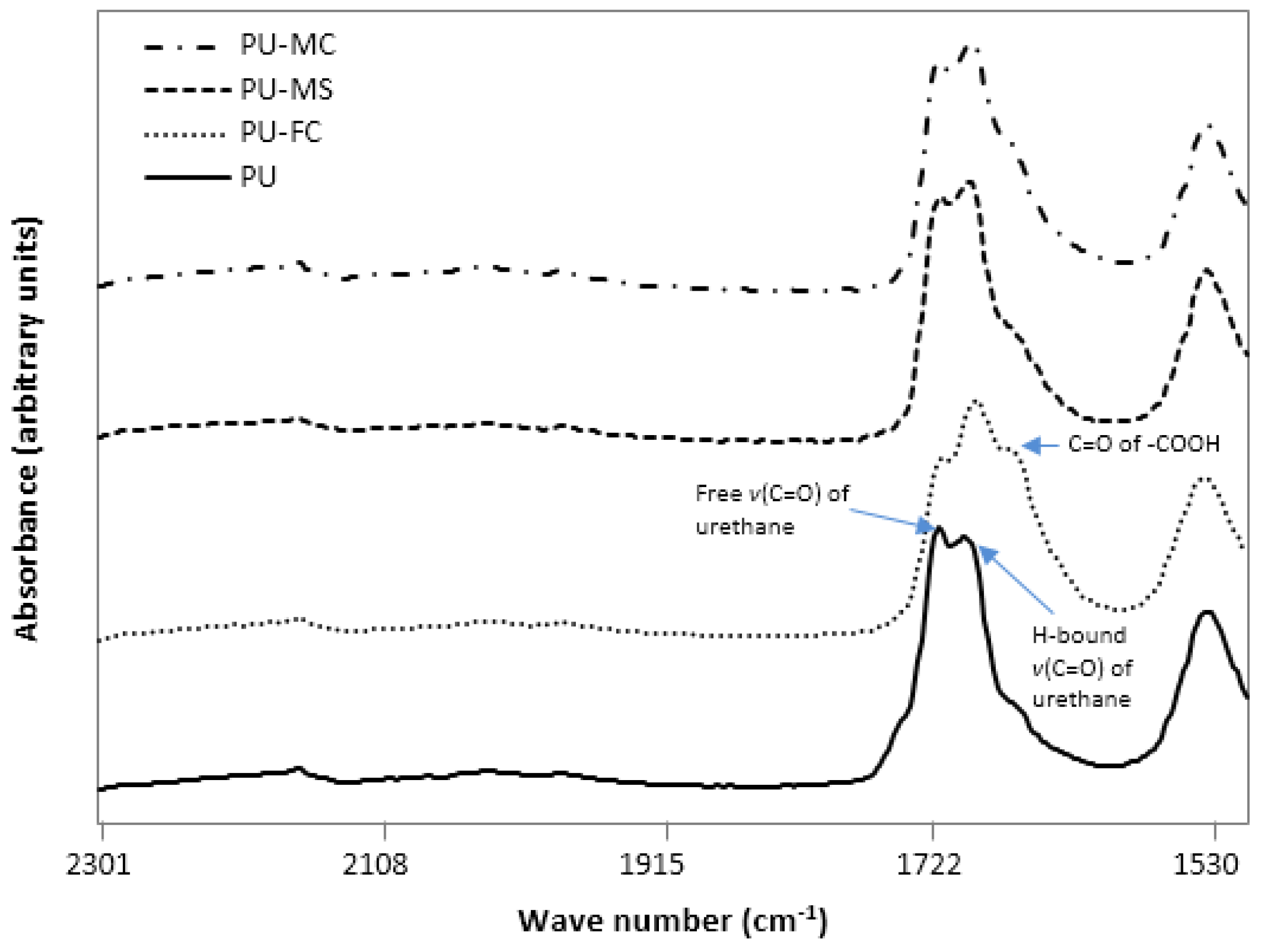
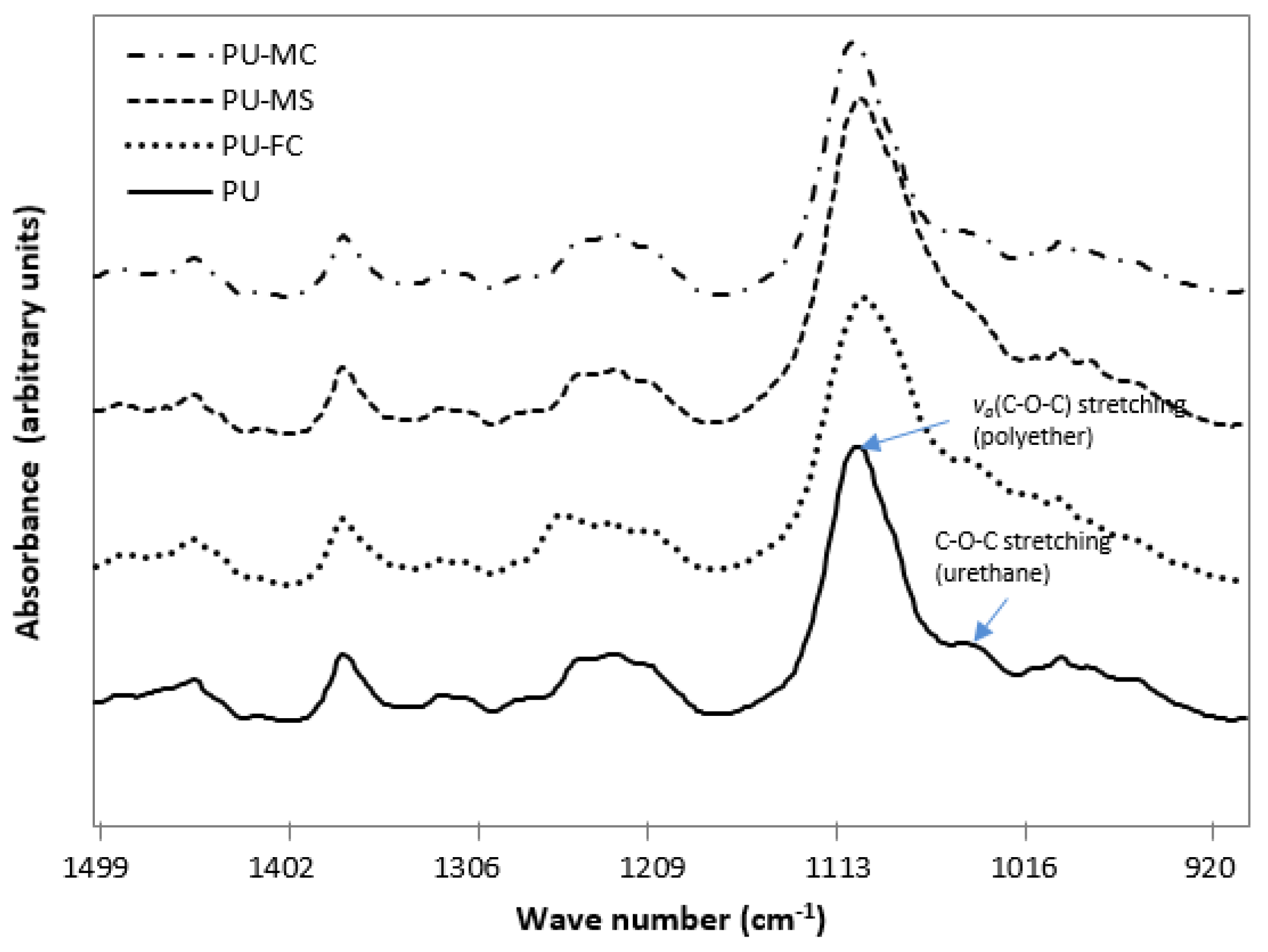
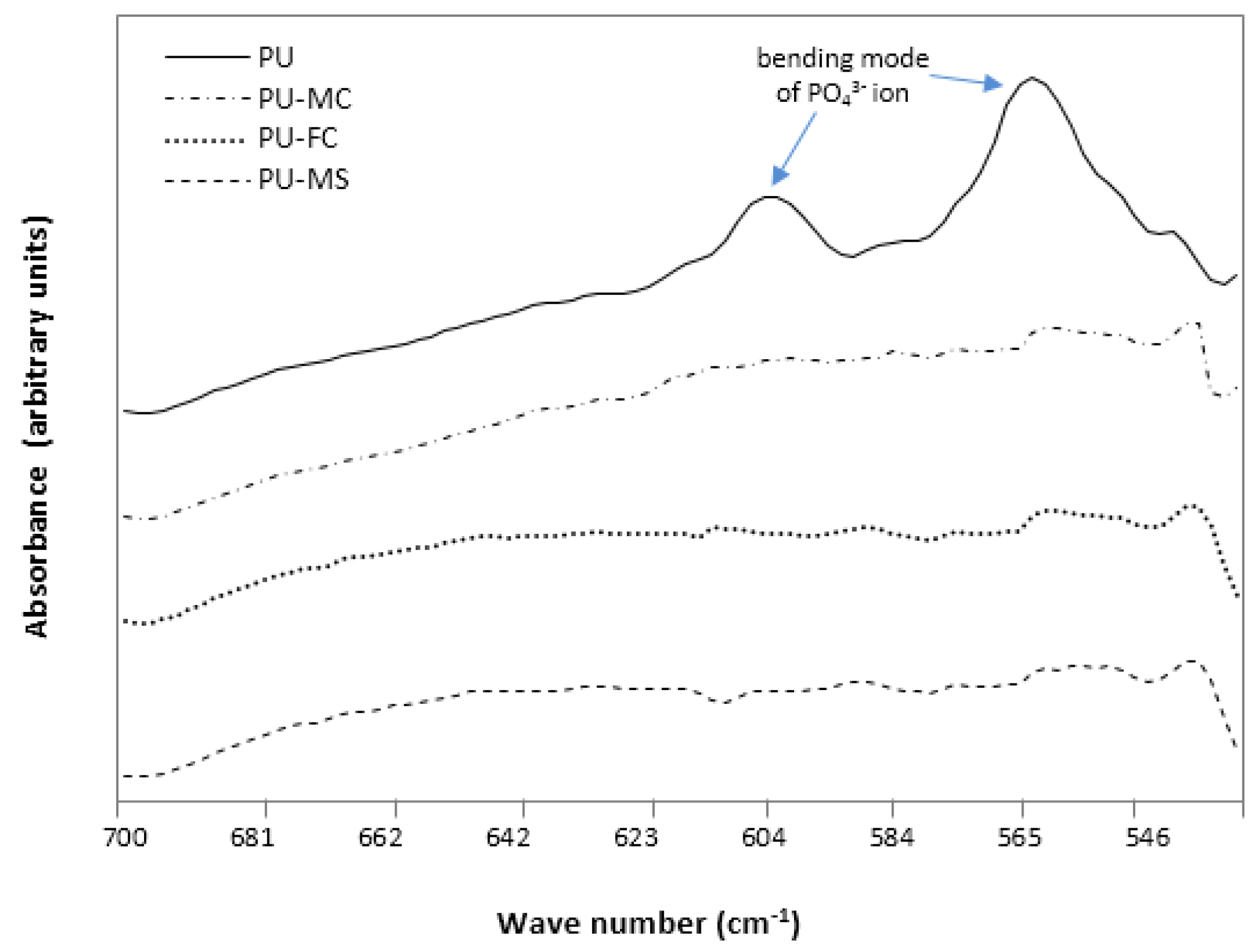
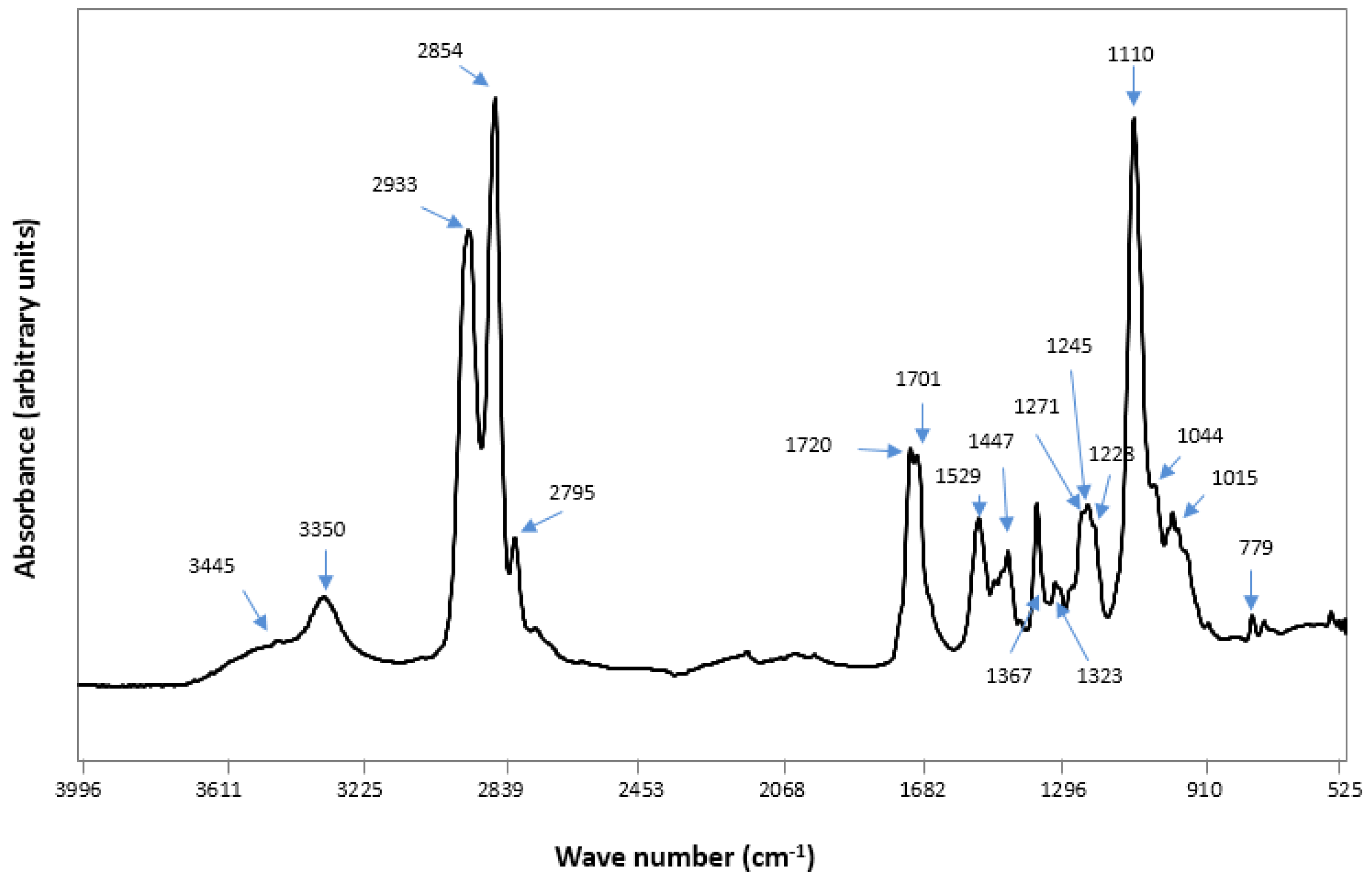
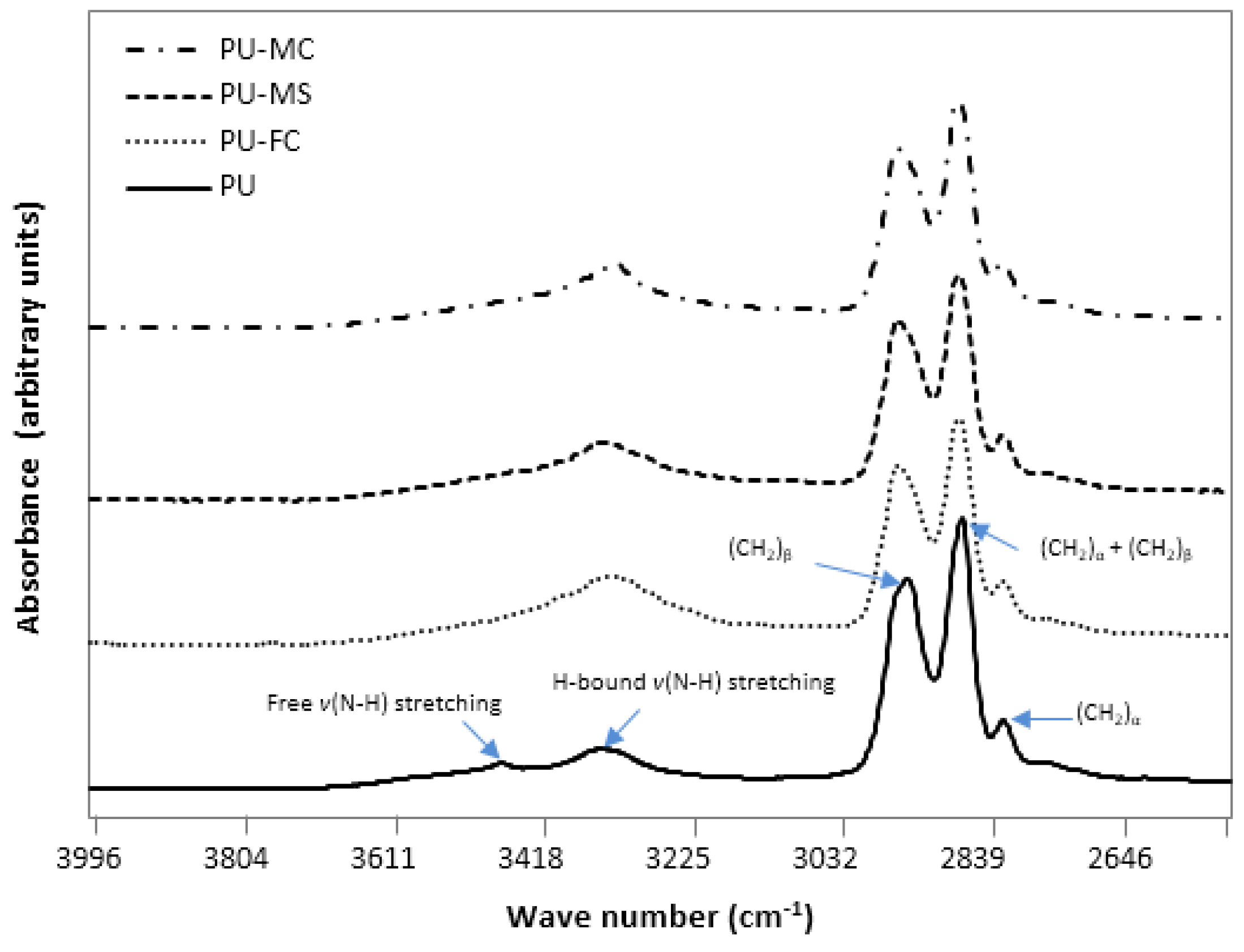
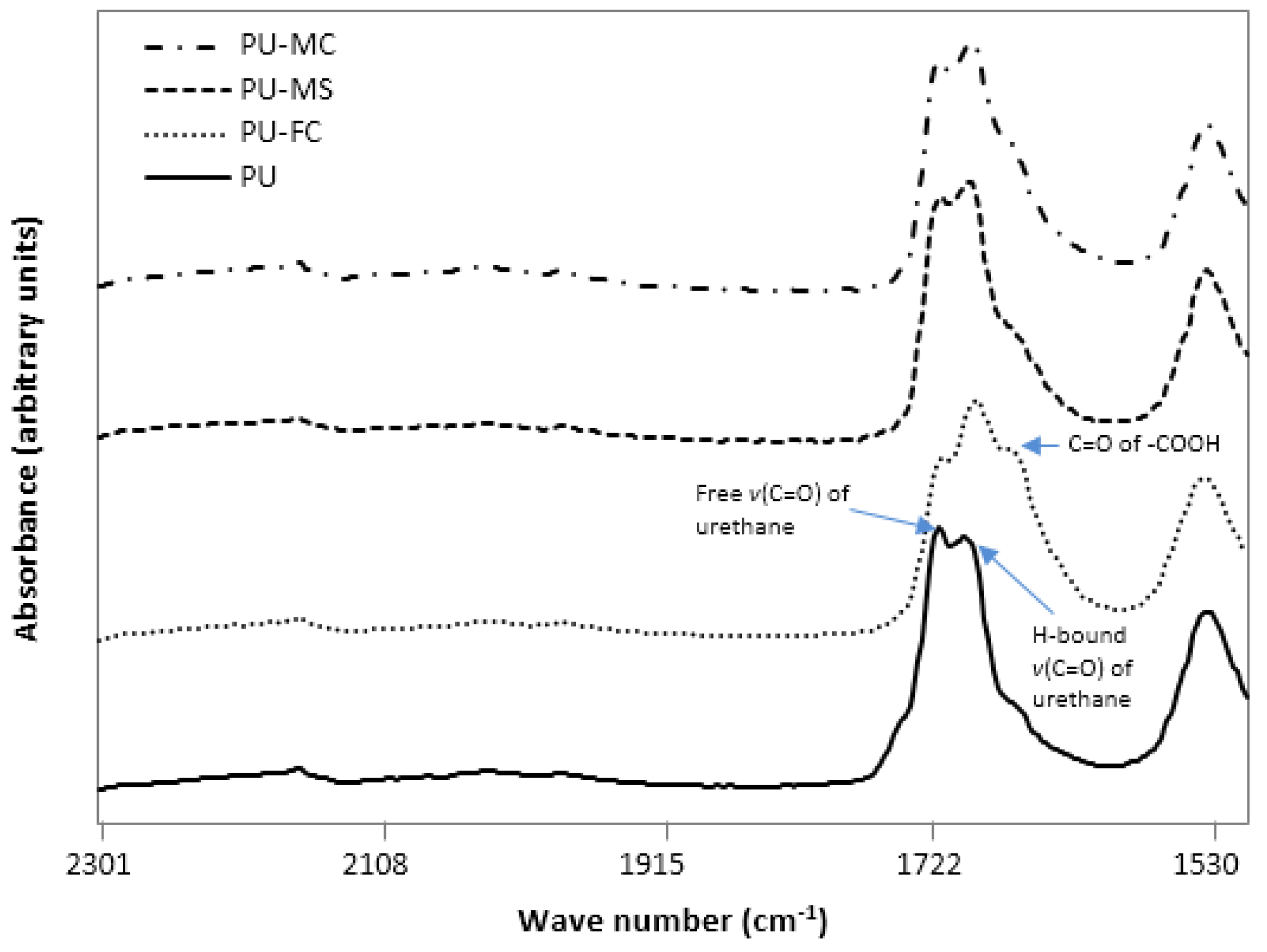
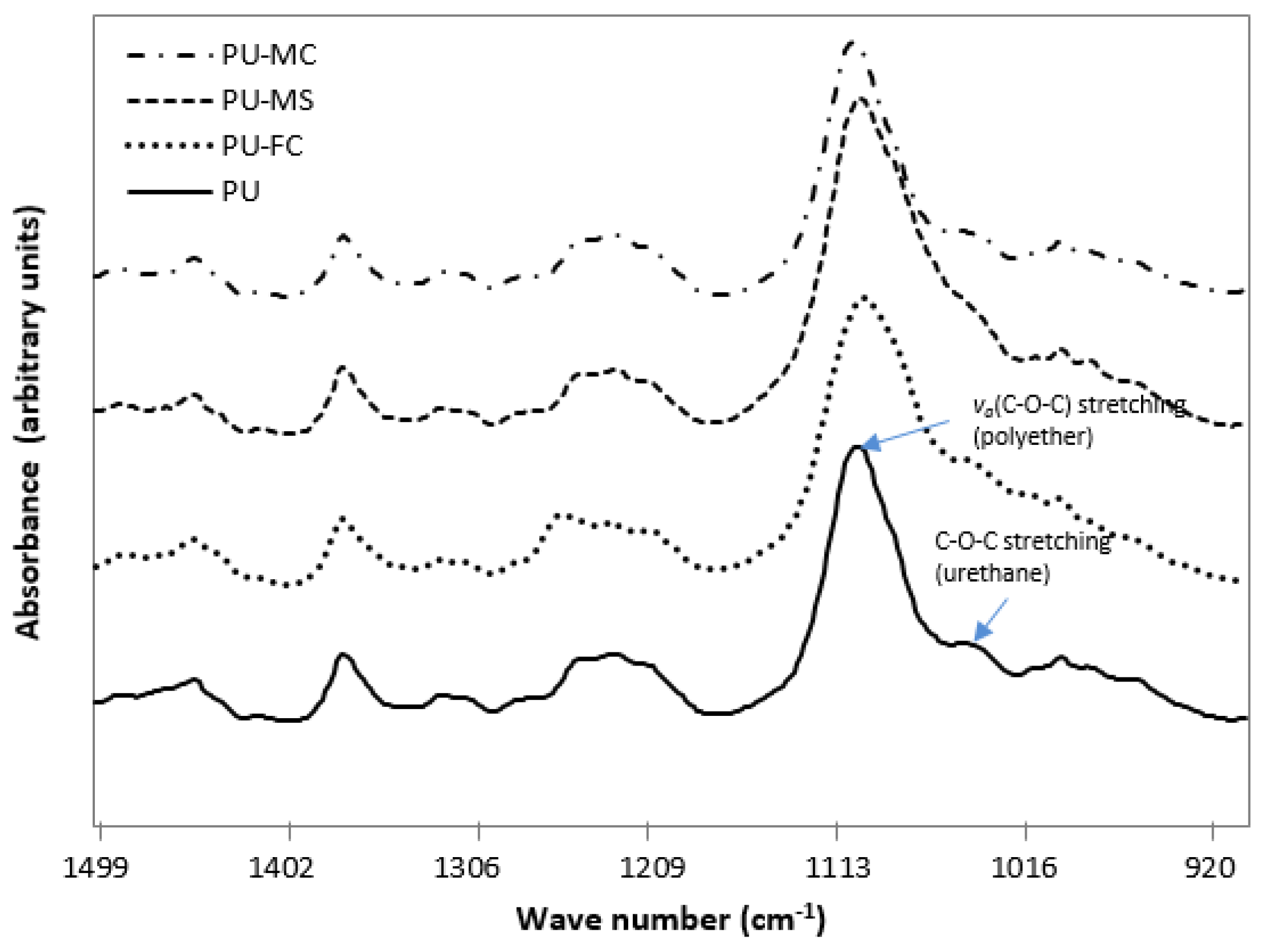
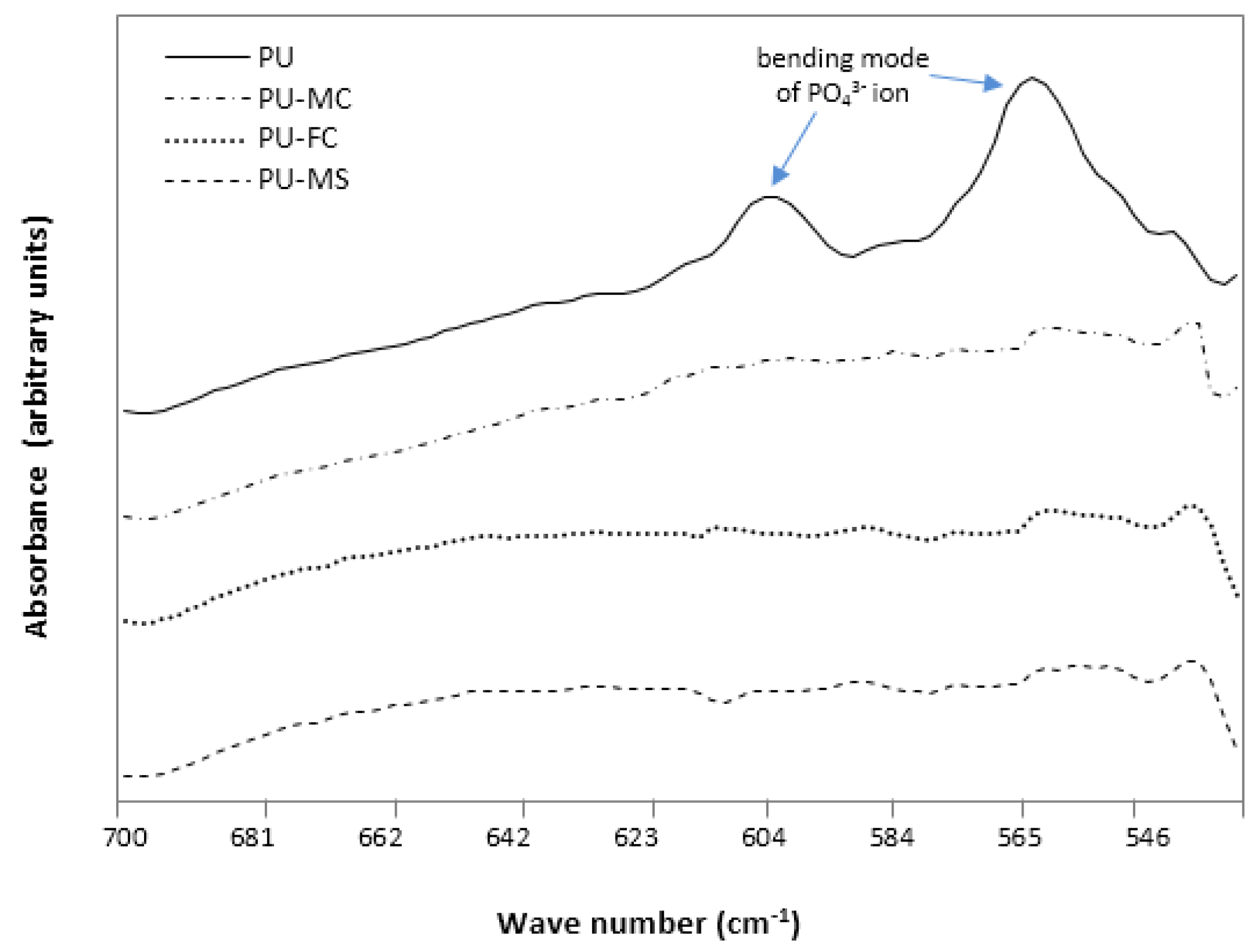
2.2. Fourier Transform Infra RedAnalysis
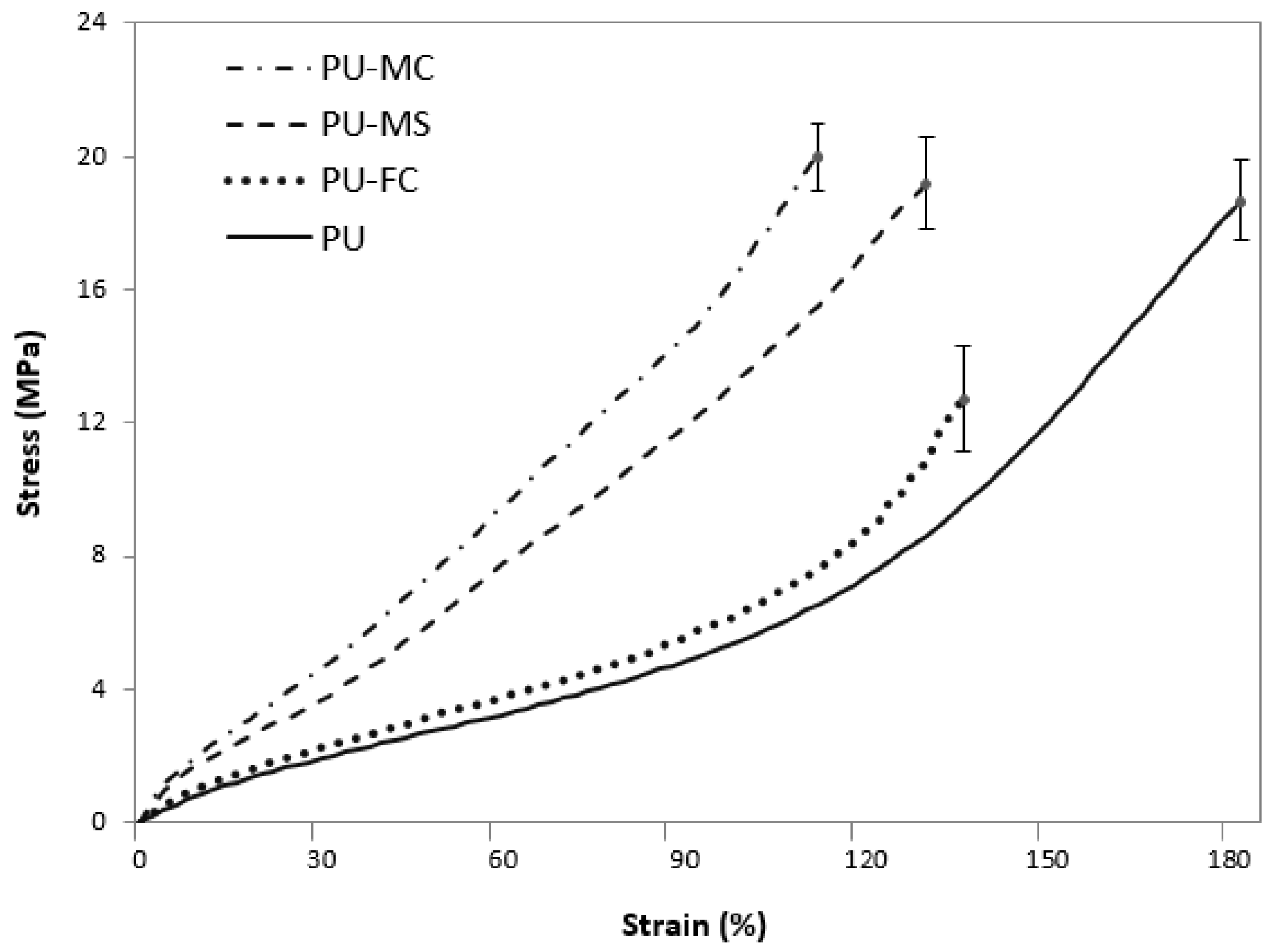
2.3. Mechanical Properties
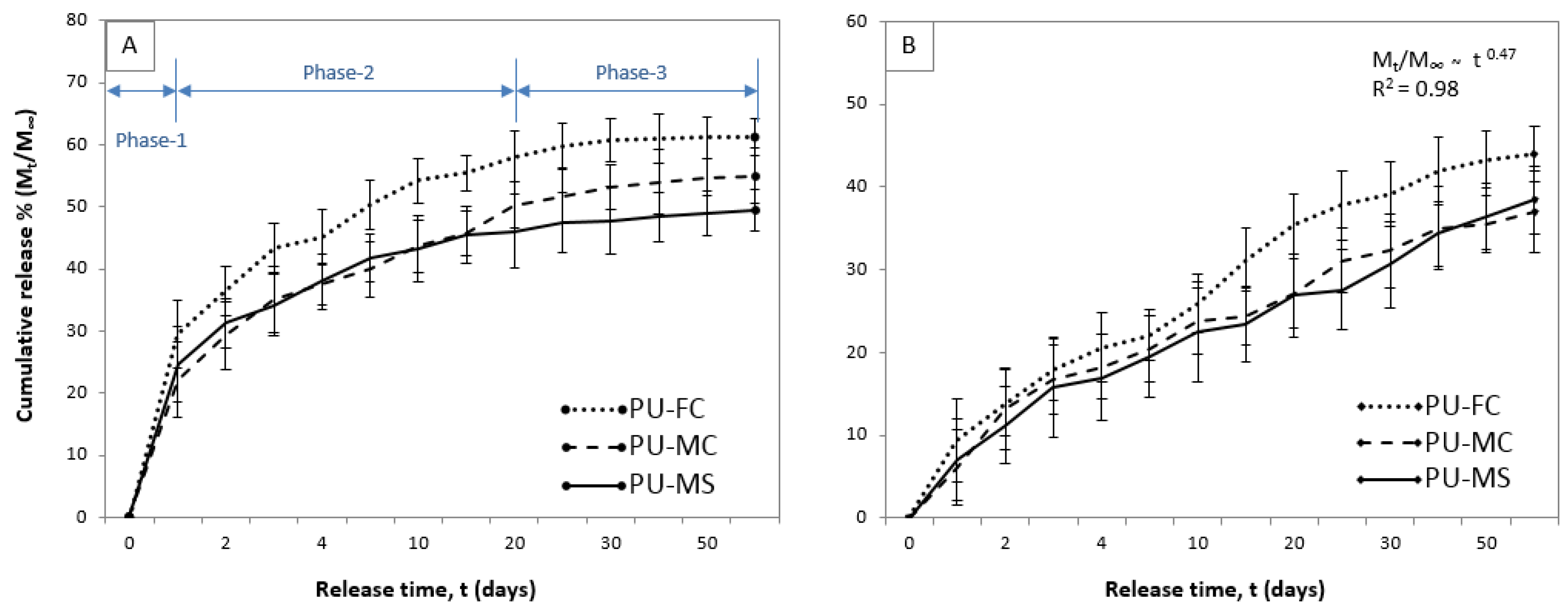
2.4. Metal Ion Release
2.5. Calcification
3. Materials and Methods
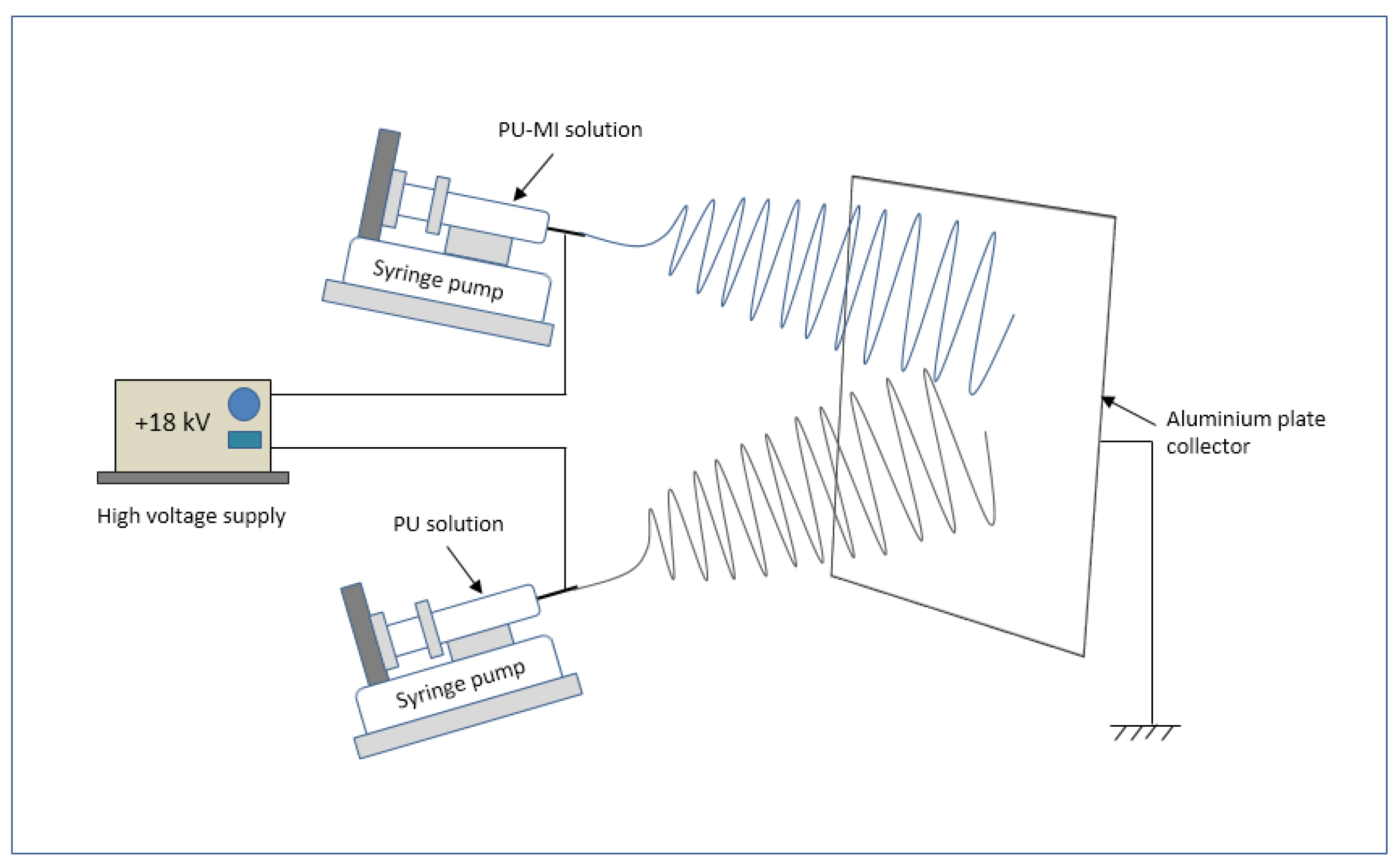
3.1. Electrospinning and MI Loading
3.2. Surface Morphology Analysis
3.3. Mechanical Characterisation
3.4. MI Release
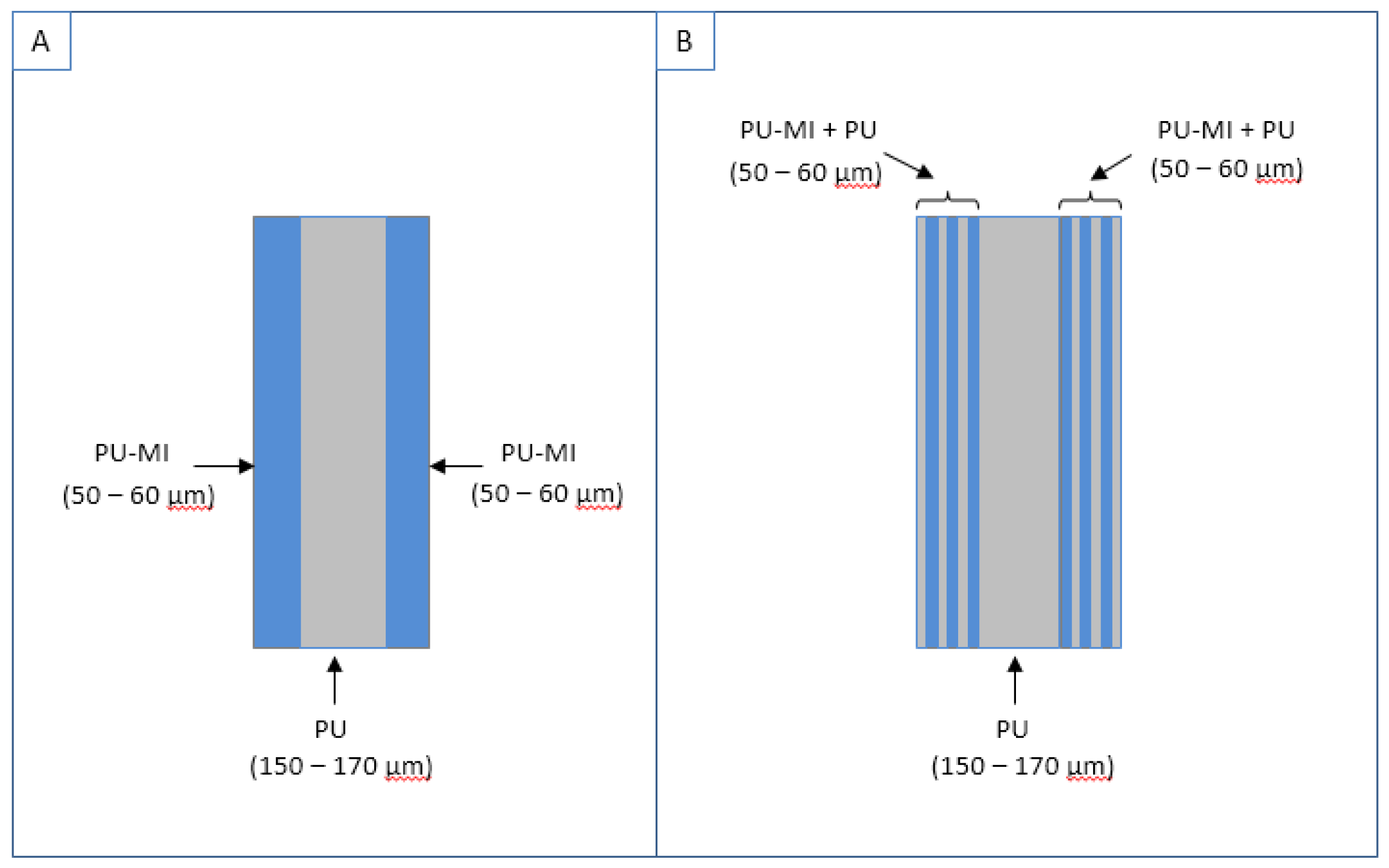
3.5. In-Vitro Calcification
3.5.1. Degree of Calcification
3.6. Infrared Spectroscopy Analysis
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Chandy, T.; Vasudev, S.C.; Sharma, C.P. Changes in polyurethane calcification due to antibiotics. Artif. Organs 1996, 20, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T. Determinants of Pathologic Mineralization. Curr. Opin. Rheumatol. 2008, 18, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Shanahan, C.M. Biology of calcification in vascular cells: Intima versus media. Herz 2001, 26, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Lord, R.S.A.; Warren, B.A. Calcified deposit formation in intimal thickenings of the human aorta. Atherosclerosis 1995, 118, 9–21. [Google Scholar] [CrossRef]
- Thoma, R.J.; Phillips, R.E. The role of material surface chemistry in implant device calcification: A hypothesis. J. Heart Valve Dis. 1995, 4, 214–221. [Google Scholar] [PubMed]
- Schoen, F.J.; Harasaki, H.; Kim, K.M.; Anderson, H.C.; Levy, R.J. Biomaterial-associated calcification: Pathology, mechanisms, and strategies for prevention. J. Biomed. Mater. Res. 1988, 22, 11–36. [Google Scholar] [PubMed]
- Schoen, F.J.; Levy, R.J. Chapter II.4.5—Pathological Calcification of Biomaterials. In Biomaterials Science, 3rd ed.; Buddy, D.R., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 739–754. [Google Scholar]
- Mehta, R.I.; Mukherjee, A.K.; Patterson, T.D.; Fishbein, M.C. Pathology of explanted polytetrafluoroethylene vascular grafts. Cardiovasc. Pathol. 2011, 20, 213–221. [Google Scholar] [CrossRef] [PubMed]
- De Valence, S.; Tille, J. C.; Mugnai, D.; Mrowczynski, W.; Gurny, R.; Möller, M.; Walpoth, B.H. Long term performance of polycaprolactone vascular grafts in a rat abdominal aorta replacement model. Biomaterials 2012, 33, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hayabuchi, Y.; Mori, K.; Kitagawa, T.; Sakata, M.; Kagami, S. Polytetrafluoroethylene graft calcification in patients with surgically repaired congenital heart disease: Evaluation using multidetector-row computed tomography. Am. Heart J. 2007, 153, 806.e801–806.e808. [Google Scholar] [CrossRef] [PubMed]
- Halwani, D.O.; Anderson, P.G.; Brott, B.C.; Anayiotos, A.S.; Lemons, J.E. The role of vascular calcification in inducing fatigue and fracture of coronary stents. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Marra, S.P.; Daghlian, C.P.; Fillinger, M.F.; Kennedy, F.E. Elemental composition, morphology and mechanical properties of calcified deposits obtained from abdominal aortic aneurysms. Acta Biomater. 2006, 2, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Nitschke, Y.; Terkeltaub, R. Genetics in Arterial Calcification: Pieces of a Puzzle and Cogs in a Wheel. Circ. Res. 2011, 109, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular Calcification Mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C. The pulsewave reflections model: Exploring for a fundamental mechanism of cardiovascular calcification. Med. Hypotheses 2007, 69, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, N.C. Mechanisms of inhibition of calcification. Clin. Orthop. Relat. Res. 1989, 247, 279–289. [Google Scholar]
- Mouriño, V.; Cattalini, J.P.; Boccaccini, A.R. Metallic ions as therapeutic agents in tissue engineering scaffolds: An overview of their biological applications and strategies for new developments. J. R. Soc. Health 2012, 9, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Khil, M.S.; Cha, D.I.; Kim, H.Y.; Kim, I.S.; Bhattarai, N. Electrospun Nanofibrous Polyurethane Membrane as Wound Dressing. J. Biomed. Mater. Res. 2003, 67, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Verreck, G.; Chun, I.; Rosenblatt, J.; Peeters, J.; Van Dijck, A.; Mensch, J.; Noppe, M.; Brewster, M.E. Incorporation of drugs in an amorphous state into electrospun nanofibers composed of a water-insoluble, nonbiodegradable polymer. J. Control. Release 2003, 92, 349–360. [Google Scholar] [CrossRef]
- Tonda-Turo, C.; Boffito, M.; Cassino, C.; Gentile, P.; Ciardelli, G. Biomimetic polyurethane—Based fibrous scaffolds. Mater. Lett. 2016, 167, 9–12. [Google Scholar] [CrossRef]
- Sartori, S.; Boffito, M.; Serafini, P.; Caporale, A.; Silvestri, A.; Bernardi, E.; Sassi, M.P.; Boccafoschi, F.; Ciardelli, G. Synthesis and structure–property relationship of polyester-urethanes and their evaluation for the regeneration of contractile tissues. React. Funct. Polym. 2013, 73, 1366–1376. [Google Scholar] [CrossRef]
- Hahn, A.; Brandes, G.; Wagener, P.; Barcikowski, S. Metal ion release kinetics from nanoparticle silicone composites. J. Control. Release 2011, 154, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Damm, C. Silver ion release from polymethyl methacrylate silver nanocomposites. Polym. Polym. Compos. 2005, 13, 649–656. [Google Scholar]
- Damm, C.; Münstedt, H. Kinetic aspects of the silver ion release from antimicrobial polyamide/silver nanocomposites. Appl. Phys. A 2008, 91, 479–486. [Google Scholar] [CrossRef]
- Reddy, M.M. Calcite growth-rate inhibition by fulvic acid and magnesium ion—Possible influence on biogenic calcite formation. J. Cryst. Growth 2012, 352, 151–154. [Google Scholar] [CrossRef]
- Rajendran, R.; Minqin, R.; Ronald, J.A.; Rutt, B.K.; Halliwell, B.; Watt, F. Does iron inhibit calcification during atherosclerosis? Free Radic. Biol. Med. 2012, 53, 1675–1679. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, S.C.; Chandy, T.; Mohanty, M.; Umasankar, P.R.; Sharma, C.P. Inhibition of bioprosthesis calcification due to synergistic effect of Fe/Mg ions to polyethylene glycol grafted bovine pericardium. J. Biomater. Appl. 2001, 16, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Sontia, B.; Touyz, R.M. Magnesium transport in hypertension. Pathophysiology 2007, 14, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Ueshima, K. Magnesium and ischemic heart disease: A review of epidemiological, experimental, and clinical evidences. Magnes. Res. 2005, 18, 275–284. [Google Scholar] [PubMed]
- Hartwig, A. Role of magnesium in genomic stability. Mutat. Res. 2001, 475, 113–121. [Google Scholar] [CrossRef]
- Laurant, P.; Touyz, R.M. Physiological and pathophysiological role of magnesium in the cardiovascular system: Implications in hypertension. J. Hypertens. 2000, 18, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Iseri, L.T.; French, J.H. Magnesium: Nature’s physiologic calcium blocker. Am. Heart J. 1984, 108, 188–193. [Google Scholar] [CrossRef]
- Gorgels, T.M.F.; Waarsing, J.; Wolf, A.; Brink, J.; Loves, W.P.; Bergen, A.B. Dietary magnesium, not calcium, prevents vascular calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2010, 88, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Schwille, P.O.; Schmiedl, A.; Schwille, R.; Brunner, P.; Kissler, H.; Cesnjevar, R.; Gepp, H. Media calcification, low erythrocyte magnesium, altered plasma magnesium, and calcium homeostasis following grafting of the thoracic aorta to the infrarenal aorta in the rat—Differential preventive effects of long-term oral magnesium supplementation alone and in combination with alkali. Biomed. Pharmacother. 2003, 57, 88–97. [Google Scholar] [PubMed]
- Ishimura, E.; Okuno, S.; Kitatani, K.; Tsuchida, T.; Yamakawa, T.; Shioi, A.; Inaba, M.; Nishizawa, Y. Significant association between the presence of peripheral vascular calcification and lower serum magnesium in hemodialysis patients. Clin. Nephrol. 2007, 68, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Esbaei, K.; Bargman, J.; Oreopoulos, D.G. Relationship between serum magnesium, parathyroid hormone, and vascular calcification in patients on dialysis: A literature review. Perit. Dial. Int. 2006, 26, 366–373. [Google Scholar] [PubMed]
- Meema, H.E.; Oreopoulos, D.G.; Rapoport, A. Serum magnesium level and arterial calcification in end-stage renal disease. Kidney Int. 1987, 32, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Turgut, F.; Kanbay, M.; Metin, M.R.; Uz, E.; Akcay, A.; Covic, A. Magnesium supplementation helps to improve carotid intima media thickness in patients on hemodialysis. Int. Urol. Nephrol. 2008, 40, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Tzanakis, I.; Pras, A.; Kounali, D.; Mamali, V.; Kartsonakis, V.; Mayopoulou-Symvoulidou, D.; Kallivretakis, N. Mitral annular calcifications in haemodialysis patients: A possible protective role of magnesium. Nephrol. Dial. Transplant. 1997, 12, 2036–2037. [Google Scholar] [CrossRef] [PubMed]
- Kircelli, F.; Peter, M.E.; Sevinc Ok, E.; Celenk, F.G.; Yilmaz, M.; Steppan, S.; Asci, G.; Ok, E.; Passlick-Deetjen, J. Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol. Dial. Transplant. 2012, 27, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, D.M. The role of magnesium binders in chronic kidney disease. Semin. Dial. 2007, 20, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Tzanakis, I.P.; Oreopoulos, D.G. Beneficial effects of magnesium in chronic renal failure: A foe no longer. Int. Urol. Nephrol. 2009, 41, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Altura, B.M.; Altura, B.T.; Gebrewold, A.; Ising, H.; Günther, T. Magnesium deficiency and hypertension: Correlation between magnesium-deficient diets and microcirculatory changes in situ. Science 1984, 223, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L.; Posner, A.S. Effect of magnesium on lipid-induced calcification: An in vitro model for bone mineralization. Calcif. Tissue Int. 1980, 32, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Termine, J.D.; Peckauskas, R.A.; Posner, A.S. Calcium phosphate formation in vitro: II. Effects of environment on amorphous-crystalline transformation. Arch. Biochem. Biophys. 1970, 140, 318–325. [Google Scholar] [CrossRef]
- Bennett, R.M.; Lehr, J.R.; McCarty, D.J. Factors affecting the solubility of calcium pyrophosphate dihydrate crystals. J. Clin. Invest. 1975, 56, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Salem, S.; Bruck, H.; Bahlmann, F.H.; Peter, M.; Passlick-Deetjen, J.; Kretschmer, A.; Steppan, S.; Volsek, M.; Kribben, A.; Nierhaus, M.; et al. Relationship between magnesium and clinical biomarkers on inhibition of vascular calcification. Am. J. Nephrol. 2012, 35, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Seymour, R.W.; Estes, G.M.; Cooper, S.L. Infrared Studies of Segmented Polyurethan Elastomers. I. Hydrogen Bonding. Macromolecules 1970, 3, 579–583. [Google Scholar] [CrossRef]
- Chan-Chan, L.H.; Solis-Correa, R.; Vargas-Coronado, R.F.; Cervantes-Uc, J.M.; Cauich-Rodríguez, J.V.; Quintana, P.; Bartolo-Pérez, P. Degradation studies on segmented polyurethanes prepared with HMDI, PCL and different chain extenders. Acta Biomater. 2010, 6, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sellitti, C.; Anderson, J.M.; Hiltner, A.; Lodoen, G.A.; Payet, C.R. An FTIR–ATR investigation of in vivo poly(ether urethane) degradation. J. Appl. Polym. Sci. 1992, 46, 201–211. [Google Scholar] [CrossRef]
- Hamon, R.F.; Khan, A.S.; Chow, A. The cation-chelation mechanism of metal-ion sorption by polyurethanes. Talanta 1982, 29, 313–326. [Google Scholar] [CrossRef]
- Francolini, I.; D'Ilario, L.; Guaglianone, E.; Donelli, G.; Martinelli, A.; Piozzi, A. Polyurethane anionomers containing metal ions with antimicrobial properties: Thermal, mechanical and biological characterization. Acta Biomater. 2010, 6, 3482–3490. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Parikh, J.; Engineer, C. Dexamethasone eluting biodegradable polymeric matrix coated stent for intravascular drug delivery. Chem. Eng. Res. Des. 2010, 88, 1479–1484. [Google Scholar] [CrossRef]
- Cherng, J.Y.; Hou, T.Y.; Shih, M.F.; Talsma, H.; Hennink, W.E. Polyurethane-based drug delivery systems. Int. J. Pharm. 2013, 450, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Modeling of diffusion controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Golomb, G.; Wagner, D. Characterization and anticalcification effects of implantable polyurethane matrices containing amorphous dispersion of bisphosphonic acid. Clin. Mater. 1991, 8, 33–42. [Google Scholar] [CrossRef]
- Gupta, K.M.; Pearce, S.M.; Poursaid, A.E.; Aliyar, H.A.; Tresco, P.A.; Mitchnik, M.A.; Kiser, P.F. Polyurethane intravaginal ring for controlled delivery of dapivirine, a nonnucleoside reverse transcriptase inhibitor of HIV-1. J. Pharm. Sci. 2008, 97, 4228–4239. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Gupta, K.M.; Fabian, J.; Albright, T.H.; Kiser, P.F. Segmented polyurethane intravaginal rings for the sustained combined delivery of antiretroviral agents dapivirine and tenofovir. Eur. J. Pharm. Sci. 2010, 39, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Yui, N.; Kataoka, K.; Sakurai, Y.; Katono, H.; Sanui, K.; Ogata, N. Novel Design of Microreservoir-Dispersed Matrices for Drug Delivery Formulations: Drug Release from Polybutadiene- and Poly (ethylene oxide)-Based Segmented Polyurethanes in Relation to Their Microdomain Structures. J. Bioact. Compat. Polym. 1988, 3, 106–125. [Google Scholar] [CrossRef]
- Golomb, G.; Wagner, D. Development of a new in vitro model for studying implantable polyurethane calcification. Biomaterials 1991, 12, 397–405. [Google Scholar] [CrossRef]
- Suh, S.W.; Han, D.K.; Kim, Y.H.; Min, B.G. Effect of Mechanical Deformation on in Vitro Calcification of Segmented Polyurethane. Seoul J. Med. 1994, 35, 275–280. [Google Scholar]
- Vasudev, S.C.; Chandy, T. Controlled release of ferric-magnesium ions from chitosan/polyethylene vinyl acetate comatrix for preventing pericardial calcification. Drug Deliv. 1999, 6, 117–126. [Google Scholar] [CrossRef]
- Chandy, T.; Kumar, B.A.; Sharma, C.P. Inhibition of in vitro calcium phosphate precipitation in presence of polyurethane via surface modification and drug delivery. J. Appl. Biomater. 1994, 5, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, H.; Kidane, A.G.; Burriesci, G.; Ramesh, B.; Darbyshire, A.; Seifalian, A.M. The anti-calcification potential of a silsesquioxane nanocomposite polymer under in vitro conditions: Potential material for synthetic leaflet heart valve. Acta Biomater. 2010, 6, 4249–4260. [Google Scholar] [PubMed]
- Park, J.C.; Hwang, Y.S.; Han, D.W.; Suh, H. A novel in vitro assessment of tissue valve calcification by a continuous flow type method. Artif. Organs 2000, 24, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Bernacca, G.M.; Mackay, T.G.; Wilkinson, R.; Wheatley, D.J. Calcification and fatigue failure in a polyurethane heart valve. Biomaterials 1995, 16, 279–285. [Google Scholar] [CrossRef]
- Nguyen, C.; Ea, H.K.; Thiaudiere, D.; Reguer, S.; Hannouche, D.; Daudon, M.; Liote, F.; Bazin, D. Calcifications in human osteoarthritic articular cartilage: Ex vivo assessment of calcium compounds using XANES spectroscopy. J. Synchrotron Radiat. 2011, 18, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.G.; Teoh, S.H.; McFarlane, W.; Poole-Warren, L.A.; Umezu, M. In vitro calcification of UHMWPE/PU composite membrane. Mater. Sci. Eng. C Mater. Biol. Appl. 2002, 20, 149–152. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, Z.; Hahn, C.; King, M.W.; Guidoin, R. Assessing the resistance to calcification of polyurethane membranes used in the manufacture of ventricles for a totally implantable artificial heart. J. Biomed. Mater. Res. 1999, 48, 648–659. [Google Scholar] [CrossRef]
- Coury, A.J.; Cahalan, P.T.; Schultz, E.L.; Stokes, K.B. In Vitro Aging of Implantable polyurethanes in Metal Ion Solutions. In Proceedings of the 2nd Transactions World Congress on Biomaterials, Washington, DC, USA, 27 April–1 May 1984. [Google Scholar]
- Stokes, K.B.; Berthelson, W.A.; Davis, M.W. Metal Catalyzed Oxidative Degradation of Implanted Polyurethane Devices. In Advances in Biomedical Polymers; Gebelein, C., Ed.; Springer: New York, NY, USA, 1987; pp. 159–169. [Google Scholar]
- Thoma, R.J.; Hung, T.Q.; Nyilas, E.; Haubold, A.D.; Phillips, R.E. Metal Ion Complexation of Poly(Ether)Urethanes. In Advances in Biomedical Polymers; Gebelein, C., Ed.; Springer: New York, NY, USA, 1987; pp. 131–145. [Google Scholar]
- Stokes, K.; Urbanski, P.; Upton, J. The in vivo auto-oxidation of polyether polyurethane by metal ions. J. Biomater. Sci. Polym. Ed. 1990, 1, 207–230. [Google Scholar] [PubMed]
- Hu, Y.J.; Wei, X.; Zhao, W.; Liu, Y.S.; Chen, G.Q. Biocompatibility of poly(3-hydroxybutyrate-co-3-hydroxyvalerate-co-3-hydroxyhexanoate) with bone marrow mesenchymal stem cells. Acta Biomater. 2009, 5, 1115–1125. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | GSM (gm/m2) | Thickness (μm) | Fibre Diameter (nm) | PU % (w/v) | Metal Salt | Metal Ion Content in Salt (w/w) | Metal Ion Content in PU (w/w) |
|---|---|---|---|---|---|---|---|
| Control PU | 130 ± 12 | 150 ± 10 | 820 ± 90 | 11 | - | - | - |
| PU-MC | 125 ± 11 | 140 ± 12 | 800 ± 105 | MgCl2 | 0.26 | 0.108 | |
| PU-MS | 134 ± 10 | 135 ± 12 | 795 ± 100 | MgSO4 | 0.2 | 0.109 | |
| PU-FC | 124 ± 15 | 145 ± 10 | 750 ± 180 | FeCl3 | 0.34 | 0.107 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, C.; Wang, X. Metal Ion-Loaded Nanofibre Matrices for Calcification Inhibition in Polyurethane Implants. J. Funct. Biomater. 2017, 8, 22. https://doi.org/10.3390/jfb8030022
Singh C, Wang X. Metal Ion-Loaded Nanofibre Matrices for Calcification Inhibition in Polyurethane Implants. Journal of Functional Biomaterials. 2017; 8(3):22. https://doi.org/10.3390/jfb8030022
Chicago/Turabian StyleSingh, Charanpreet, and Xungai Wang. 2017. "Metal Ion-Loaded Nanofibre Matrices for Calcification Inhibition in Polyurethane Implants" Journal of Functional Biomaterials 8, no. 3: 22. https://doi.org/10.3390/jfb8030022