Metabolite Identification through Machine Learning— Tackling CASMI Challenge Using FingerID

Abstract
:
1. Introduction
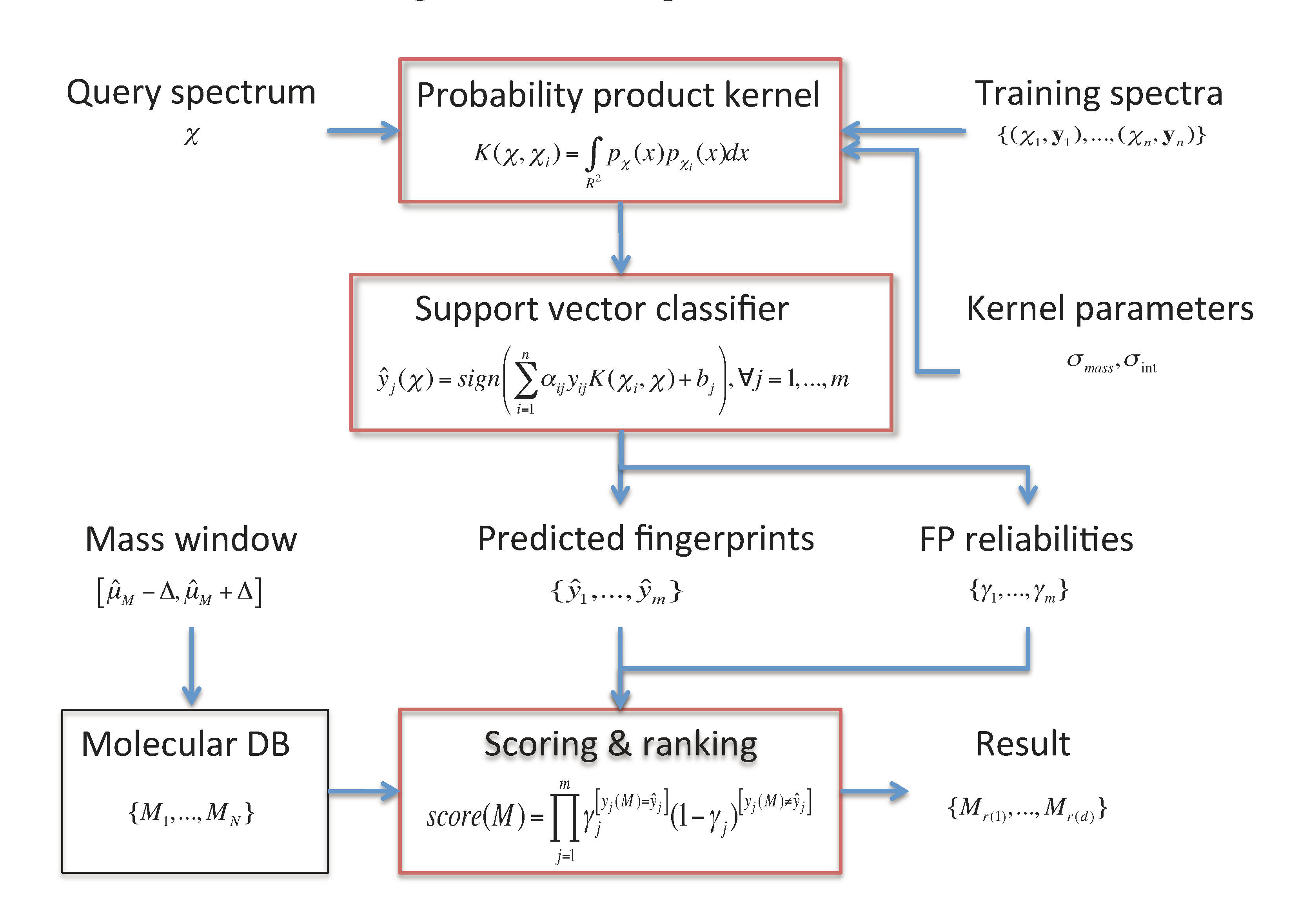
2. Metabolite Identification through FingerID
- A molecular fingerprint prediction module that relies on support vector machines (SVM) equipped with a probability product kernel representation of mass spectra.
- A molecule scoring and ranking module uses the predicted fingerprints to retrieve the best matching candidate molecules from a molecular database such as KEGG or PubChem.
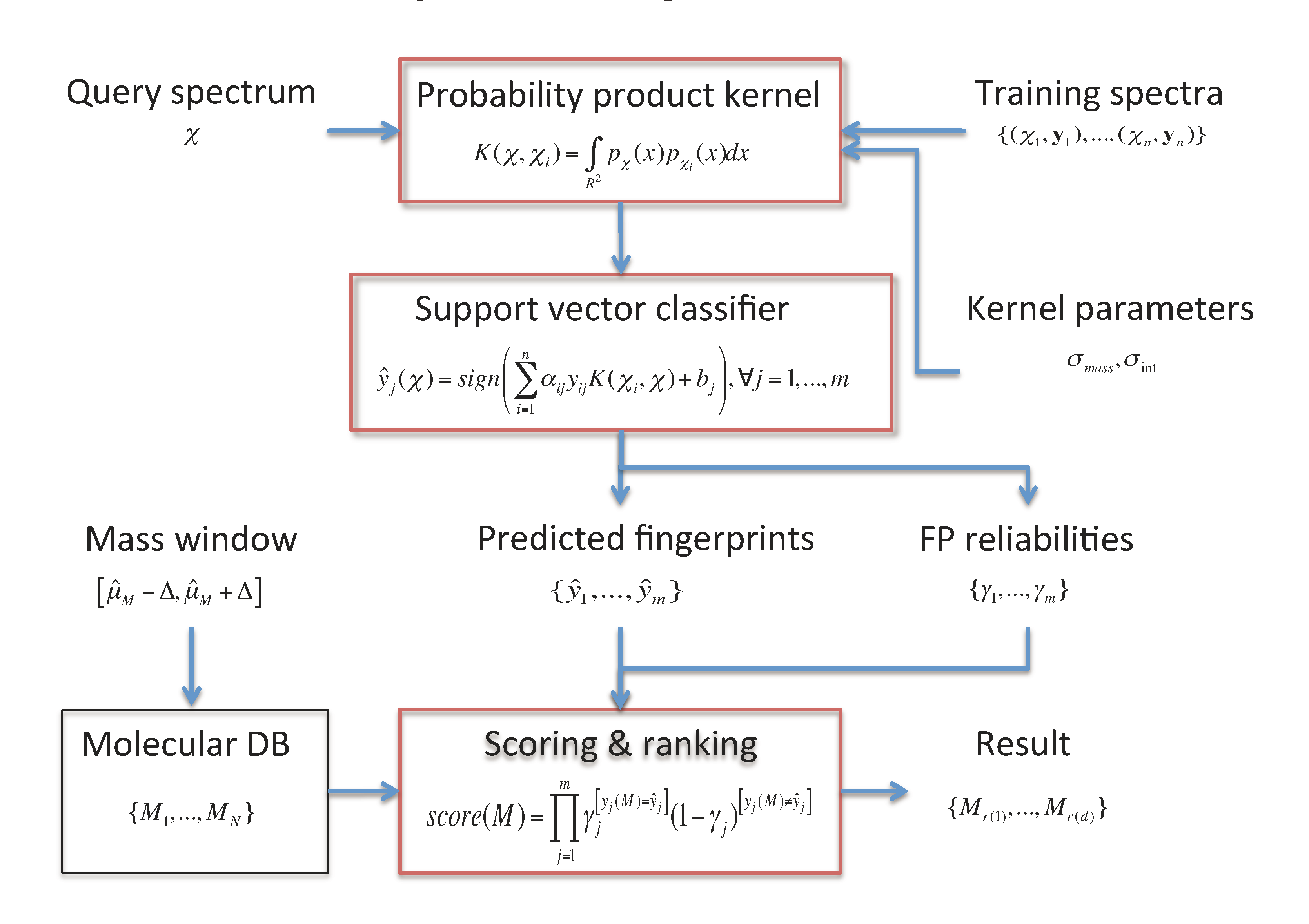
2.1. Fingerprint Prediction through SVM
 , (k = 1,...,lχ) consisting of the peak mass µ(k) and the normalized peak intensity ι(k). Our goal is to learn a mapping between the mass spectra χ ∈
, (k = 1,...,lχ) consisting of the peak mass µ(k) and the normalized peak intensity ι(k). Our goal is to learn a mapping between the mass spectra χ ∈  and a set of m molecular fingerprints
and a set of m molecular fingerprints  . The fingerprints encode molecular properties with the value yj = +1 denoting the presence of jth property in the corresponding molecule. →{+1, −1}m using support vector machines (SVM) [17]. For each fingerprint, a separate SVM model
. The fingerprints encode molecular properties with the value yj = +1 denoting the presence of jth property in the corresponding molecule. →{+1, −1}m using support vector machines (SVM) [17]. For each fingerprint, a separate SVM model

2.2. Probability Product Kernel
 centered around the peak measurement and with covariance shared with all peaks
centered around the peak measurement and with covariance shared with all peaks

 and
and  for the mass and the intensity, respectively, are both estimated from data and no covariance is assumed between them. The spectrum χ is finally represented as a mixture of its peak distributions
for the mass and the intensity, respectively, are both estimated from data and no covariance is assumed between them. The spectrum χ is finally represented as a mixture of its peak distributions  .
.
- Mass loss kernel Kmloss records the difference between a fragment peak and the theoretical precursor peak by centering a Gaussian at the difference, giving the probabilitywhere χ̃ = (µprec, ι(k)) is a dummy peak with the precursor mass and the same intensity as the peak χ(k). This kernel can be interpreted as capturing putative cleaved fragments or combinations of them.
![Metabolites 03 00484 i011]()
- Mass difference kernel Kdiff computes the difference of all pairs of peaks and centers the Gaussian at the peak difference
![Metabolites 03 00484 i012]() . This kernel can be seen as a generalization of the mass loss kernel by not fixing a precursor mass but instead recording all possible fragmentation reactions between the peaks of two mass spectra. The kernel computation has quadratically higher complexity compared with the other two variants.
. This kernel can be seen as a generalization of the mass loss kernel by not fixing a precursor mass but instead recording all possible fragmentation reactions between the peaks of two mass spectra. The kernel computation has quadratically higher complexity compared with the other two variants.


2.3. Candidate Retrieval
 of the fingerprints as the reliability scores. Given the reliability scores and the predicted fingerprints ŷ, the model assigns the Poisson-binomial probability for the fingerprint vector y as follows:
of the fingerprints as the reliability scores. Given the reliability scores and the predicted fingerprints ŷ, the model assigns the Poisson-binomial probability for the fingerprint vector y as follows:


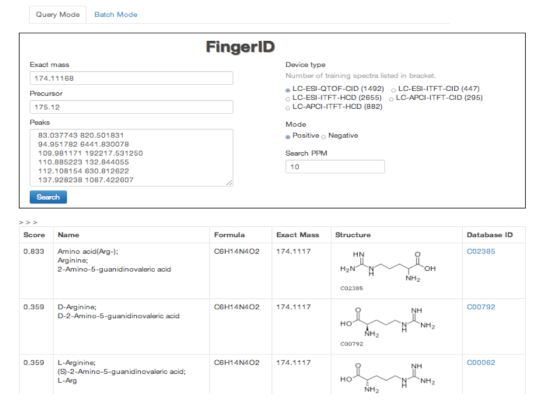
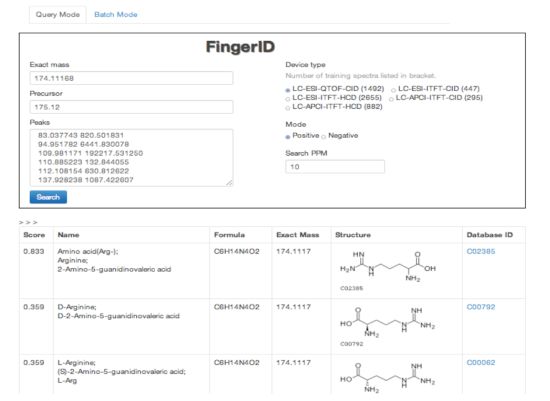
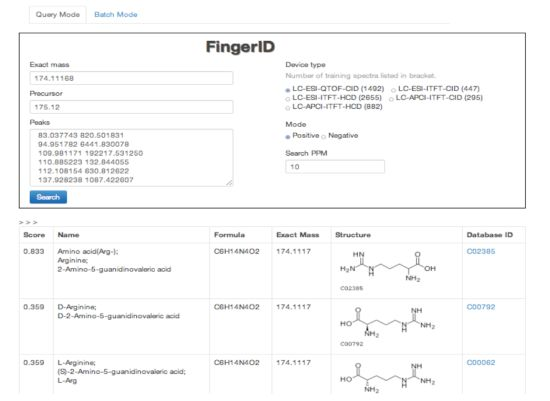
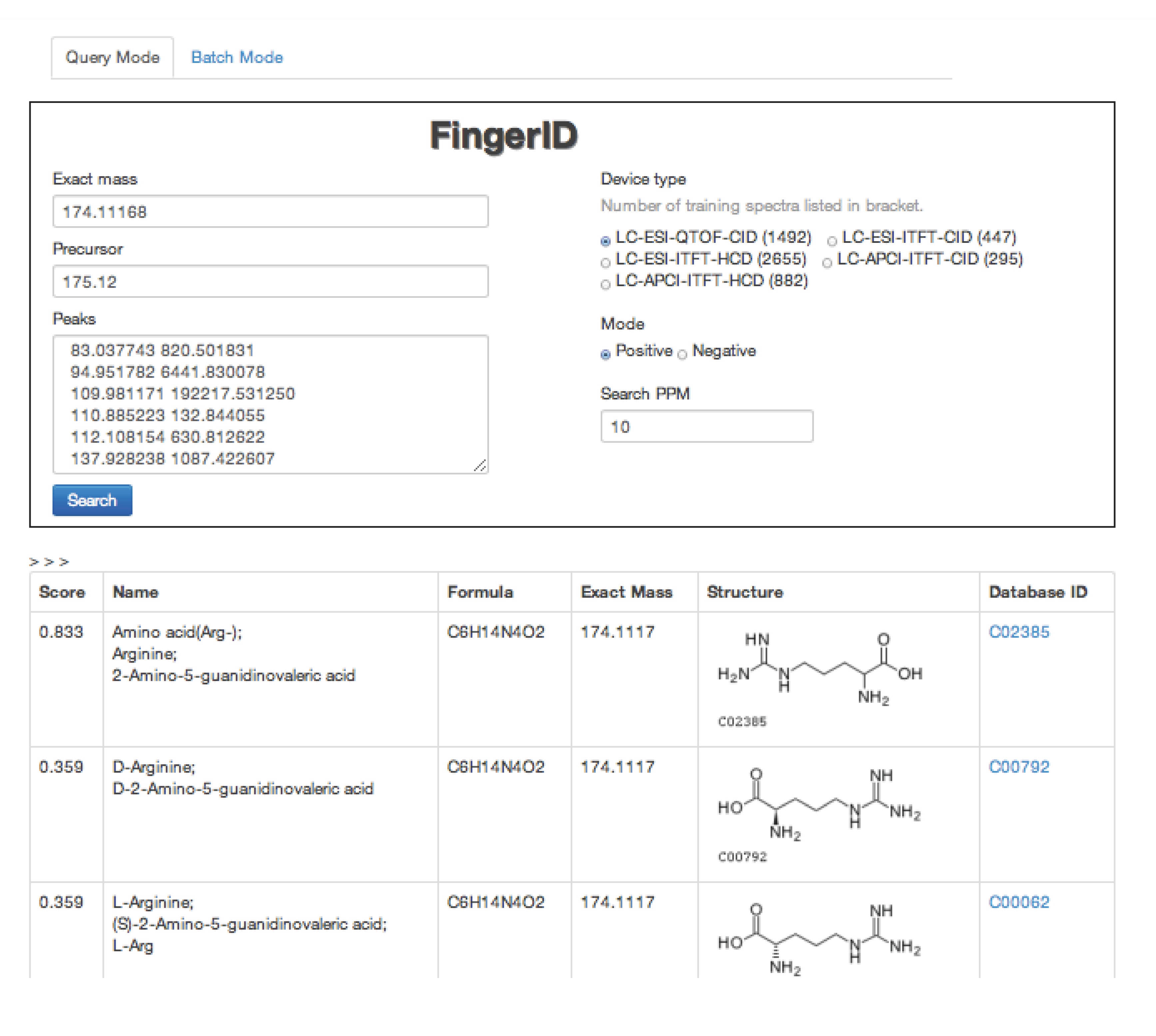
2.4. FingerID Web Server
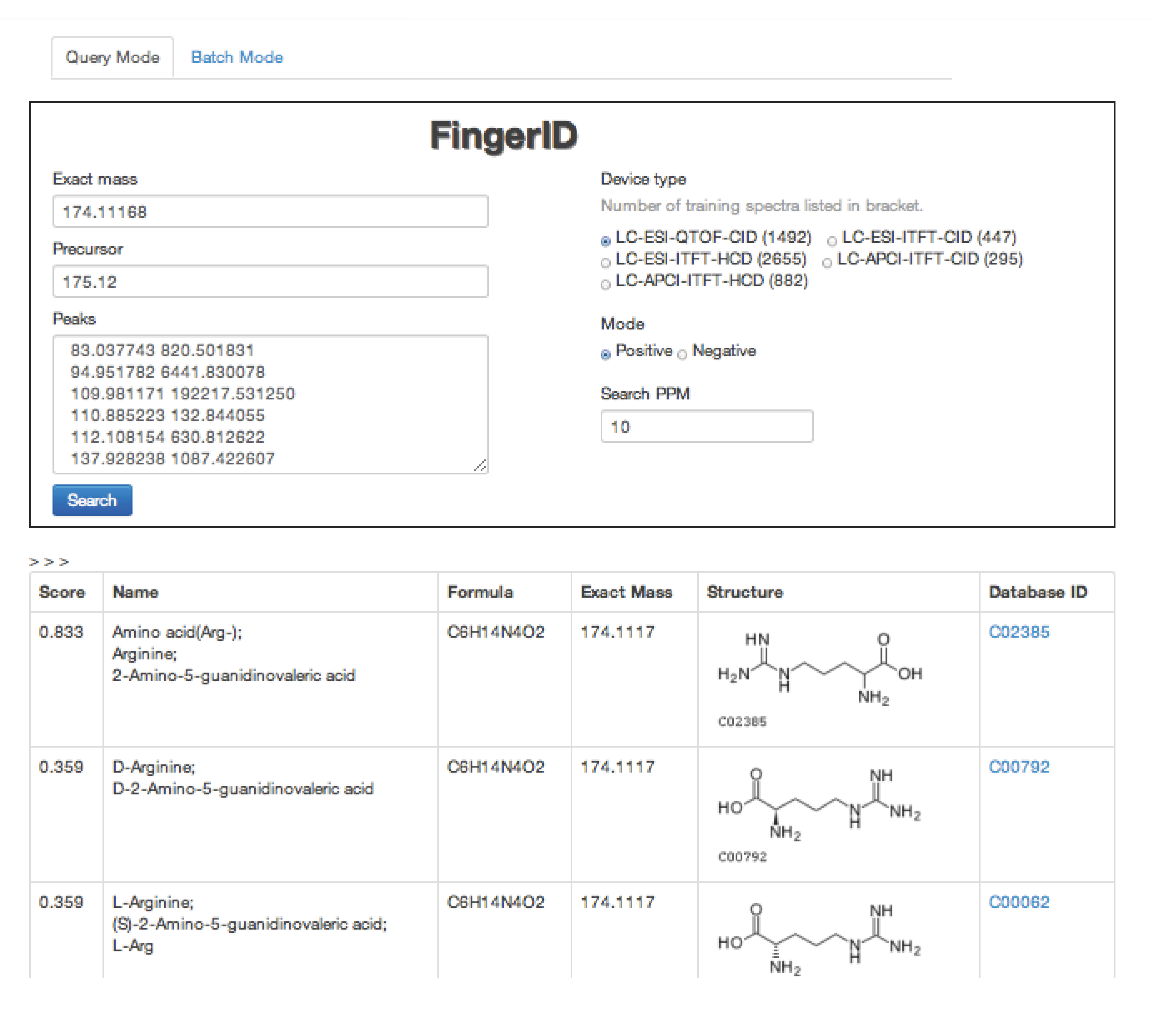
2.5. FingerID Software Distribution
3. Materials and Methods
3.1. CASMI Challenge Data
3.2. Mass Spectral Training Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS type | Instrument type | No. of spectra | No. of molecules | Fingerprints |
|---|---|---|---|---|
| MS2 | (1) LC-APCI-ITFT-CID | 295 | 65 | 179 |
| (2) LC-APCI-ITFT-HCD | 882 | 86 | 181 | |
| (3) LC-ESI-ITFT-CID | 447 | 224 | 281 | |
| (4) LC-ESI-ITFT-HCD | 2655 | 225 | 281 | |
| (5) LC-ESI-QTOF-CID | 1027 | 523 | 290 | |
| MS1 | LC-ESI-ITFT | 41 | 41 | - |
| LC-ESI-QTOF | 62 | 62 | - |
3.3. Molecular Fingerprints
3.4. Molecular Databases
3.5. SVM Model Training and Evaluation
4. Results and Discussion
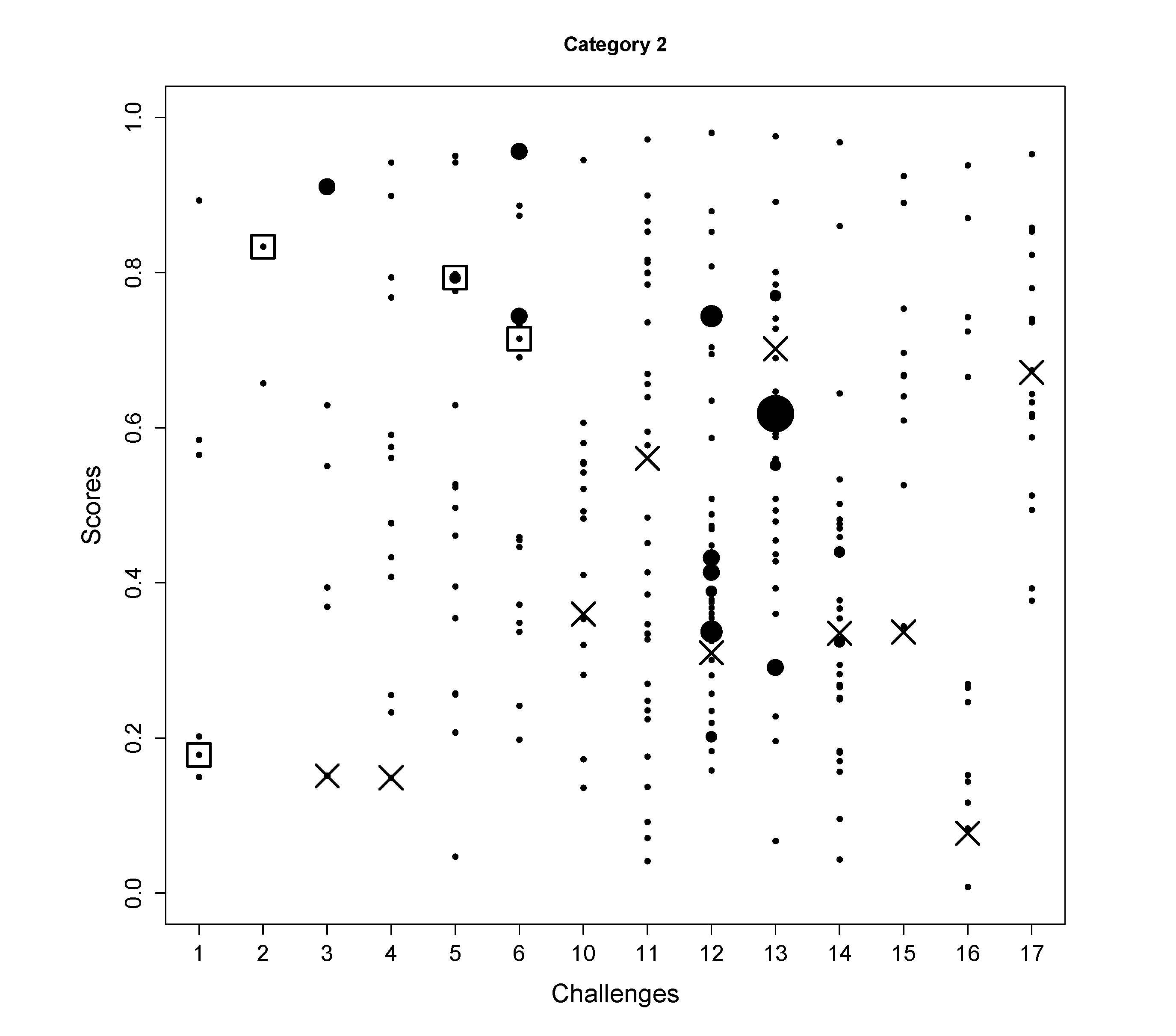
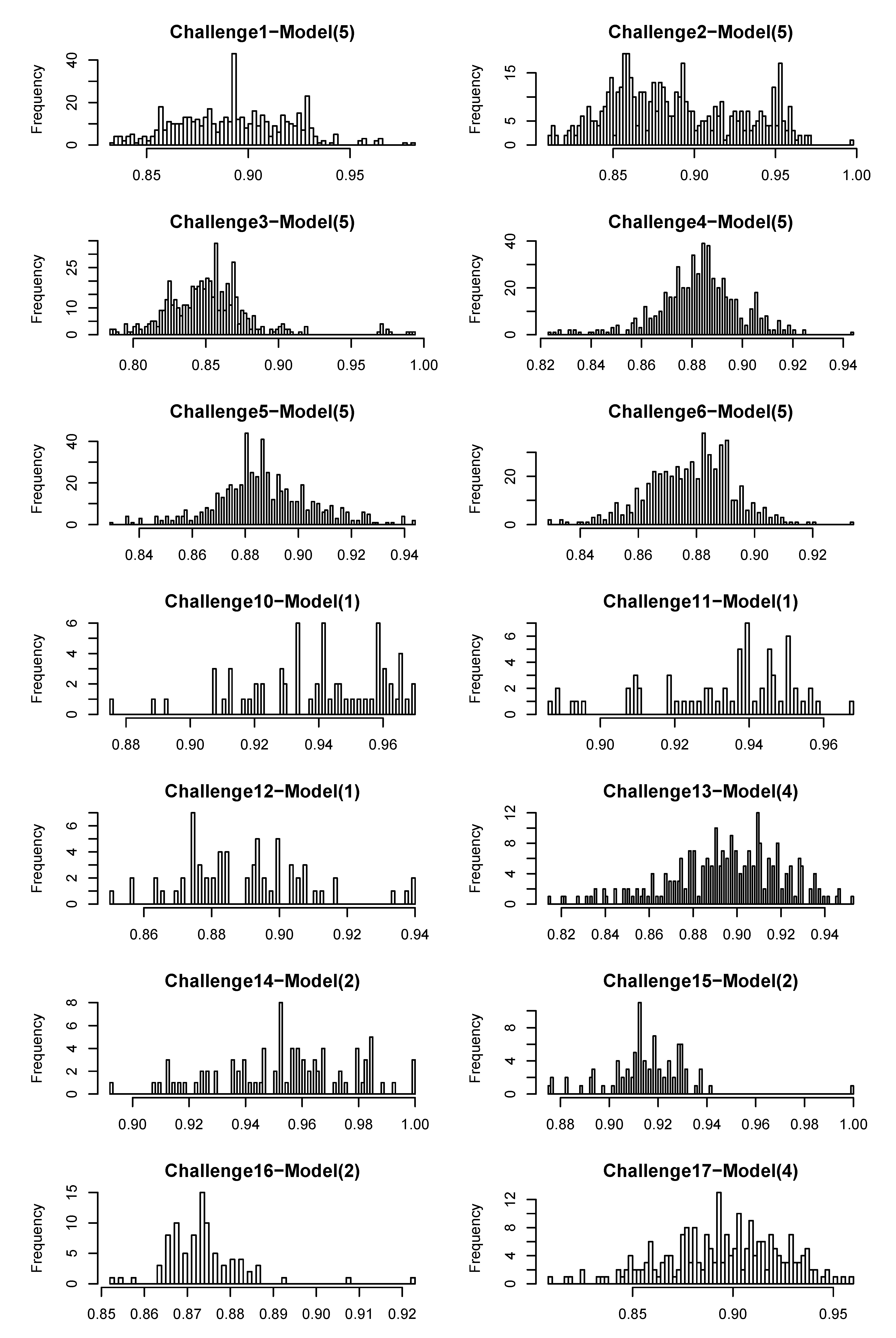
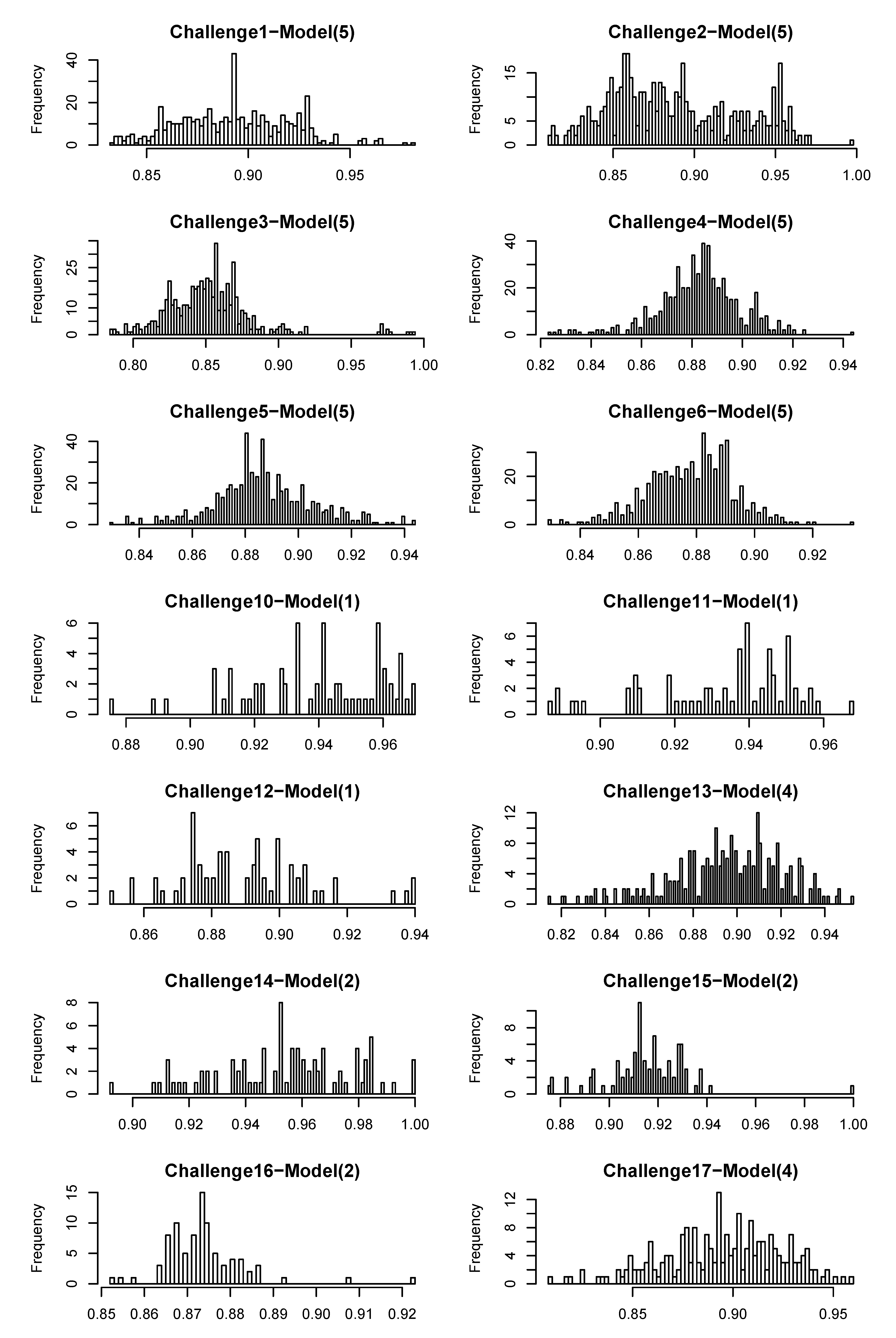
4.1. CASMI Challenge Results
| Challenge | 1 | 2 | 3 | 4 | 5 | 6 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| Model | (5) | (5) | (5) | (5) | (5) | (5) | (1) | (1) | (1) | (4) | (2) | (2) | (2) | (4) |
| Category 1 rank
# candidates | 4 | 1 | - | 3 | 4 | 4 | 1 | - | - | - | 1 | 5 | - | - |
| 5 | 4 | 6 | 12 | 11 | 15 | 11 | 15 | 21 | 15 | 12 | 6 | 12 | 11 | |
| Category 2 rank
# candidates | 5 | 1 | - | - | 5 | 11 | - | - | - | - | - | - | - | - |
| 6 | 6 | 8 | 14 | 17 | 20 | 15 | 32 | 47 | 38 | 28 | 10 | 13 | 18 | |
| Wrong FP(%) | 25 | 33 | 32 | 33 | 30 | 34 | 23 | 25 | 32 | 19 | 21 | 31 | 41 | 24 |
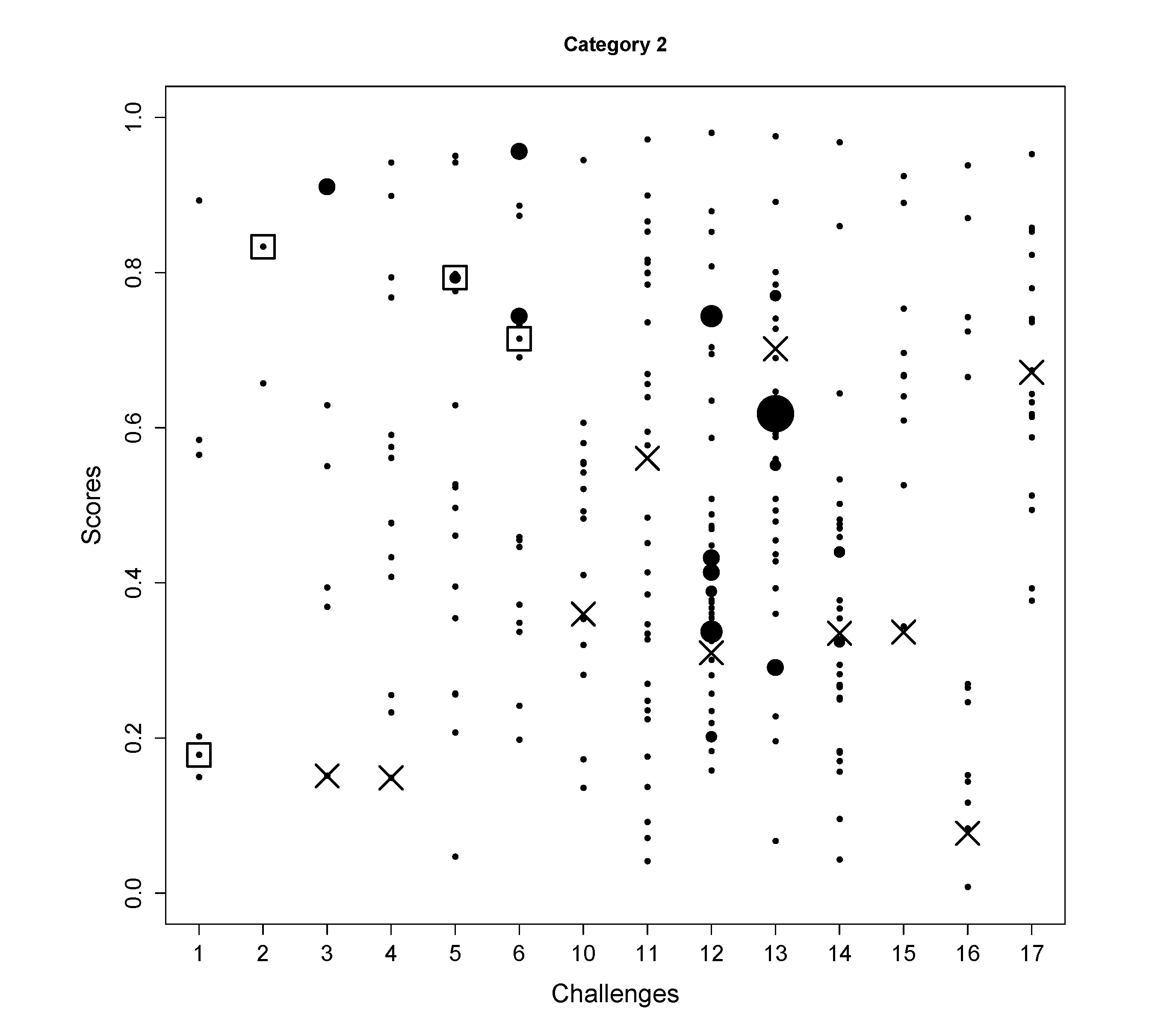
- In Challenge 1, the correct molecule was in the fifth position out of six. The score of the correct molecule was rather low; the high rank is probably a consequence of KEGG not having many molecules with a similar molecular weight, rather than good fingerprint prediction.
- In Challenge 2, the search ppm was set to 200 and the correct solution was obtained despite the 30 ppm error in the original challenge data. In the version of KEGG, only three entries have the mass around 592.1792 within 200 ppm and only two of them had molecular fingerprint generated using OpenBabel. Thus, the identification is simply choosing one from the two and the FingerID made the right choice. Incidentally, after correcting the 30 ppm error, FingerID still ranked the true molecule at the top, which surpassed other CASMI participants.
- In Challenge 5, the correct molecule ranks fifth and had the same confidence score as the fourth one. If the CASMI organizers took the rank of the score as criterion, this would have been the winning entry for this challenge.
- In Challenge 6, half of the candidates had better scores than the correct one, which means the molecular fingerprint prediction was not perfect. However, comparing the absolute rank to the other participants, this was sufficient to win this challenge.
- In Challenge 11, the highest intensity peak in MS1 is not the molecule with an adduct. Thus, the exact mass of the molecule was estimated incorrectly.
4.2. Evaluation of the FingerID Framework in the CASMI Contest
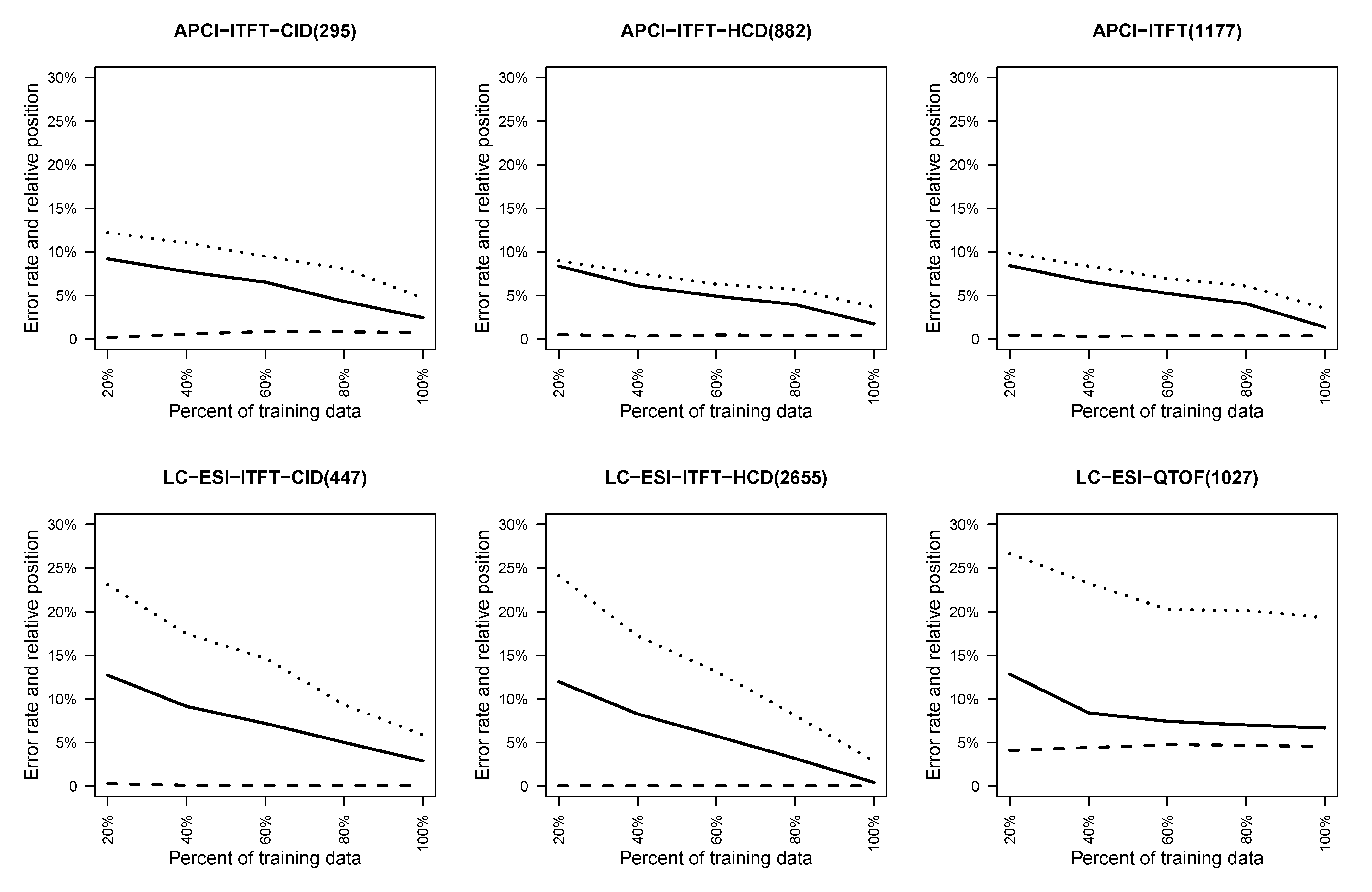
4.2.1. Effect of Training Set Size on Fingerprint Prediction Reliability
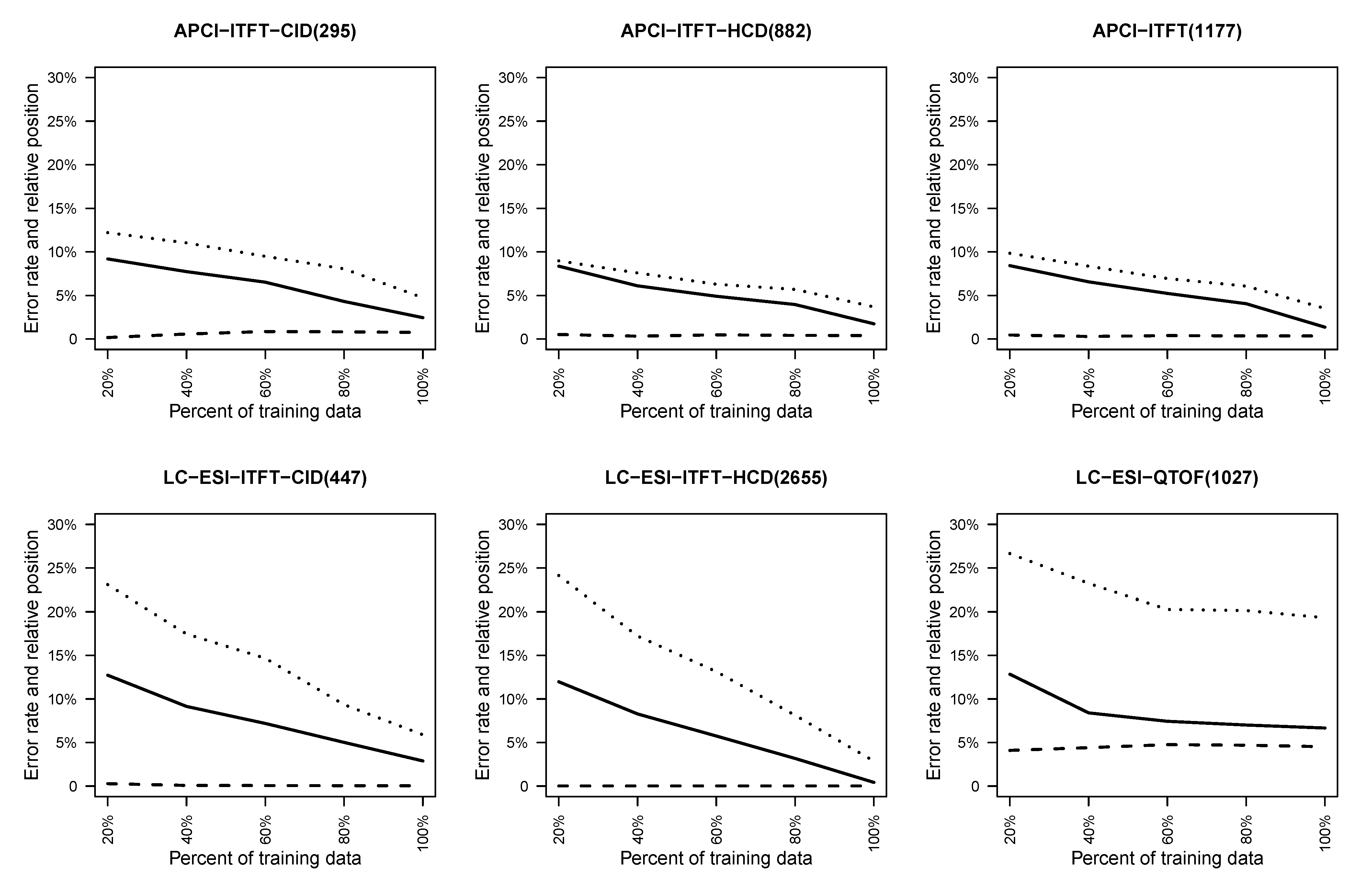
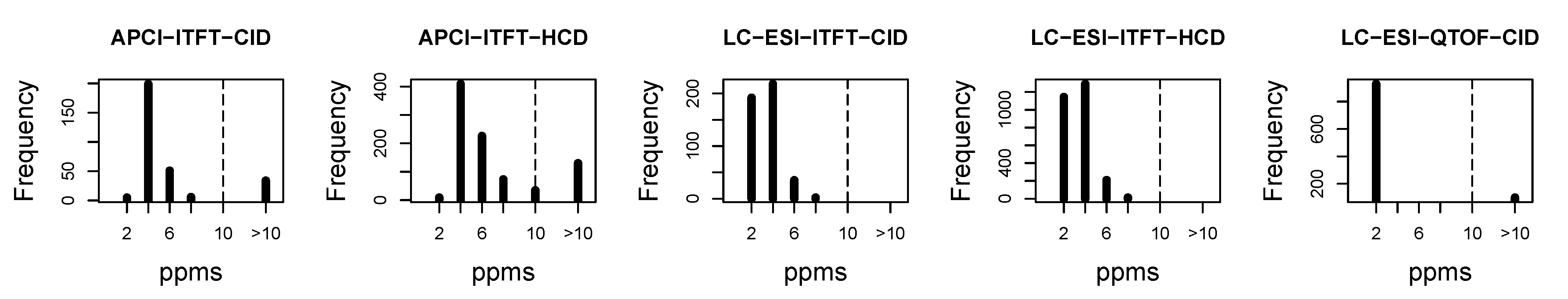
4.2.2. Quality of Exact Mass Prediction

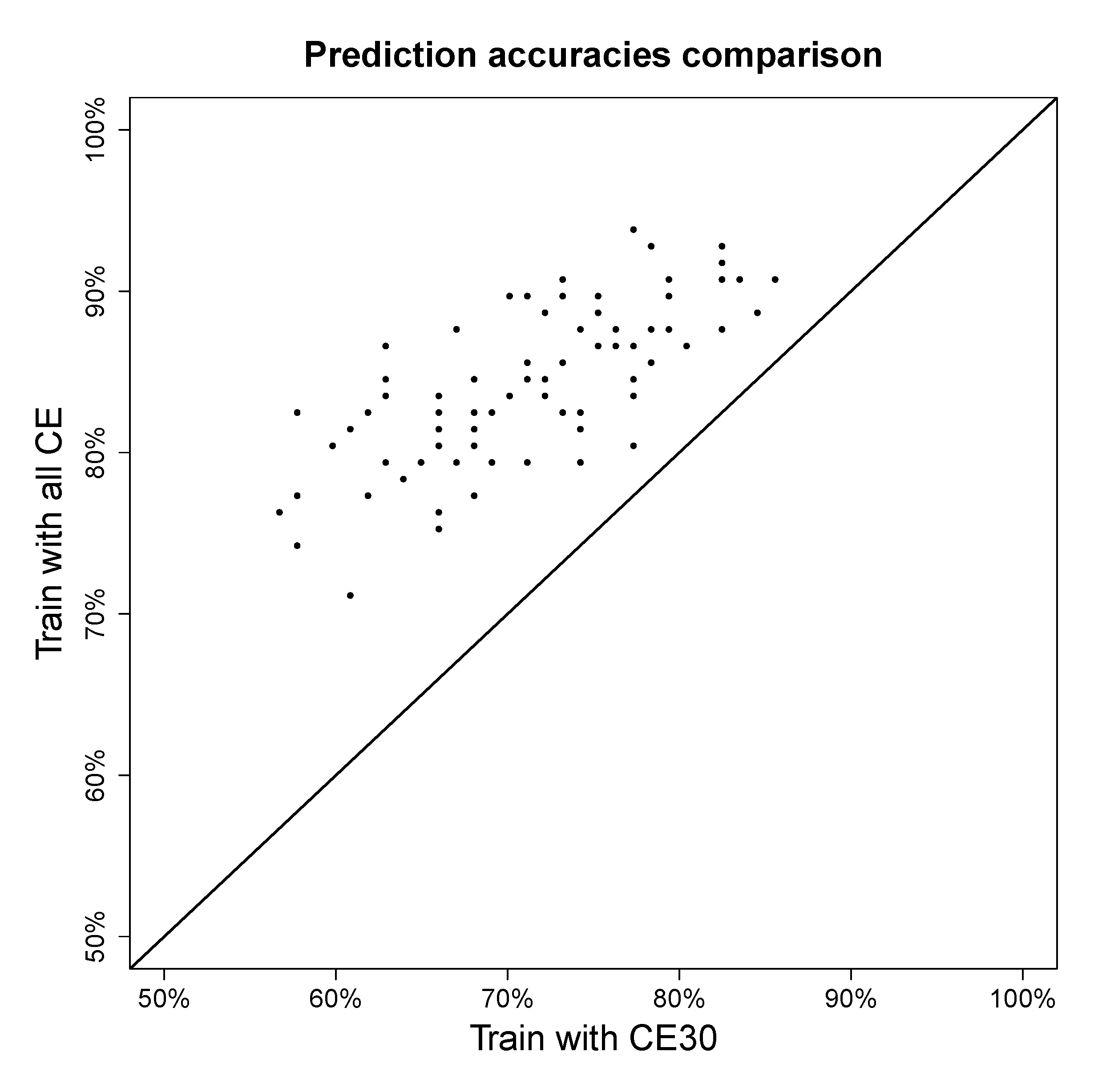
4.2.3. Effect of Using Multiple Collision Energies
4.2.4. Degree of Uniqueness of Fingerprints
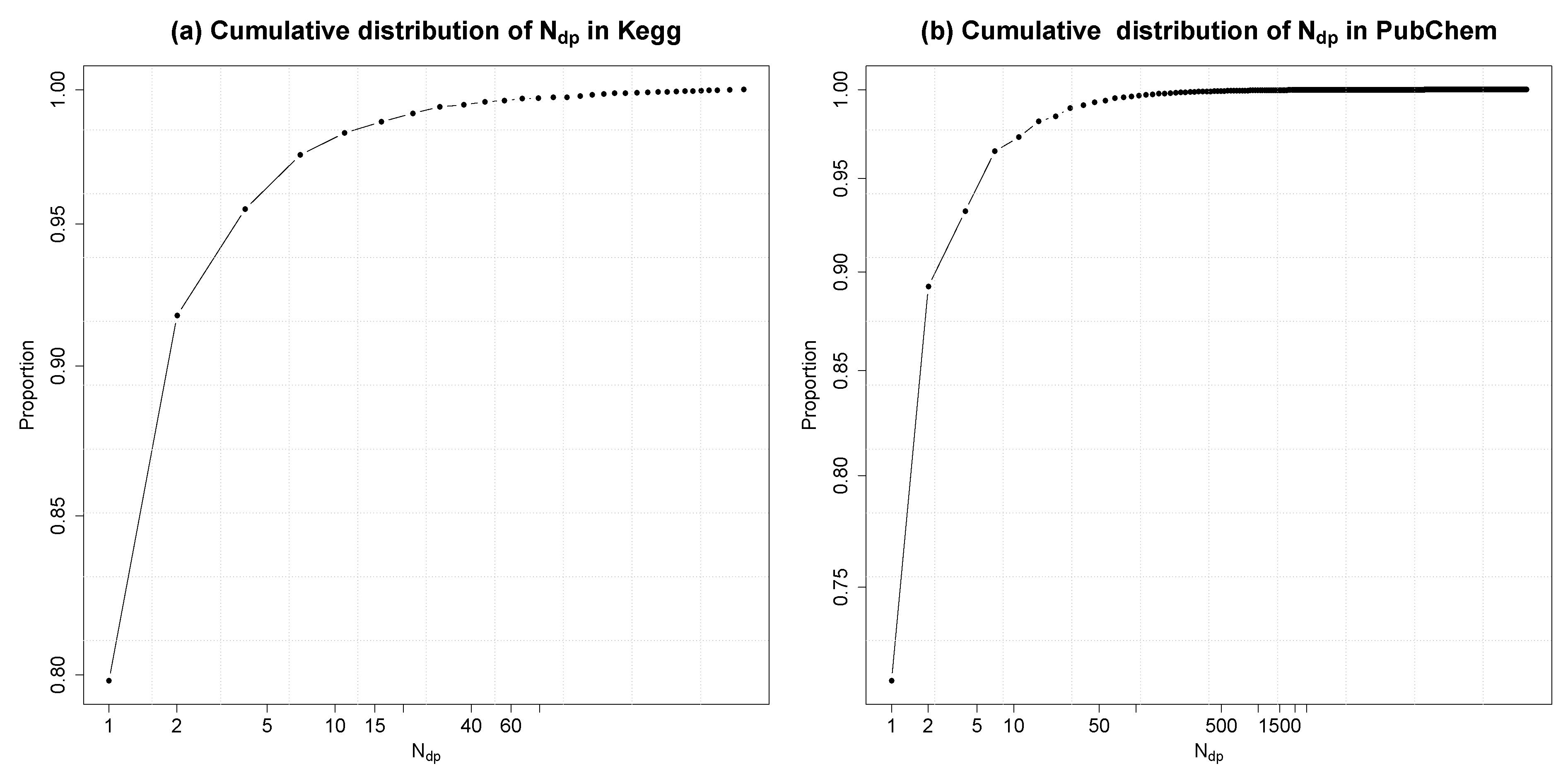
4.3. Extensions
4.3.1. Isotopic Distribution Matching
 different isotopologues caused by that element [22]. All of the isotopologues also have distinct abundances, which can be derived by applying multinomial probability over the isotope abundances [23,24]. Consequently, the theoretical mass spectrum that arises from the set of isotopologues can be simulated and compared with the observed spectrum. This information is more informative for metabolite identification than using the mass alone. Many methods and tools have been published for this purpose [25,26,27].
different isotopologues caused by that element [22]. All of the isotopologues also have distinct abundances, which can be derived by applying multinomial probability over the isotope abundances [23,24]. Consequently, the theoretical mass spectrum that arises from the set of isotopologues can be simulated and compared with the observed spectrum. This information is more informative for metabolite identification than using the mass alone. Many methods and tools have been published for this purpose [25,26,27].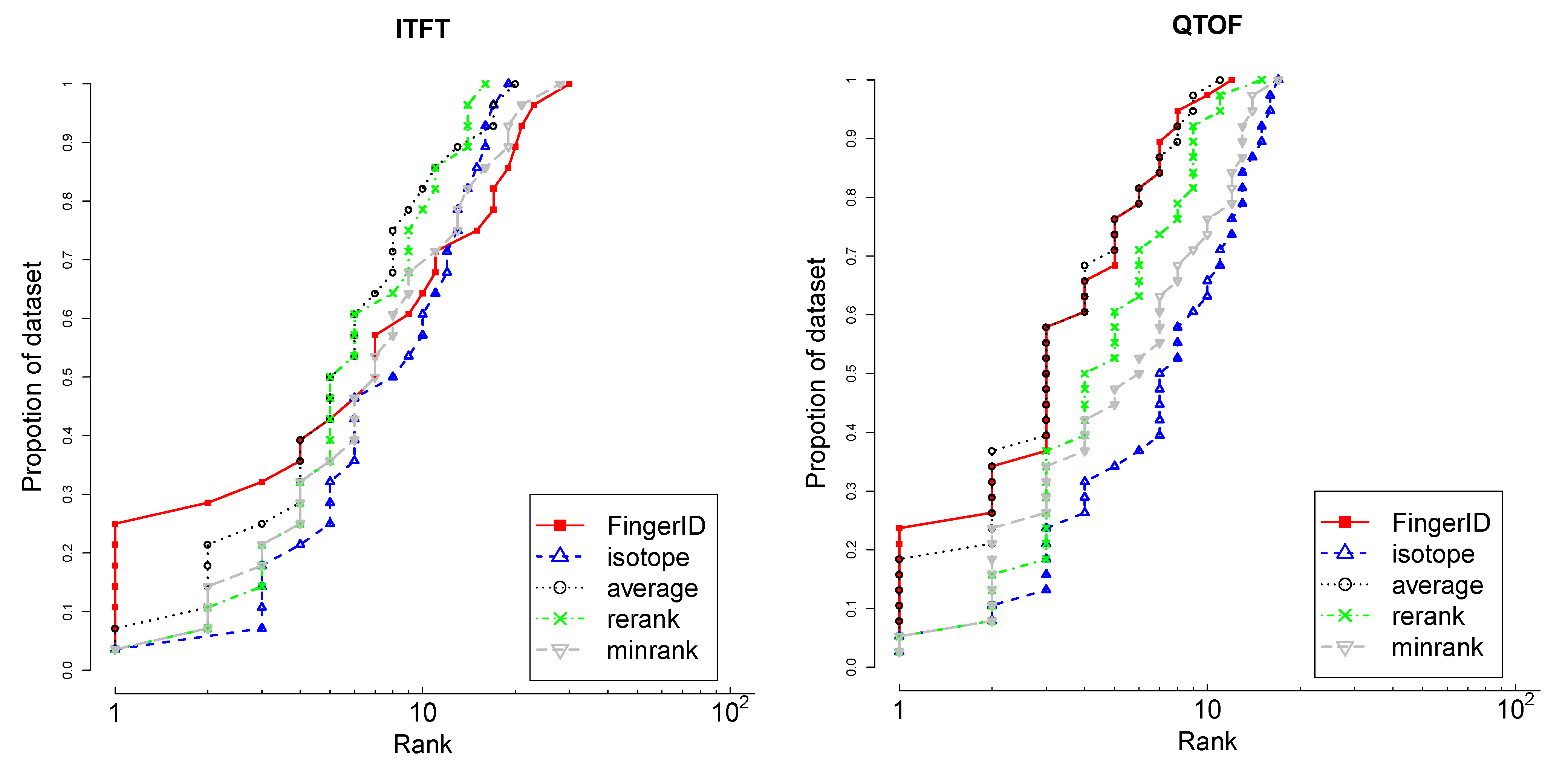
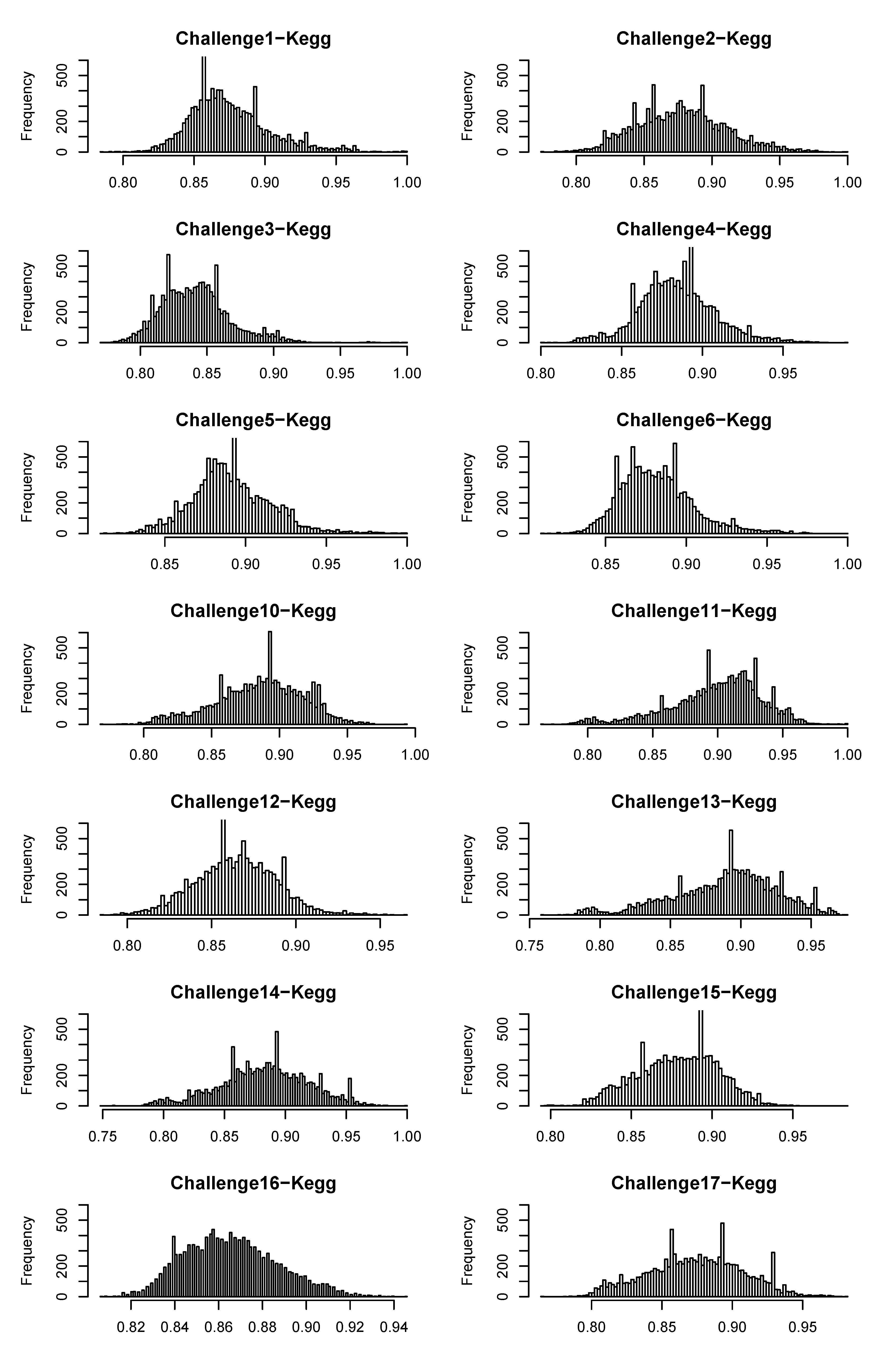
4.3.2. Using PubChem as the Molecular Database
| Challenge | 1 | 2 | 3 | 4 | 5 | 6 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 |
| Mass window (ppm) | 300 | 200 | 500 | 300 | 300 | 300 | 500 | 500 | 500 | 500 | 600 | 500 | 300 | 500 |
| Candidates | 55,242 | 9,931 | 87,514 | 75,205 | 75,320 | 115,639 | 26,701 | - | - | 69,708 | 10,390 | 18,669 | 97,567 | 19,718 |
| Cat 2 rank | 70 | 2 | 355 | 12,699 | 4,050 | 6,881 | 11 | - | - | 13,796 | 341 | 13,689 | 71,410 | 8,350 |
| 10 ppm rank | 25 | - | 5 | 2,225 | 1,343 | 1,048 | 7 | - | - | 166 | 57 | 1,815 | 2,416 | 231 |
| Ideal ppm | 2.34 | 50 | 3.90 | 9.38 | 1.17 | 9.38 | 0.98 | - | - | 0.98 | 9.38 | 7.81 | 0.98 | 1.95 |
| Ideal rank | 23 | 2 | 4 | 2,225 | 1,337 | 1,048 | 7 | - | - | 15 | 57 | 1,780 | 775 | 198 |
5. Conclusions
Acknowledgements
Conflict of Interest
Appendix
Appendix A
 and
and  , the similarity between them, if no prior knowledge exists on the weight of each individual fingerprint, is defined by:
, the similarity between them, if no prior knowledge exists on the weight of each individual fingerprint, is defined by:

References
- Kell, D. Metabolomics and systems biology: Making sense of the soup. Curr. Opin. Microbiol. 2004, 7, 296–307. [Google Scholar] [CrossRef]
- Pitkänen, E.; Rousu, J.; Ukkonen, E. Computational methods for metabolic reconstruction. Curr. Opin. Biotechnol. 2010, 21, 70–77. [Google Scholar] [CrossRef]
- Neumann, S.; Böcker, S. Computational mass spectrometry for metabolomics: Identification of metabolites and small molecules. Anal. Bioanal. Chem. 2010, 398, 2779–2788. [Google Scholar] [CrossRef]
- Wishart, D. Computational strategies for metabolite identification in metabolomics. Bioanalysis 2009, 1, 1579–1596. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- AtMetExpress LCMS. Available online: http://prime.psc.riken.jp/lcms/AtMetExpress/ (accessed on 3 June 2013).
- PlantMetabolomics. Available online: http://www.plantmetabolomics.org/ (accessed on 3 June 2013).
- Heinonen, M.; Rantanen, A.; Mielikäinen, T.; Pitkänen, E.; Kokkonen, J.; Rousu, J. Ab Initio Prediction of Molecular Fragments from Tandem Mass Spectrometry Data. In Proceedings of the German Conference on Bioinformatics, Tübingen, Germany, September 2006; Gesellschaft für Informatik: Bonn, Germany, 2006; Volume P-83, pp. 40–53. [Google Scholar]
- Heinonen, M.; Rantanen, A.; Mielikäinen, T.; Kokkonen, J.; Kiuru, J.; Ketola, R.; Rousu, J. FiD: A software for ab initio structural identification of product ions from tandem mass spectrometric data. Rapid Commun. Mass Spectrom. 2008, 22, 3043–3052. [Google Scholar] [CrossRef]
- Wolf, S.; Schmidt, S.; Müller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinforma. 2010, 11, 148. [Google Scholar] [CrossRef]
- Böcker, S.; Letzel, M.; Liptak, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef]
- FingerID. Available online: http://sourceforge.net/p/fingerid/ (accessed on 3 June 2013).
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. Ann. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar] [CrossRef]
- Critical Assessment of Small Molecule Identification. Available online: http://www.casmicontest.org/ (accessed on 3 June 2013).
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar]
- Jebara, T.; Kondor, R.; Howard, A. Probability product kernels. J. Mach. Learn. Res. 2004, 5, 819–844. [Google Scholar]
- FingerID web server. Available online: http://research.ics.aalto.fi/kepaco/fingerid/ (accessed on 3 June 2013).
- Isaac, G.; Jeannotte, R.; Esch, S.; Welti, R. New Mass-Spectrometry-Based Strategies for Lipids. In Genetic Engineering; Setlow, J., Ed.; Springer: New York, NY, USA, 2007; Volume 28, Genetic Engineering; pp. 129–157. [Google Scholar]
- O’Boyle, N.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open babel: An open chemical toolbox. J. Cheminf. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Böcker, S.; Letzel, M.C.; Lipták, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef]
- Rousu, J.; Rantanen, A.; Ketola, R.; Kokkonen, J. Isotopomer distribution computation from tandem mass spectrometric data with overlapping fragment spectra. Spectroscopy 2005, 19, 53–67. [Google Scholar] [CrossRef]
- Rantanen, A.; Rousu, J.; Ketola, R.; Kokkonen, J.; Tarkiainen, V. Computing positional isotopomer distributions from tandem mass spectrometric data. Metab. Eng. 2002, 4, 285–294. [Google Scholar] [CrossRef]
- Yergey, J.A. A general approach to calculating isotopic distributions for mass spectrometry. Int. J. Mass Spectrom. Ion Phys. 1983, 52, 337–349. [Google Scholar] [CrossRef]
- Kubinyi, H. Calculation of isotope distributions in mass spectrometry. A trivial solution for a non-trivial problem. Anal. Chim. Acta 1991, 247, 107–119. [Google Scholar] [CrossRef]
- Patiny, L.; Borel, A. ChemCalc: A building block for tomorrow’s chemical infrastructure. J. Chem. Inf. Model. 2013, 53, 1223–1228. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shen, H.; Zamboni, N.; Heinonen, M.; Rousu, J. Metabolite Identification through Machine Learning— Tackling CASMI Challenge Using FingerID. Metabolites 2013, 3, 484-505. https://doi.org/10.3390/metabo3020484
Shen H, Zamboni N, Heinonen M, Rousu J. Metabolite Identification through Machine Learning— Tackling CASMI Challenge Using FingerID. Metabolites. 2013; 3(2):484-505. https://doi.org/10.3390/metabo3020484
Chicago/Turabian StyleShen, Huibin, Nicola Zamboni, Markus Heinonen, and Juho Rousu. 2013. "Metabolite Identification through Machine Learning— Tackling CASMI Challenge Using FingerID" Metabolites 3, no. 2: 484-505. https://doi.org/10.3390/metabo3020484
APA StyleShen, H., Zamboni, N., Heinonen, M., & Rousu, J. (2013). Metabolite Identification through Machine Learning— Tackling CASMI Challenge Using FingerID. Metabolites, 3(2), 484-505. https://doi.org/10.3390/metabo3020484