Electrodeposition of Two-Dimensional Pt Nanostructures on Highly Oriented Pyrolytic Graphite (HOPG): The Effect of Evolved Hydrogen and Chloride Ions

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
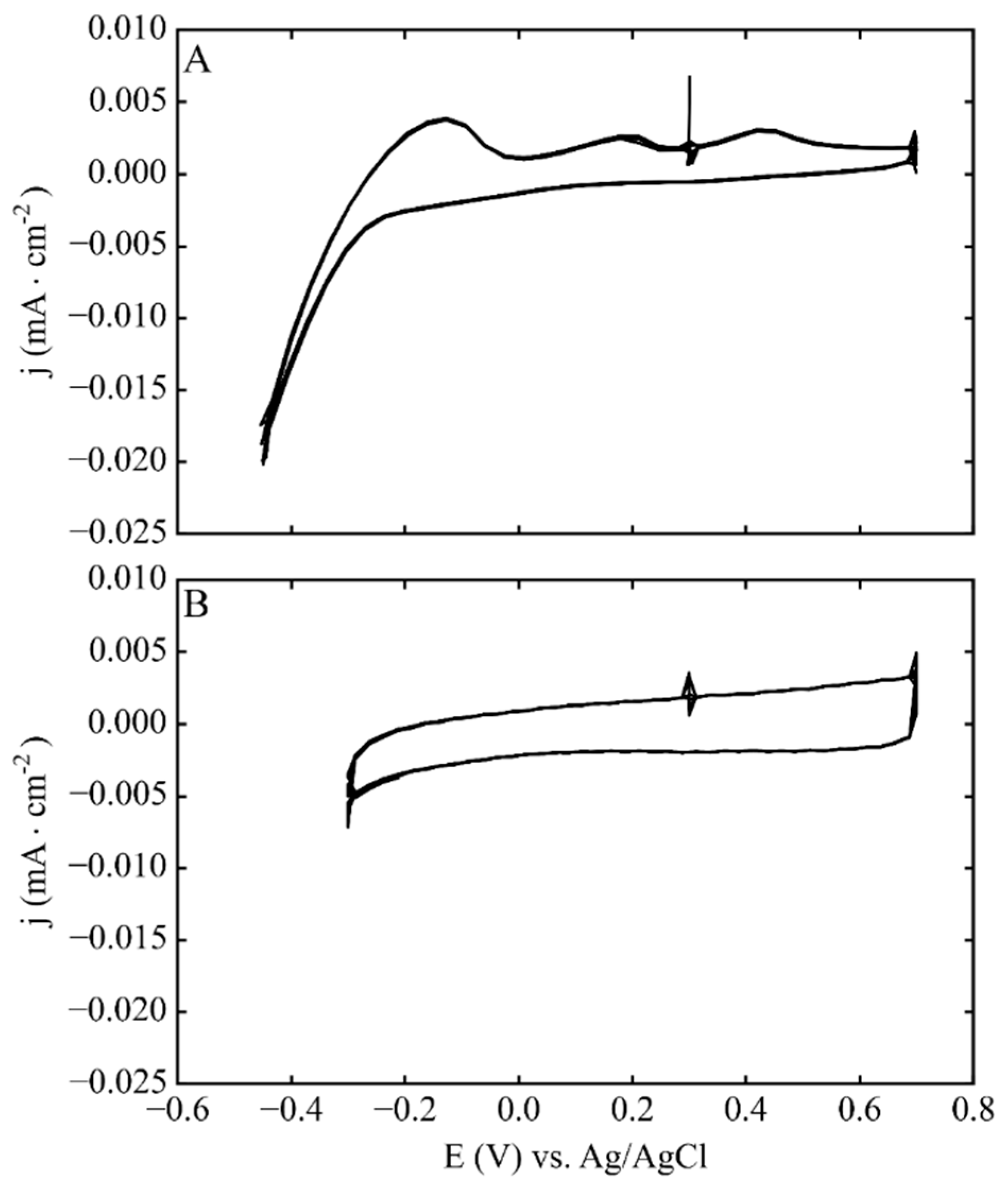{kind=link}
{kind=link}
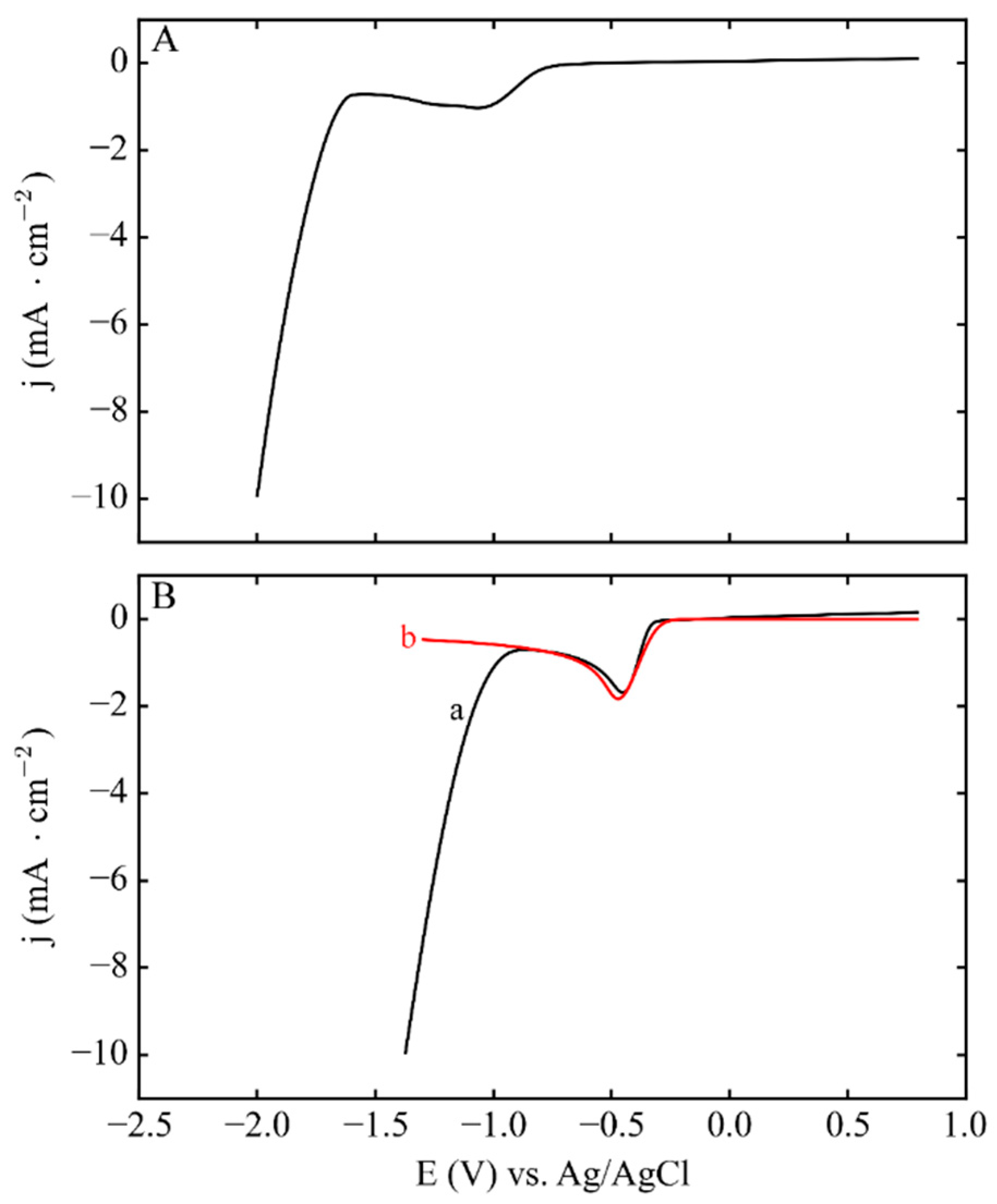{kind=link}
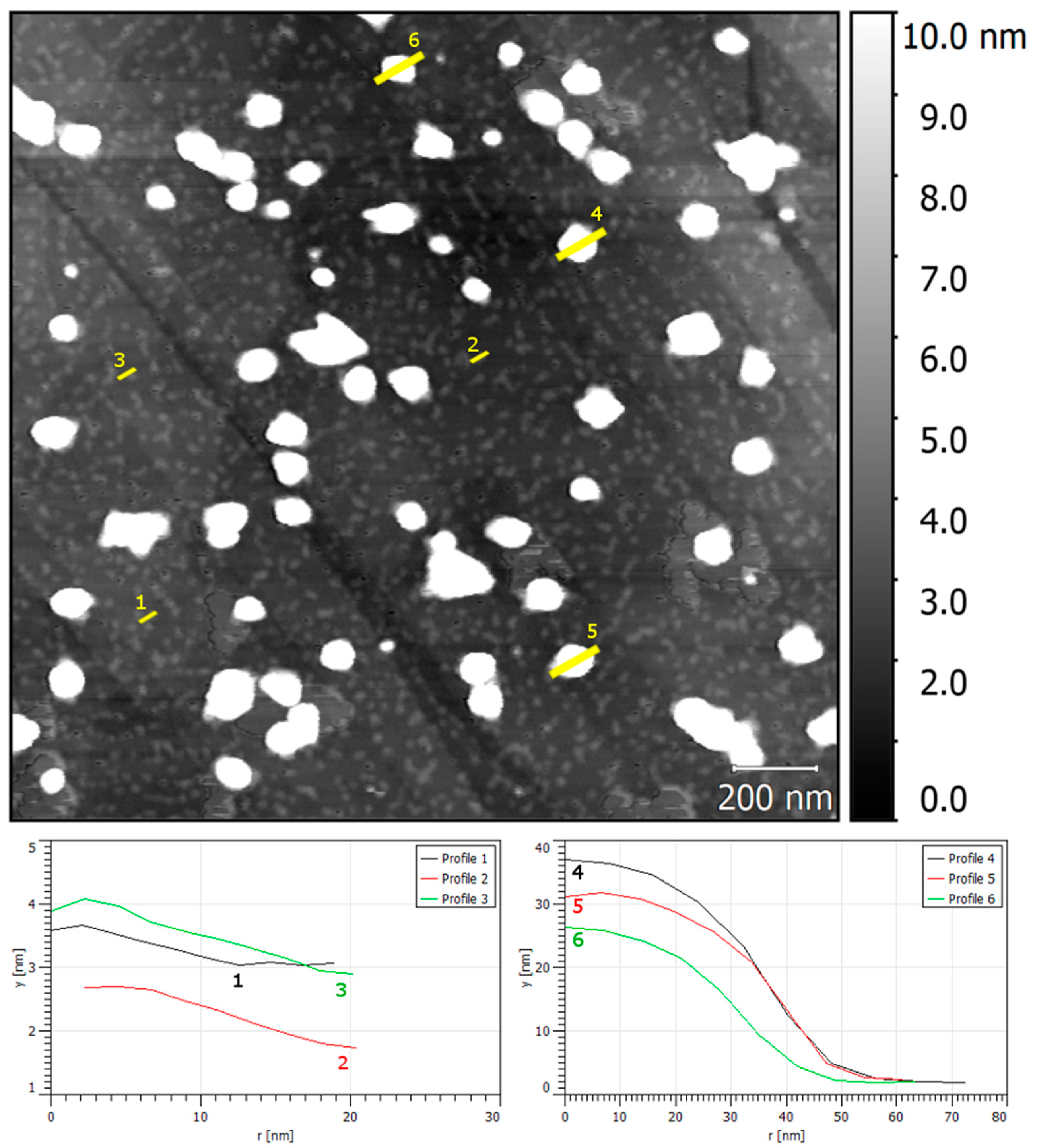{kind=link}
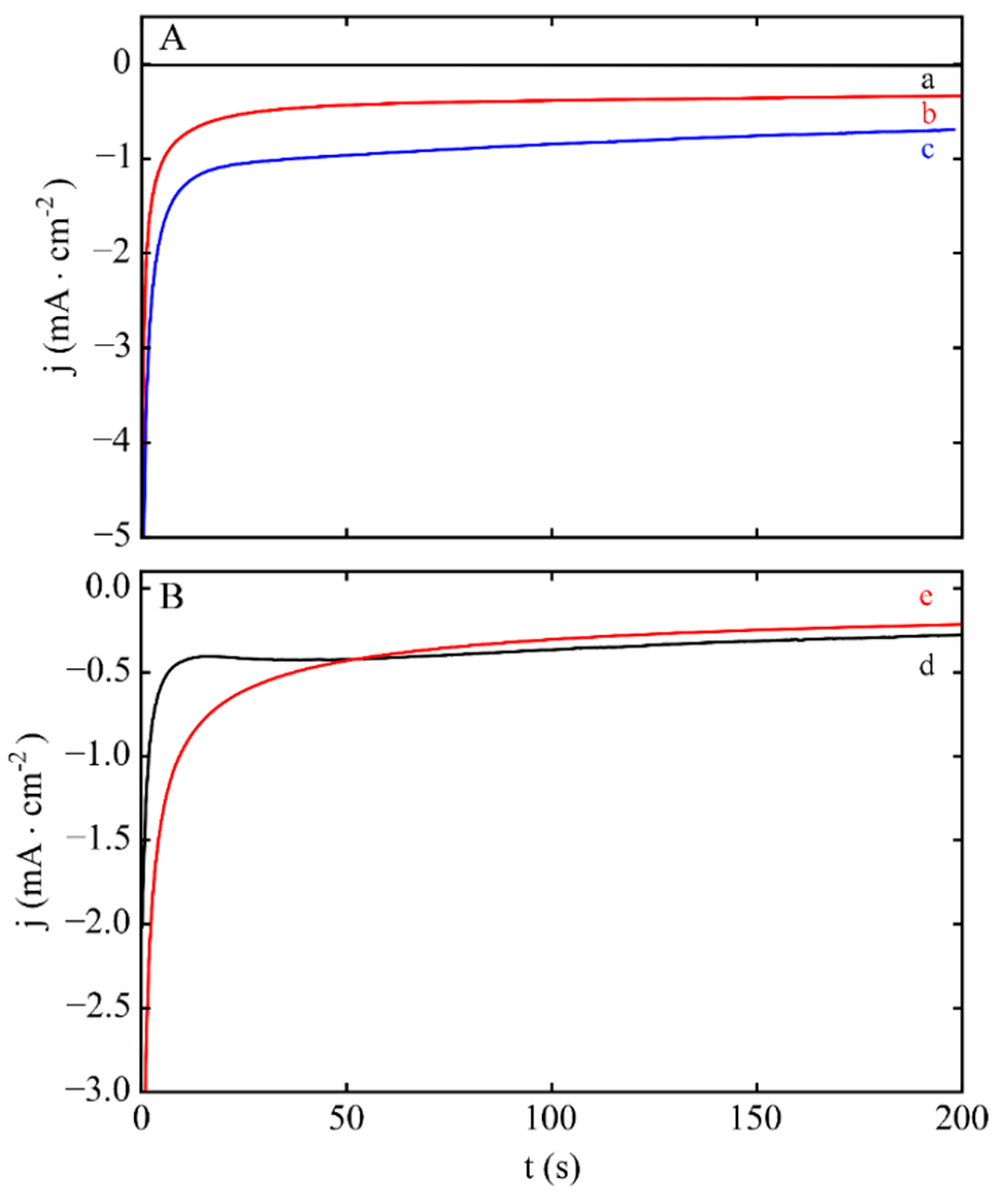{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
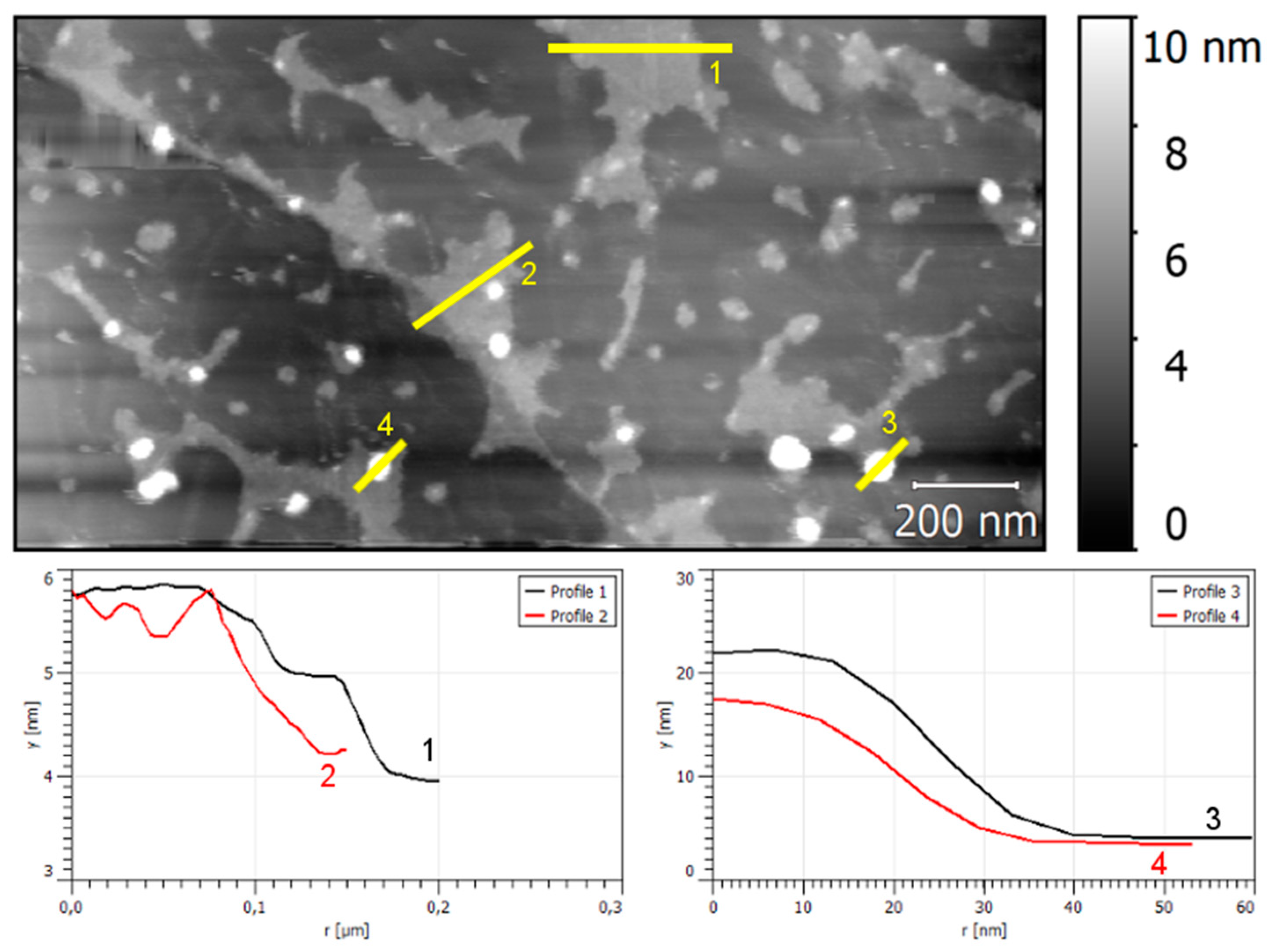
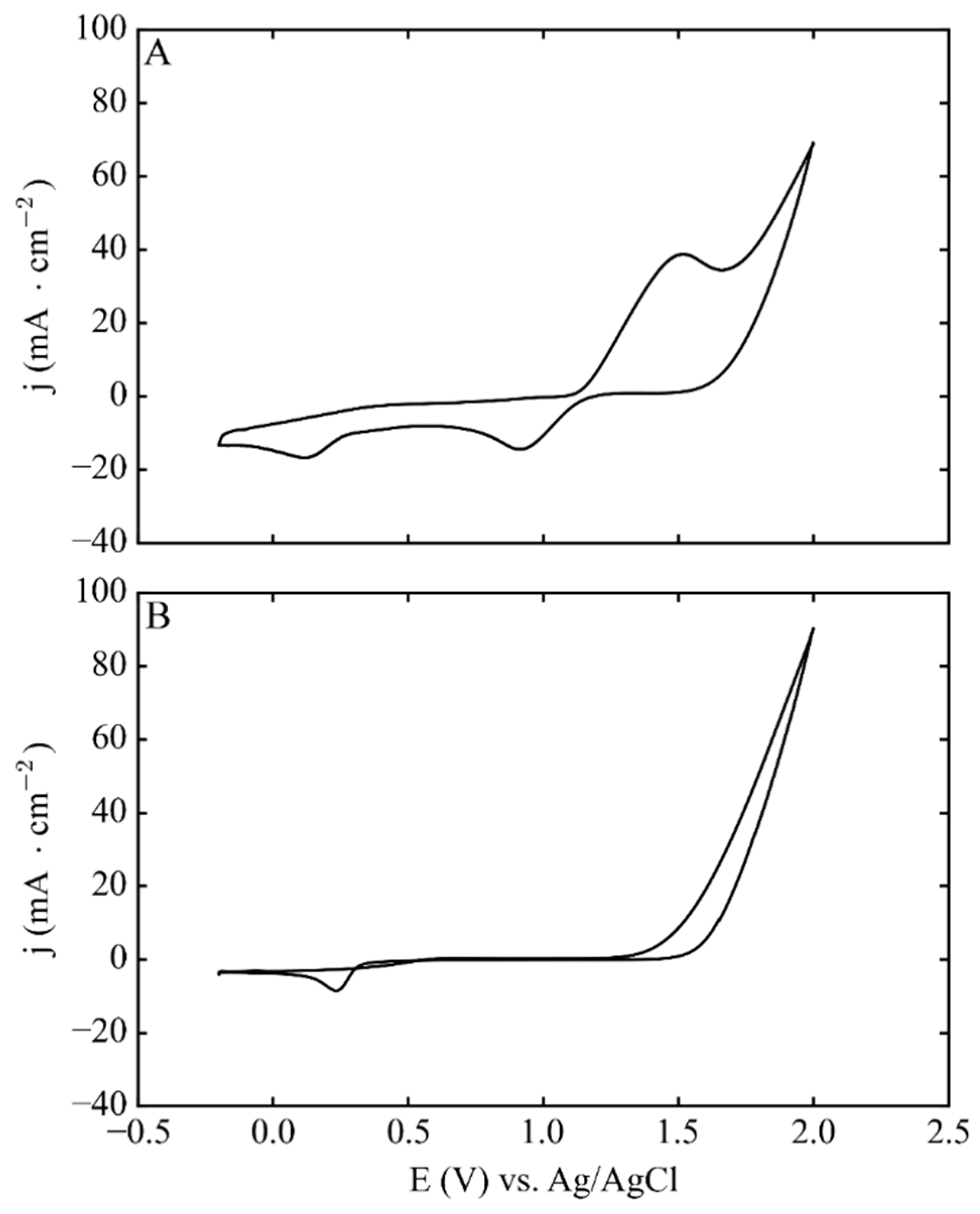
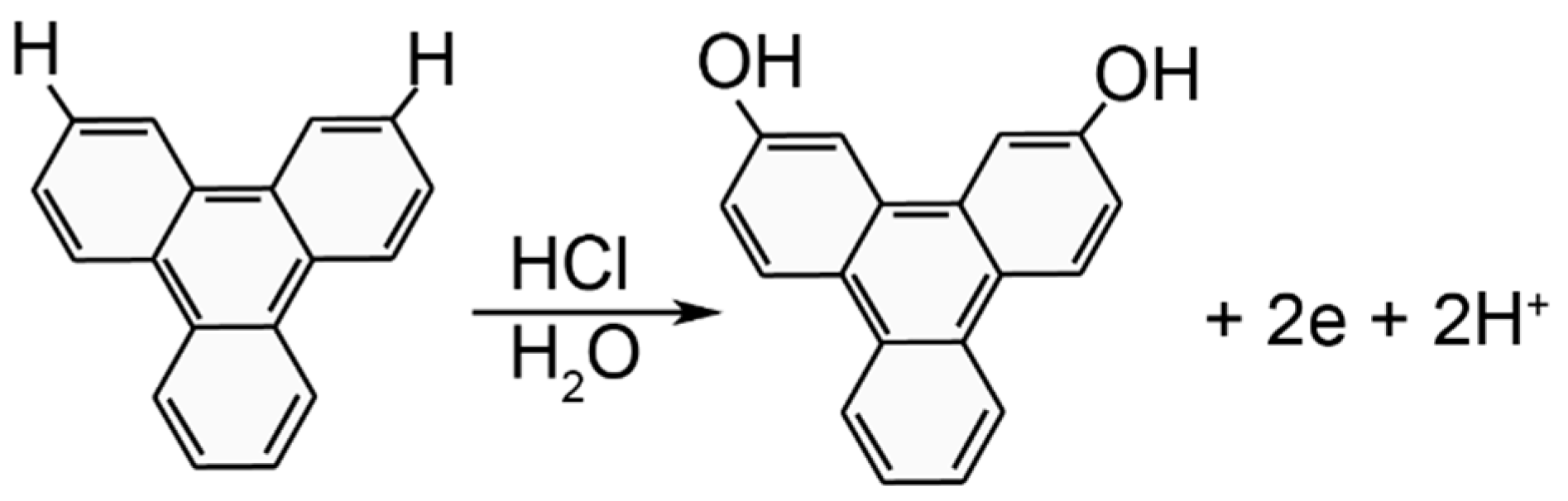
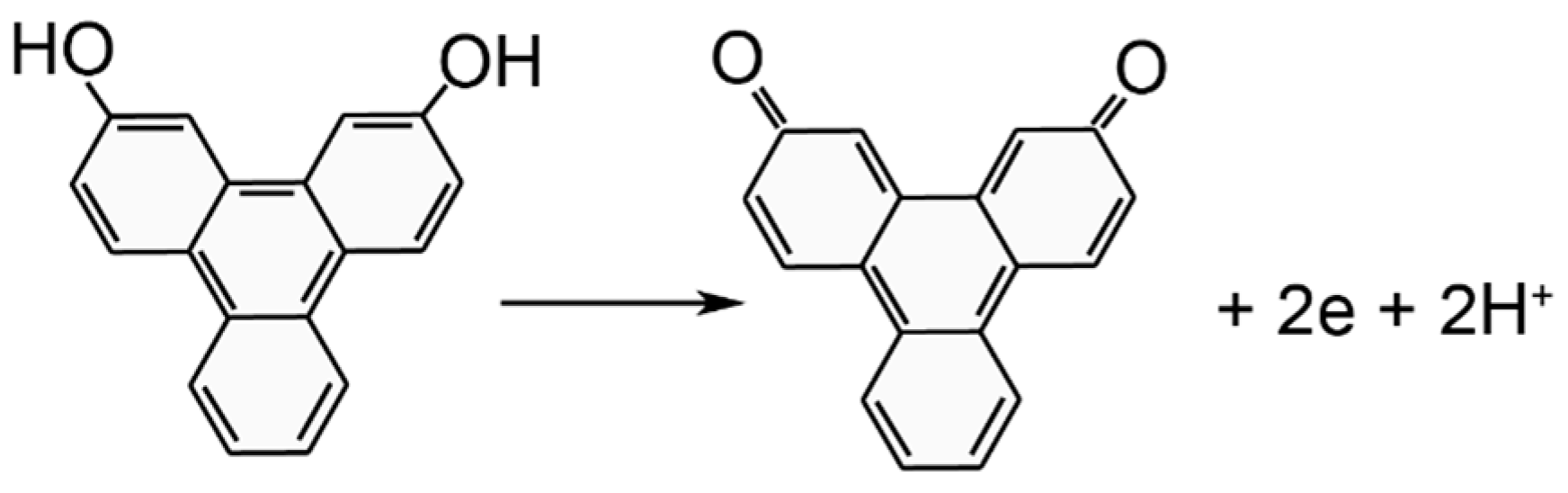
3. Results and Discussion
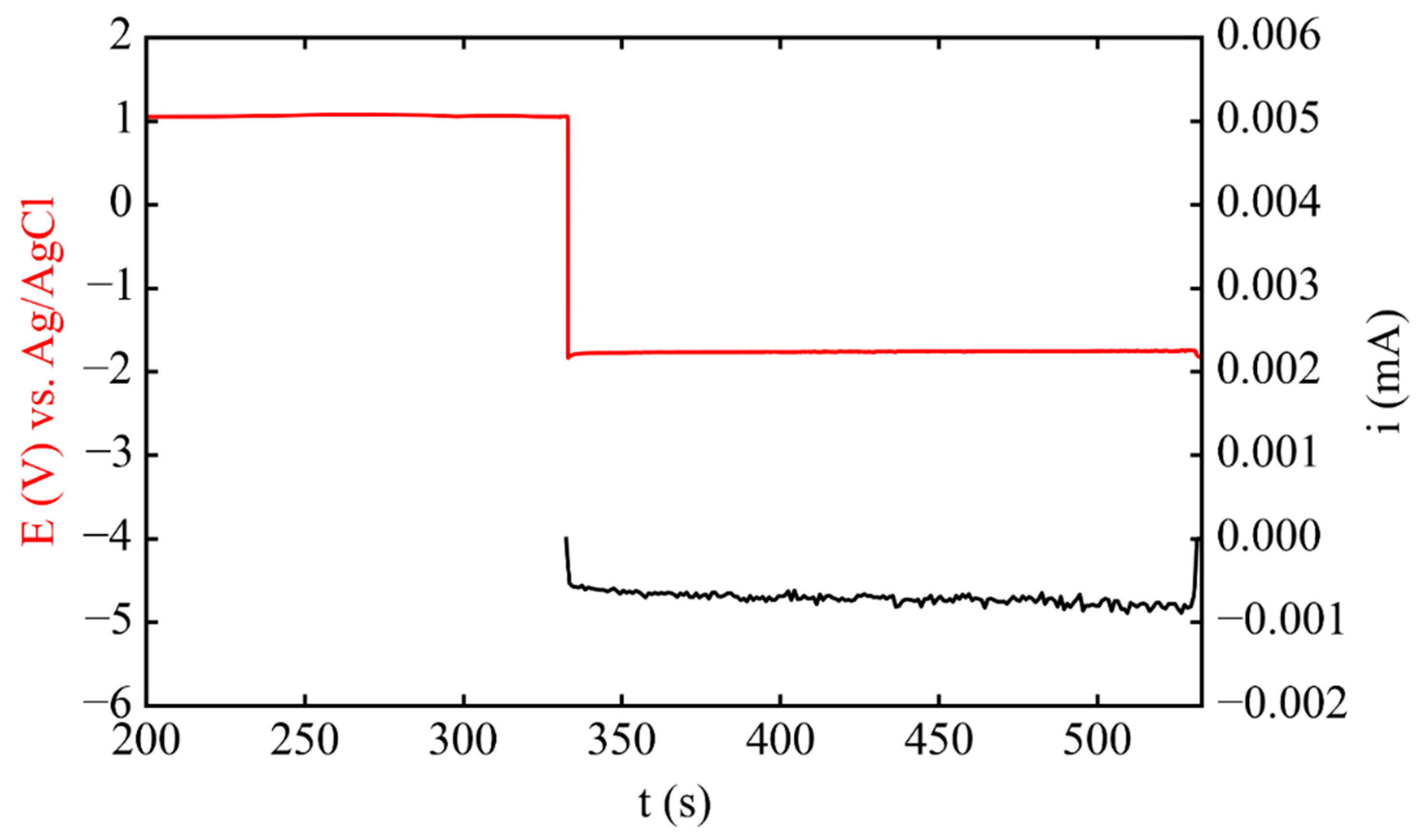
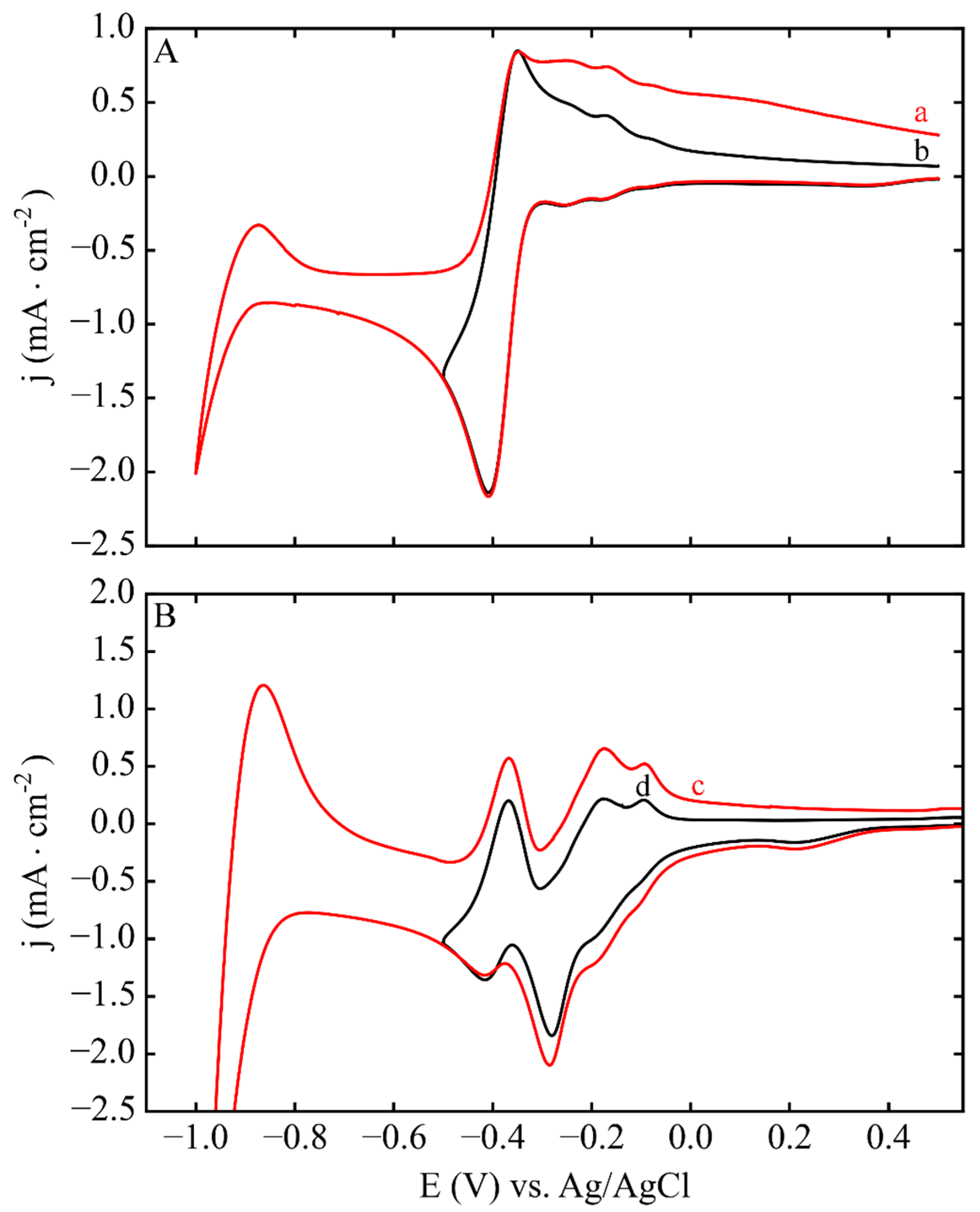
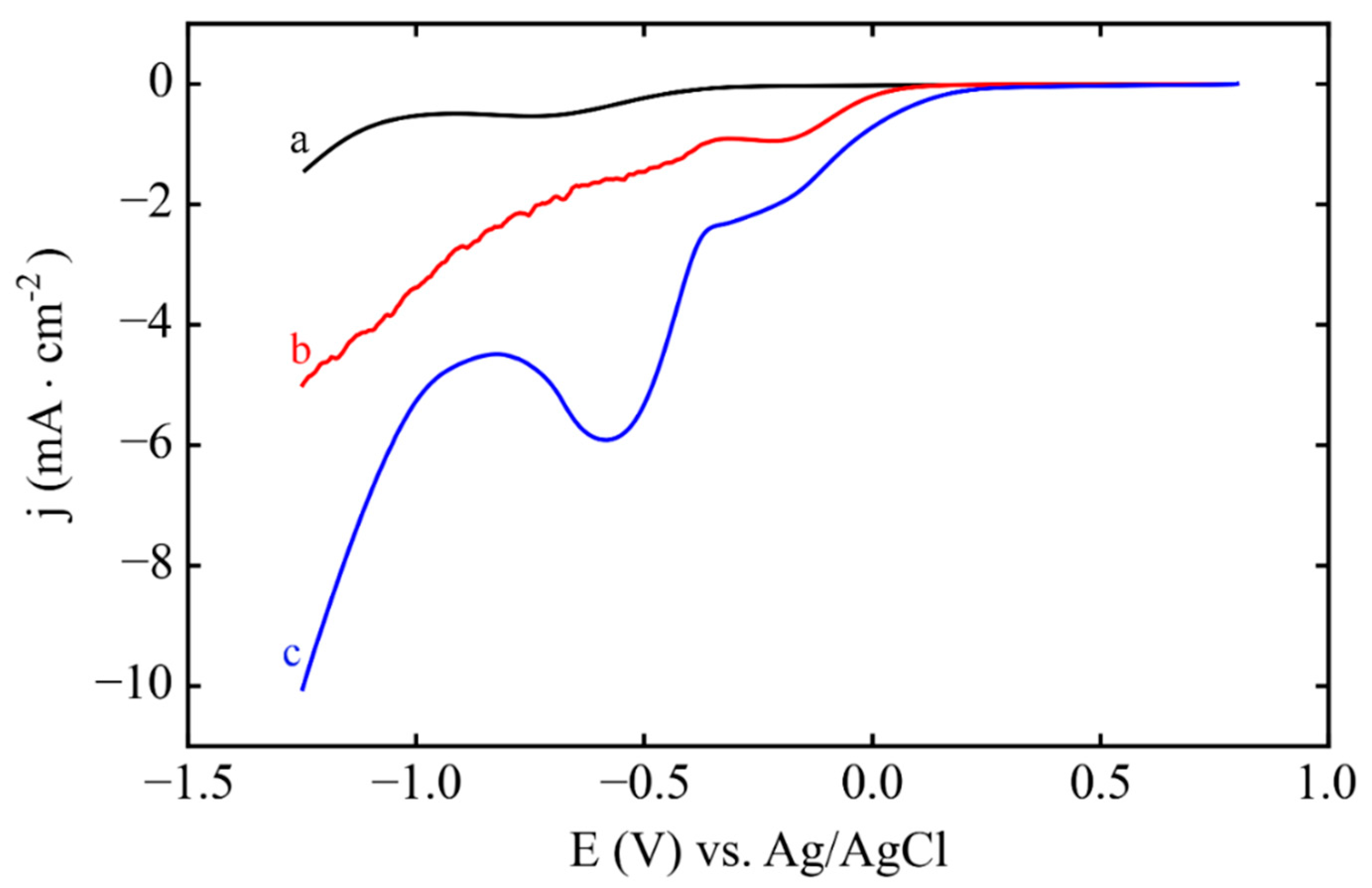
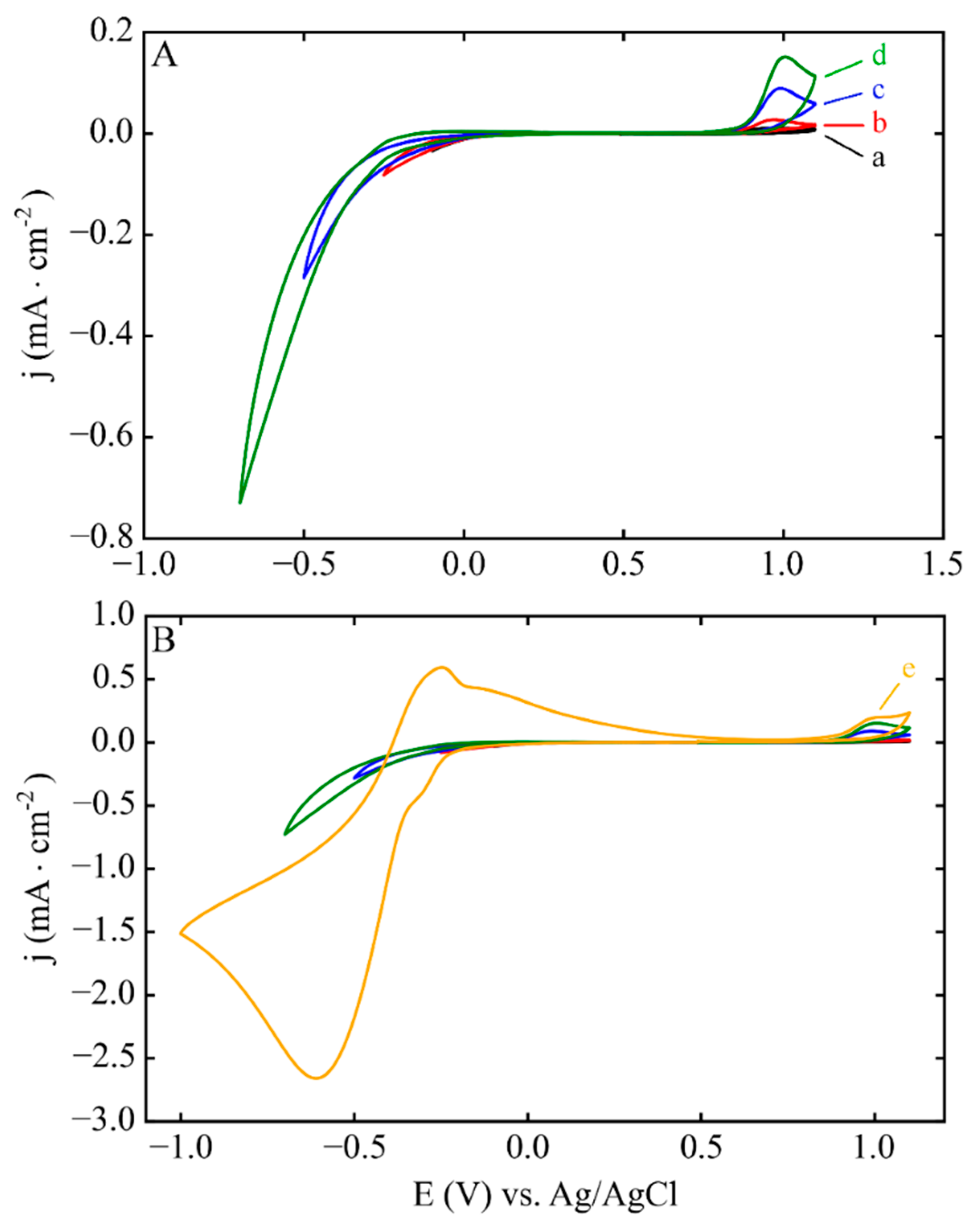
3.1. Electrodeposition Process Characterization
3.2. Phase 1: Electrode Immersion
3.3. Phase 2. Electrodeposition
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ercolano, G.; Cavaliere, S.; Rozière, J.; Jones, D.J. Recent developments in electrocatalyst design thrifting noble metals in fuel cells. Curr. Opin. Electrochem. 2018, 9, 271–277. [Google Scholar] [CrossRef]
- Lv, H.; Li, D.; Strmcnik, D.; Paulikas, A.P.; Markovic, N.M.; Stamenkovic, V.R. Recent advances in the design of tailored nanomaterials for efficient oxygen reduction reaction. Nano Energy 2016, 29, 149–165. [Google Scholar] [CrossRef]
- Cui, C.; Gan, L.; Heggen, M.; Rudi, S.; Strasser, P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 2013, 12, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Rudi, S.; Cui, C.; Heggen, M.; Strasser, P. Size-controlled synthesis of sub-10 nm PtNi3 alloy nanoparticles and their unusual volcano-shaped size effect on ORR electrocatalysis. Small 2016, 12, 3189–3196. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Miao, L.; Guo, W.; Yao, X.; Qin, F.; Wang, Z.; Du, H.; Li, J.; Kang, F.; Gan, L. Modulating surface composition and oxygen reduction reaction activities of Pt–Ni octahedral nanoparticles by microwave-enhanced surface diffusion during solvothermal synthesis. Chem. Mater. 2018, 30, 4355–4360. [Google Scholar] [CrossRef]
- Xiong, Y.; Xiao, L.; Yang, Y.; DiSalvo, F.J.; Abruña, H.D. High-loading intermetallic Pt3Co/C core-shell nanoparticles as enhanced activity electrocatalyst towards the oxygen reduction reaction (ORR). Chem. Mater. 2018, 30, 1532–1539. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Di Noto, V.; Negro, E.; Polizzi, S.; Vezzù, K.; Toniolo, L.; Cavinato, G. Synthesis, studies and fuel cell performance of “core-shell” electrocatalysts for oxygen reduction reaction based on a PtNix carbon nitride “shell” and a pyrolyzed polyketone nanoball “core”. Int. J. Hydrog. Energy 2014, 39, 2812–2827. [Google Scholar] [CrossRef]
- Gan, L.; Cui, C.; Rudi, S.; Strasser, P. Core-shell and nanoporous particle architectures and their effect on the activity and stability of Pt ORR electrocatalysts. Top. Catal. 2014, 57, 236–244. [Google Scholar] [CrossRef]
- Gong, H.; Cao, X.; Li, F.; Gong, Y.; Gu, L. PdAuCu nanobranch as self-repairing electrocatalyst for oxygen reduction reaction. ChemSusChem 2017, 10, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Ding, J.; Guo, S.; Zhang, X.; Su, D.; Zhu, X.; Yao, J.; Guo, J.; Lu, G.; Huang, X. A general method for multimetallic platinum alloy nanowires as highly active and stable oxygen reduction catalysts. Adv. Mater. 2015, 27, 7204–7212. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Guo, S.; Zhang, X.; Shen, X.; Su, D.; Lu, G.; Zhu, X.; Yao, J.; Guo, J.; Huang, X. Surface engineering of hierarchical platinum-cobalt nanowires for efficient electrocatalysis. Nat. Commun. 2016, 7, 11850. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, Z.; Cheng, T.; Fortunelli, A.; Chen, C.-Y.; Yu, R.; Zhang, Q.; Gu, L.; Merinov, B.V.; Lin, Z.; et al. Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction. Science 2016, 354, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.-W.; Cao, X.; Zhou, F.; Cui, C.-H.; Zhang, W.-J.; Yu, S.-H. A free-standing Pt-nanowire membrane as a highly stable electrocatalyst for the oxygen reduction reaction. Adv. Mater. 2011, 23, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Chen, W.; He, D.; Wan, J.; Pei, J.; Dong, J.; Wang, Y.; An, P.; Jin, Z.; Xing, W.; et al. Design of ultrathin Pt-Mo-Ni nanowire catalysts for ethanol electrooxidation. Sci. Adv. 2017, 3, e1603068. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Zhu, E.; Chen, Y.; Lin, Z.; Huang, X.; Duan, X.; Huang, Y. Biomimetic synthesis of an ultrathin platinum nanowire network with a high twin density for enhanced electrocatalytic activity and durability. Angew. Chem. Int. Ed. 2013, 52, 12577–12581. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xin, H.L.; Hovden, R.; Wang, H.; Yu, Y.; Muller, D.A.; DiSalvo, F.J.; Abruña, H.D. Structurally ordered intermetallic platinum–cobalt core–shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 2013, 12, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Alia, S.M.; Pylypenko, S.; Dameron, A.; Neyerlin, K.C.; Kocha, S.S.; Pivovar, B.S. Oxidation of platinum nickel nanowires to improve durability of oxygen-reducing electrocatalysts. J. Electrochem. Soc. 2016, 163, 296–301. [Google Scholar] [CrossRef]
- Ercolano, G.; Farina, F.; Cavaliere, S.; Jones, D.J.; Rozière, J. Towards ultrathin Pt films on nanofibres by surface-limited electrodeposition for electrocatalytic applications. J. Mater. Chem. A 2017, 5, 3974–3980. [Google Scholar] [CrossRef]
- Parsonage, E.E.; Debe, M.K. Nanostructured Electrode Membranes. U.S. Patent US5338430A, 16 August 1994. [Google Scholar]
- Alia, S.M.; Ngo, C.; Shulda, S.; Ha, M.A.; Dameron, A.A.; Weker, J.N.; Neyerlin, K.C.; Kocha, S.S.; Pylypenko, S.; Pivovar, B.S. Exceptional oxygen reduction reaction activity and durability of platinum-nickel nanowires through synthesis and post-treatment optimization. ACS Omega 2017, 2, 1408–1418. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Alia, S.M.; Yan, Y.S.; Pivovar, B.S. Galvanic displacement as a route to highly active and durable extended surface electrocatalysts. Catal. Sci. Technol. 2014, 4, 3589–3600. [Google Scholar] [CrossRef]
- Papaderakis, A.; Mintsouli, I.; Georgieva, J.; Sotiropoulos, S. Electrocatalysts prepared by ggalvanic replacement. Catalysts 2017, 7, 80. [Google Scholar] [CrossRef]
- Gruber, D.; Ponath, N.; Müller, J.; Lindstaedt, F. Sputter-deposited ultra-low catalyst loadings for PEM fuel cells. J. Power Sources 2005, 150, 67–72. [Google Scholar] [CrossRef]
- Milchev, A. Electrocrystallization: Fundamentals of Nucleation and Growth; Kluwer Academic Publishers: New York, NY, USA, 2002. [Google Scholar]
- Liu, Y.; Gokcen, D.; Bertocci, U.; Moffat, T.P. Self-terminating growth of platinum films by electrochemical deposition. Science 2012, 338, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; Farina, F.; Cavaliere, S.; Jones, D.J.; Rozière, J. Multilayer hierarchical nanofibrillar electrodes with tuneable lacunarity with 2D like Pt deposits for PEMFC. ECS Trans. 2017, 80, 757–762. [Google Scholar] [CrossRef]
- Farina, F.; Ercolano, G.; Cavaliere, S.; Jones, D.J.; Rozière, J. Surface-limited electrodeposition of continuous platinum networks on highly ordered pyrolytic graphite. Nanomaterials 2018. under review. [Google Scholar]
- Zoval, J.V.; Lee, J.; Gorer, S.; Penner, R.M. Electrochemical preparation of platinum nanocrystallites with size selectivity on basal plane oriented graphite surfaces. J. Phys. Chem. B 1998, 102, 1166–1175. [Google Scholar] [CrossRef]
- Lu, G.; Zangari, G. Electrodeposition of platinum on highly oriented pyrolyticgraphite. Part I: electrochemical characterization. J. Phys. Chem. B 2005, 109, 7998–8007. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zangari, G. Electrodeposition of platinum nanoparticles on highly oriented pyrolitic graphite: Part II: Morphological characterization by atomic force microscopy. Electrochim. Acta 2006, 51, 2531–2538. [Google Scholar] [CrossRef]
- Simonov, A.N.; Cherstiouk, O.V.; Vassiliev, S.Y.; Zaikovskii, V.I.; Filatov, A.Y.; Rudina, N.A.; Savinova, E.R.; Tsirlina, G.A. Potentiostatic electrodeposition of Pt on GC and on HOPG at low loadings: Analysis of the deposition transients and the structure of Pt deposits. Electrochim. Acta 2014, 150, 279–289. [Google Scholar] [CrossRef]
- Arroyo Gómez, J.J.; García Silvana, G. Spontaneous deposition of Pt-nanoparticles on HOPG surfaces. Surf. Interface Anal. 2015, 47, 1127–1131. [Google Scholar] [CrossRef]
- Shen, P.K.; Chi, N.; Chan, K.Y.; Phillips, D.L. Platinum nanoparticles spontaneously formed on HOPG. Appl. Surf. Sci. 2001, 172, 159–166. [Google Scholar] [CrossRef]
- Quaino, P.M.; Gennero de Chialvo, M.R.; Vela, M.E.; Salvarezza, R.C. Self-assembly of platinum nanowires on HOPG. J. Argent. Chem. Soc. 2005, 93, 215–224. [Google Scholar]
- Juarez, M.F.; Fuentes, S.; Soldano, G.J.; Avalle, L.; Santos, E. Spontaneous formation of metallic nanostructures on highly oriented pyrolytic graphite (HOPG): An ab initio and experimental study. Faraday Discuss. 2014, 172, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Bowden, B.; Davies, M.; Davies, P.R.; Guan, S.; Morgan, D.J.; Roberts, V.; Wotton, D. The deposition of metal nanoparticles on carbon surfaces: The role of specific functional groups. Faraday Discuss. 2018. [Google Scholar] [CrossRef] [PubMed]
- Buono, C.; Davies, P.R.; Davies, R.J.; Jones, T.; Kulhavý, J.; Lewis, R.; Morgan, D.J.; Robinson, N.; Willock, D.J. Spectroscopic and atomic force studies of the functionalisation of carbon surfaces: New insights into the role of the surface topography and specific chemical states. Faraday Discuss. 2014, 173, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.; Buono, C.; Davies, P.R.; Davies, R.J.; Legge, T.; Lai, A.; Lewis, R.; Morgan, D.J.; Robinson, N.; Willock, D.J. The functionalisation of graphite surfaces with nitric acid: Identification of functional groups and their effects on gold deposition. J. Catal. 2015, 323, 10–18. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; John Wiley and Sons: New York, NY, USA, 2001; p. 833. [Google Scholar]
- Zhou, J.; Zu, Y.; Bard, A.J. Scanning electrochemical microscopy: Part 39. The proton/hydrogen mediator system and its application to the study of the electrocatalysis of hydrogen oxidation. J. Electroanal. Chem. 2000, 491, 22–29. [Google Scholar] [CrossRef]
- Himmelblau, D.M. Diffusion of dissolved gases in liquids. Chem. Rev. 1964, 64, 527–550. [Google Scholar] [CrossRef]
- Colom, F., II. Palladium and platinum. In Standard Potentials in Aqueous Solution; Bard, A.J., Parsons, R., Eds.; IUPAC-Marcel Dekker, Inc.: New York, NY, USA, 1985. [Google Scholar]
- Ginstrup, O. The redox system platinum(0)/platinum(II)/platinum(IV) with chloro and bromo ligands. Acta Chem. Scand. 1972, 26, 1527–1541. [Google Scholar] [CrossRef]
- Uosaki, K.; Ye, S.; Naohara, H.; Oda, Y.; Haba, T.; Kondo, T. Electrochemical epitaxial growth of a Pt(111) phase on an Au(111) electrode. J. Phys. Chem. B 1997, 101, 7566–7572. [Google Scholar] [CrossRef]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpuche-Aviles, M.A.; Farina, F.; Ercolano, G.; Subedi, P.; Cavaliere, S.; Jones, D.J.; Rozière, J. Electrodeposition of Two-Dimensional Pt Nanostructures on Highly Oriented Pyrolytic Graphite (HOPG): The Effect of Evolved Hydrogen and Chloride Ions. Nanomaterials 2018, 8, 668. https://doi.org/10.3390/nano8090668
Alpuche-Aviles MA, Farina F, Ercolano G, Subedi P, Cavaliere S, Jones DJ, Rozière J. Electrodeposition of Two-Dimensional Pt Nanostructures on Highly Oriented Pyrolytic Graphite (HOPG): The Effect of Evolved Hydrogen and Chloride Ions. Nanomaterials. 2018; 8(9):668. https://doi.org/10.3390/nano8090668
Chicago/Turabian StyleAlpuche-Aviles, Mario A., Filippo Farina, Giorgio Ercolano, Pradeep Subedi, Sara Cavaliere, Deborah J. Jones, and Jacques Rozière. 2018. "Electrodeposition of Two-Dimensional Pt Nanostructures on Highly Oriented Pyrolytic Graphite (HOPG): The Effect of Evolved Hydrogen and Chloride Ions" Nanomaterials 8, no. 9: 668. https://doi.org/10.3390/nano8090668
APA StyleAlpuche-Aviles, M. A., Farina, F., Ercolano, G., Subedi, P., Cavaliere, S., Jones, D. J., & Rozière, J. (2018). Electrodeposition of Two-Dimensional Pt Nanostructures on Highly Oriented Pyrolytic Graphite (HOPG): The Effect of Evolved Hydrogen and Chloride Ions. Nanomaterials, 8(9), 668. https://doi.org/10.3390/nano8090668