Impact of Blood Collection Tubes and Sample Handling Time on Serum and Plasma Metabolome and Lipidome

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Metabolite Comparisons
2.2. Taxonomy Enrichment
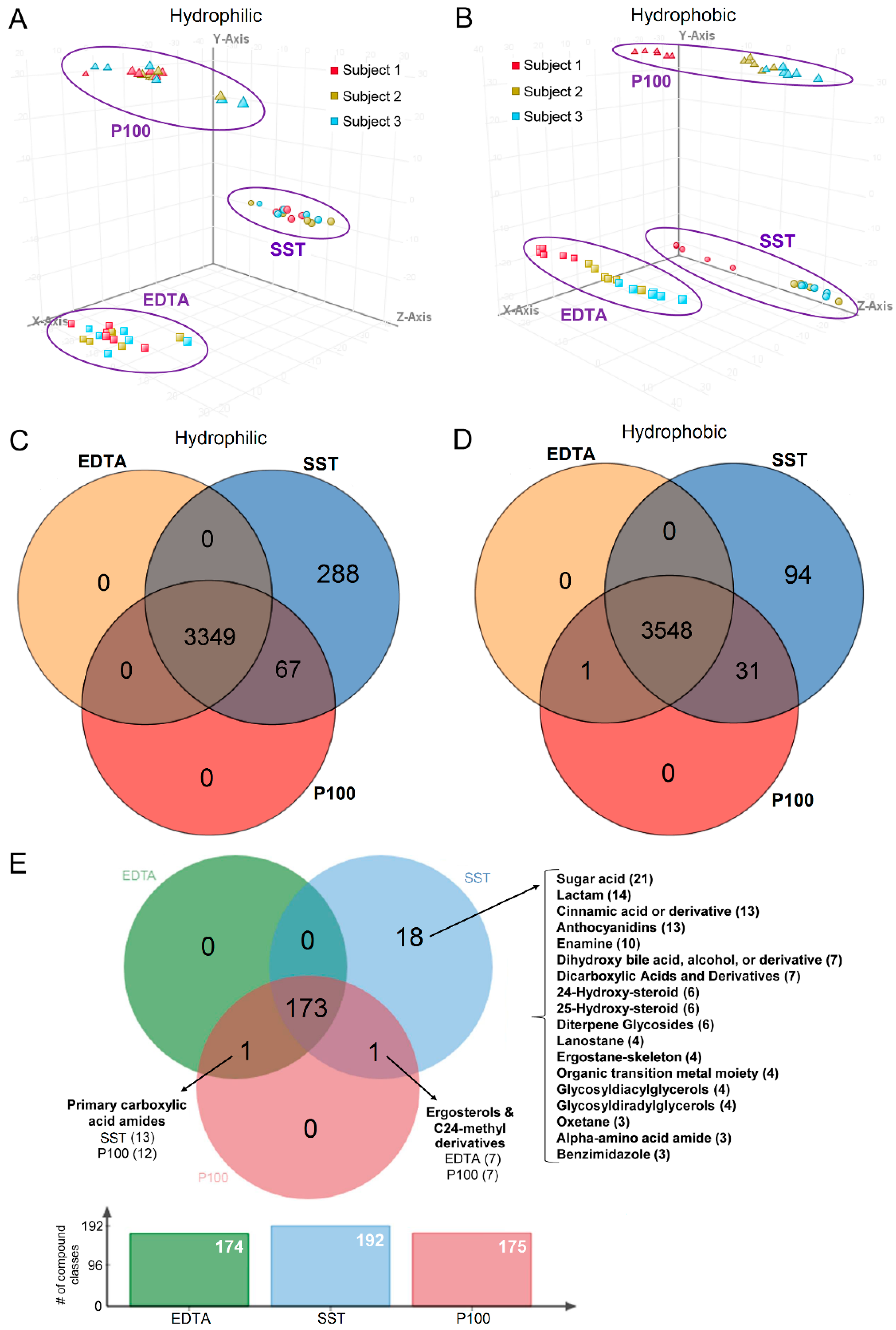
2.3. Clustering Based on Collection Tube and Subject
2.4. Tube Overlap Based on Captured Metabolites
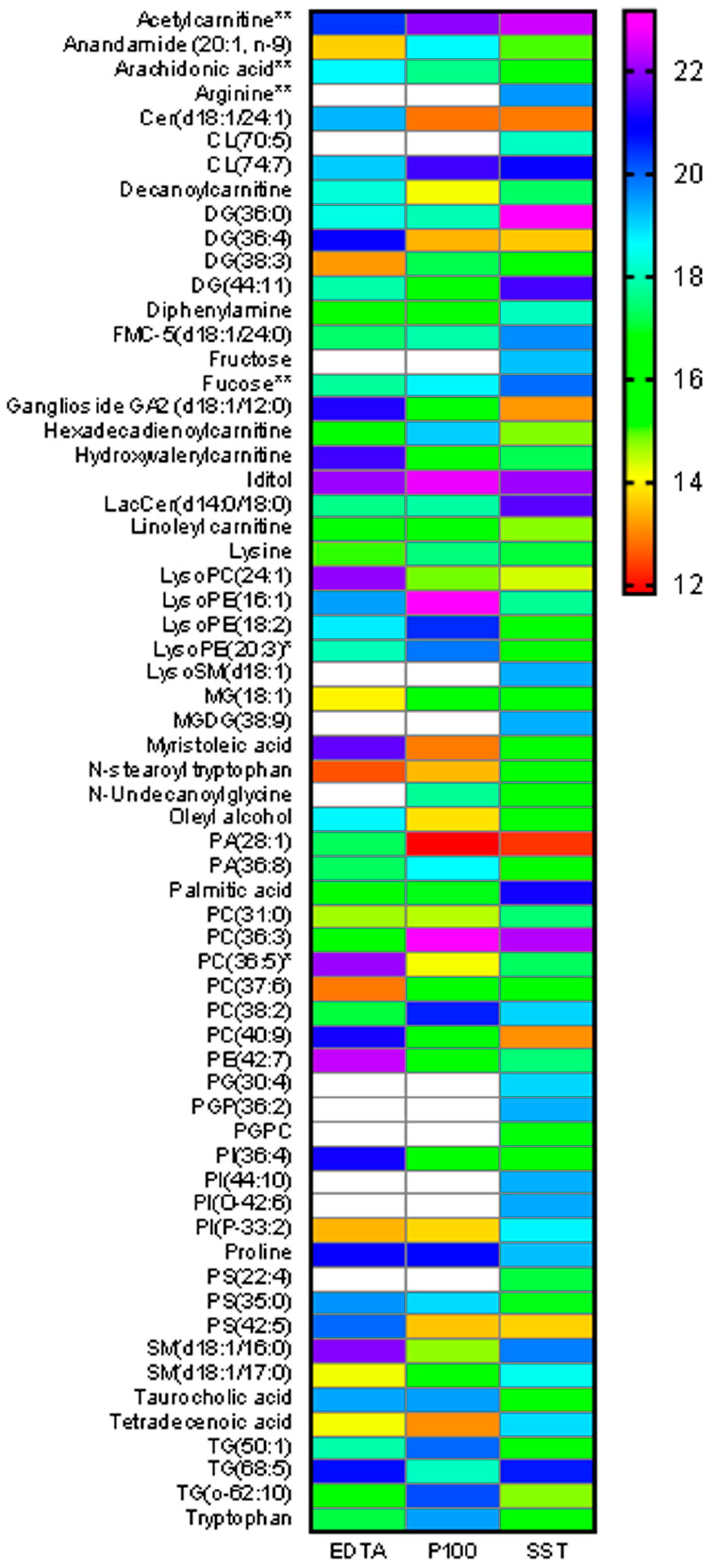
2.5. Metabolite Abundance Differences across Tubes
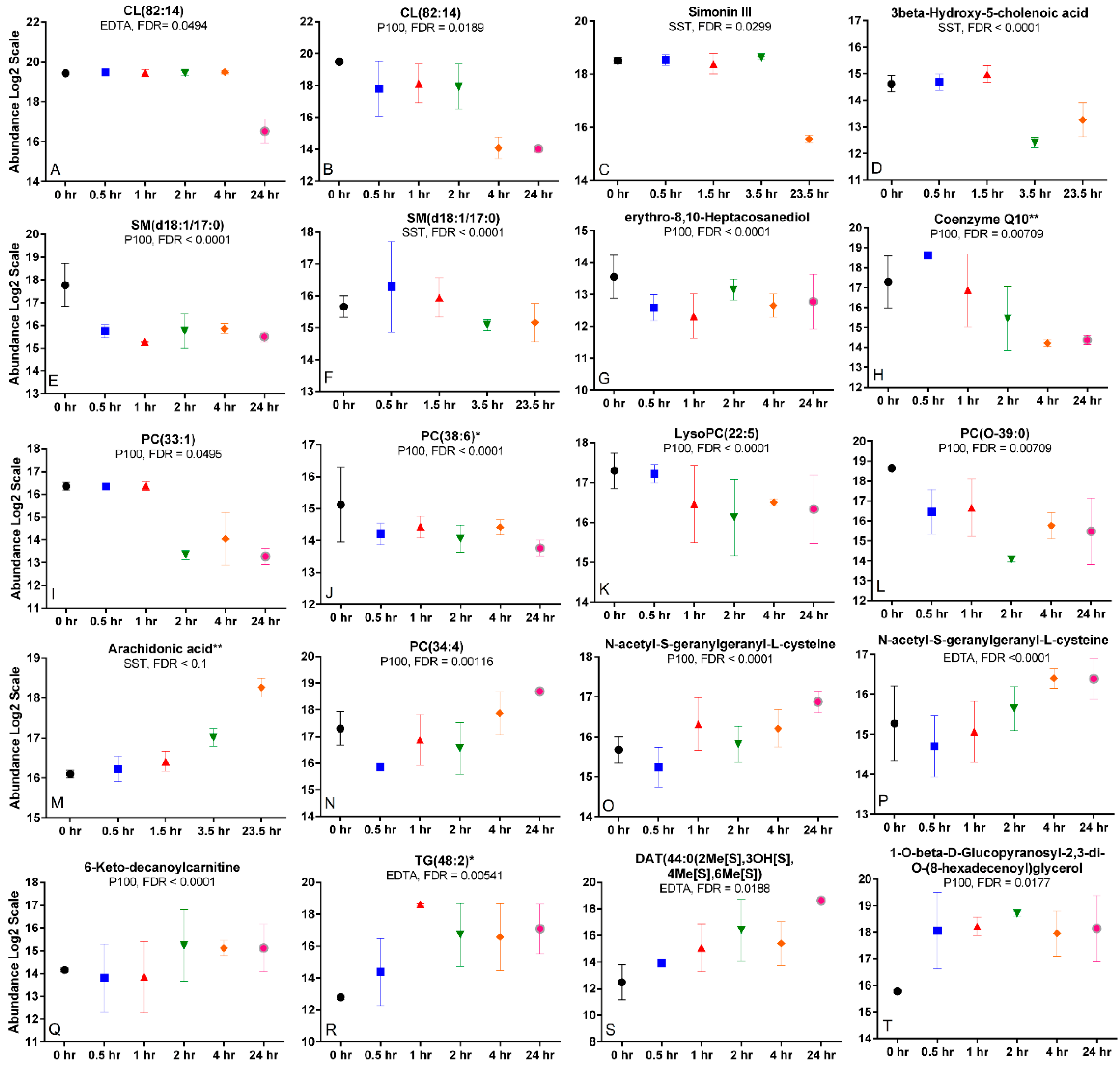
2.6. Time Trends and Changes Indicative of Metabolite Degradation, Oxidation, or Hydrolysis
2.7. Differing Time Trends across Tubes
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Study Population and Sample Collection
4.3. Reagents and Standards
4.4. Sample Preparation
4.5. Liquid Chromatography
4.6. Mass Spectrometry (MS)
4.7. Tandem Mass Spectrometry
4.8. Data Processing of QCs and Samples
4.9. Metabolite Annotation
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Milburn, M.V.; Ryals, J.A.; Lonergan, S.C.; Mitchell, M.W.; Wulff, J.E.; Alexander, D.C.; Evans, A.M.; Bridgewater, B.; Miller, L.; et al. Plasma metabolomic profiles enhance precision medicine for volunteers of normal health. Proc. Natl. Acad. Sci. USA 2015, 112, 4901–4910. [Google Scholar] [CrossRef] [PubMed]
- Klupczynska, A.; Derezinski, P.; Kokot, Z.J. Metabolomics in medical sciences—Trends, challenges and perspectives. Acta Pol. Pharm. 2015, 72, 629–641. [Google Scholar] [PubMed]
- Benjamin, R.J.; McLaughlin, L.S. Plasma components: Properties, differences, and uses. Transfusion 2012, 52, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Kim, C.; Gelfand, C.A. Inhibition of intrinsic proteolytic activities moderates preanalytical variability and instability of human plasma. J. Proteome Res. 2007, 6, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, W.K.; Anderson, W.; Basta, P.V.; Carretta, E.E.; Doerschuk, C.M.; Barr, R.G.; Bleecker, E.R.; Christenson, S.A.; Curtis, J.L.; Han, M.K.; et al. Comparison of serum, edta plasma and p100 plasma for luminex-based biomarker multiplex assays in patients with chronic obstructive pulmonary disease in the spiromics study. J. Transl. Med. 2014, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kastenmuller, G.; He, Y.; Belcredi, P.; Moller, G.; Prehn, C.; Mendes, J.; Wahl, S.; Roemisch-Margl, W.; Ceglarek, U.; et al. Differences between human plasma and serum metabolite profiles. PLoS ONE 2011, 6, e21230. [Google Scholar] [CrossRef]
- Paglia, G.; Del Greco, F.M.; Sigurdsson, B.B.; Rainer, J.; Volani, C.; Hicks, A.A.; Pramstaller, P.P.; Smarason, S.V. Influence of collection tubes during quantitative targeted metabolomics studies in human blood samples. Clin. Chim. Acta 2018, 486, 320–328. [Google Scholar] [CrossRef]
- Yin, P.; Peter, A.; Franken, H.; Zhao, X.; Neukamm, S.S.; Rosenbaum, L.; Lucio, M.; Zell, A.; Häring, H.U.; Xu, G.; et al. Preanalytical aspects and sample quality assessment in metabolomics studies of human blood. Clin. Chem. 2013, 59, 833–845. [Google Scholar] [CrossRef]
- Jobard, E.; Trédan, O.; Postoly, D.; André, F.; Martin, A.L.; Elena-Herrmann, B.; Boyault, S. A systematic evaluation of blood serum and plasma pre-analytics for metabolomics cohort studies. Int. J. Mol. Sci. 2016, 17, 2035. [Google Scholar] [CrossRef] [PubMed]
- Teahan, O.; Gamble, S.; Holmes, E.; Waxman, J.; Nicholson, J.K.; Bevan, C.; Keun, H.C. Impact of analytical bias in metabonomic studies of human blood serum and plasma. Anal. Chem. 2006, 78, 4307–4318. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Domingues, M.R.; Galhano, E.; Pita, C.; Almeida Mdo, C.; Carreira, I.M.; Gil, A.M. Human plasma stability during handling and storage: Impact on NMR metabolomics. Analyst 2014, 139, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Ilies, M.; Iuga, C.A.; Loghin, F.; Dhople, V.M.; Thiele, T.; Volker, U.; Hammer, E. Impact of blood sample collection methods on blood protein profiling studies. Clin. Chim. Acta 2017, 471, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Montalvo, L.; Chrebtow, V.; Busch, M.P. Quantitation of genomic DNA in plasma and serum samples: Higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001, 41, 276–282. [Google Scholar] [CrossRef]
- Bowen, R.A.R. Interferences from blood collection tube components on clinical chemistry assays. Biochem. Med. 2014, 24, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Barra, G.B.; Santa Rita, T.H.; de Almeida Vasques, J.; Chianca, C.F.; Nery, L.F.; Santana Soares Costa, S. Edta-mediated inhibition of dnases protects circulating cell-free DNA from ex vivo degradation in blood samples. Clin. Biochem. 2015, 48, 976–981. [Google Scholar] [CrossRef]
- Lam, N.Y.; Rainer, T.H.; Chiu, R.W.; Lo, Y.M. Edta is a better anticoagulant than heparin or citrate for delayed blood processing for plasma DNA analysis. Clin. Chem. 2004, 50, 256–257. [Google Scholar] [CrossRef]
- Barton, R.H.; Waterman, D.; Bonner, F.W.; Holmes, E.; Clarke, R.; Nicholson, J.K.; Lindon, J.C. The influence of edta and citrate anticoagulant addition to human plasma on information recovery from NMR-based metabolic profiling studies. Mol. Biosyst. 2010, 6, 215–224. [Google Scholar] [CrossRef]
- Katayama, S.; Lee, J.B. Prostaglandins and leukotrienes. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Trugo, L., Finglas, P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 4798–4804. [Google Scholar]
- Zhang, D.J.; Elswick, R.K.; Miller, W.G.; Bailey, J.L. Effect of serum-clot contact time on clinical chemistry laboratory results. Clin. Chem. 1998, 44, 1325–1333. [Google Scholar]
- Boyanton, B.L., Jr.; Blick, K.E. Stability studies of twenty-four analytes in human plasma and serum. Clin. Chem. 2002, 48, 2242–2247. [Google Scholar] [PubMed]
- Liu, L.; Aa, J.; Wang, G.; Yan, B.; Zhang, Y.; Wang, X.; Zhao, C.; Cao, B.; Shi, J.; Li, M.; et al. Differences in metabolite profile between blood plasma and serum. Anal. Biochem. 2010, 406, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Mullner, E.; Poutanen, K.; Mykkanen, H.; Moazzami, A.A. Metabolic changes in serum metabolome in response to a meal. Eur. J. Nutr. 2015, 56, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Kujala, U.M.; Makinen, V.P.; Heinonen, I.; Soininen, P.; Kangas, A.J.; Leskinen, T.H.; Rahkila, P.; Wurtz, P.; Kovanen, V.; Cheng, S.; et al. Long-term leisure-time physical activity and serum metabolome. Circulation 2013, 127, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Hadrevi, J.; Bjorklund, M.; Kosek, E.; Hallgren, S.; Antti, H.; Fahlstrom, M.; Hellstrom, F. Systemic differences in serum metabolome: A cross sectional comparison of women with localised and widespread pain and controls. Sci. Rep. 2015, 5, 15925. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Mittelstrass, K.; Do, K.T.; Stuckler, F.; Ried, J.; Adamski, J.; Peters, A.; Illig, T.; Kronenberg, F.; Friedrich, N.; et al. Gender-specific pathway differences in the human serum metabolome. Metabolomics 2015, 11, 1815–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.K.; Ang, J.E.; Revell, V.L.; Holmes, B.; Mann, A.; Robertson, F.P.; Cui, N.; Middleton, B.; Ackermann, K.; Kayser, M.; et al. Effect of sleep deprivation on the human metabolome. Proc. Natl. Acad. Sci. USA 2014, 111, 10761–10766. [Google Scholar] [CrossRef] [Green Version]
- Barton, S.; Navarro, S.L.; Buas, M.F.; Schwarz, Y.; Gu, H.; Djukovic, D.; Raftery, D.; Kratz, M.; Neuhouser, M.L.; Lampe, J.W. Targeted plasma metabolome response to variations in dietary glycemic load in a randomized, controlled, crossover feeding trial in healthy adults. Food Funct. 2015, 6, 2949–2956. [Google Scholar] [CrossRef] [Green Version]
- Courant, F.; Antignac, J.P.; Dervilly-Pinel, G.; Le Bizec, B. Basics of mass spectrometry based metabolomics. Proteomics 2014, 14, 2369–2388. [Google Scholar] [CrossRef] [Green Version]
- Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006, 7, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Cruickshank, C.; Armstrong, M.; Mahaffey, S.; Reisdorph, R.; Reisdorph, N. New sample preparation approach for mass spectrometry-based profiling of plasma results in improved coverage of metabolome. J. Chromatogr. 2013, 1300, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruickshank-Quinn, C.; Quinn, K.D.; Powell, R.; Yang, Y.; Armstrong, M.; Mahaffey, S.; Reisdorph, R.; Reisdorph, N. Multi-step preparation technique to recover multiple metabolite compound classes for in-depth and informative metabolomic analysis. J. Vis. Exp. 2014, 89, e51670. [Google Scholar] [CrossRef] [PubMed]
- Stein, S. Nist MS Search Program. Available online: https://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:nist17 (accessed on 30 October 2018).
- Yang, X.; Neta, P.; Stein, S.E. Quality control for building libraries from electrospray ionization tandem mass spectra. Anal. Chem. 2014, 86, 6393–6400. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beger, R.; Beale, M.H.; Daykin, C.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical analysis working group (cawg) metabolomics standards initiative (msi). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Polpitiya, A.D.; Qian, W.J.; Jaitly, N.; Petyuk, V.A.; Adkins, J.N.; Camp, D.G., II; Anderson, G.A.; Smith, R.D. Dante: A statistical tool for quantitative analysis of omics data. Bioinformatics 2008, 24, 1556–1558. [Google Scholar] [CrossRef] [PubMed]
- Stacklies, W.; Redestig, H.; Scholz, M.; Walther, D.; Selbig, J. Pcamethods—A bioconductor package providing pca methods for incomplete data. Bioinformatics 2007, 23, 1164–1167. [Google Scholar] [CrossRef]
- Jin, Z.; Kang, J.; Yu, T. Missing value imputation for lc-ms metabolomics data by incorporating metabolic network and adduct ion relations. Bioinformatics 2017, 34, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Chagoyen, M.; Pazos, F. Mbrole: Enrichment analysis of metabolomic data. Bioinformatics 2011, 27, 730–731. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Blood Processing Time Differences | ||||||
|---|---|---|---|---|---|---|
| SST (Serum) | P100 (Plasma) | EDTA (Plasma) | ||||
| * Number of Compounds | † Significant in SST | * Number of Compounds | † Significant in P100 | * Number of Compounds | † Significant in EDTA | |
| Hydrophilic | 3704 | 30 | 3416 | 44 | 3349 | 43 |
| Hydrophobic | 3673 | 16 | 3580 | 42 | 3548 | 7 |
| Total | 7377 | 46 | 6996 | 86 | 6897 | 50 |
| Blood Collection Tube Differences | |||||
|---|---|---|---|---|---|
| Comparison | Fraction | † Significant Compounds | † Total | Number, Regulation and Tube | Number of Classes Affected * |
| P100 vs. EDTA | Hydrophilic | 177 | 305 | 134 ↑ in P100; 43 ↑ in EDTA | 9 |
| Hydrophobic | 128 | 76 ↑ in P100; 52 ↑ in EDTA | |||
| EDTA vs. SST | Hydrophilic | 488 | 719 | 410 ↑ in SST; 78 ↑ in EDTA | 37 |
| Hydrophobic | 231 | 174 ↑ in SST; 57 ↑ in EDTA | |||
| P100 vs. SST | Hydrophilic | 433 | 622 | 343 ↑ in SST; 90 ↑ in P100 | 38 |
| Hydrophobic | 189 | 145 ↑ in SST; 44 ↑ in P100 | |||
| Comparison | Chemical Taxonomy | Category | # in Set | p-Value | FDR |
|---|---|---|---|---|---|
| P100 vs. EDTA | Primary alcohol | HMDB | 17 | 2.63 × 10−4 | 1.21 × 10−2 |
| P100 vs. EDTA | Secondary carboxylic acid amide | HMDB | 11 | 2.04 × 10−4 | 1.21 × 10−2 |
| P100 vs. EDTA | Carboxamide group | HMDB | 11 | 7.48 × 10−4 | 2.29 × 10−2 |
| P100 vs. EDTA | Allyl alcohol | HMDB | 7 | 5.93 × 10−4 | 2.18 × 10−2 |
| P100 vs. EDTA | Amino Acids, Peptides and Analogues | HMDB | 7 | 1.99 × 10−3 | 4.58 × 10−2 |
| P100 vs. EDTA | Fatty Alcohols | HMDB | 5 | 1.21 × 10−4 | 1.11 × 10−2 |
| P100 vs. EDTA | N-acyl-amine | HMDB | 5 | 1.35 × 10−3 | 3.55 × 10−2 |
| P100 vs. EDTA | Lysophosphatidylethanolamines | HMDB | 4 | 1.53 × 10−6 | 2.82 × 10−4 |
| P100 vs. EDTA | Sphingomyelins | Lipid Maps | 3 | 4.31 × 10−5 | 2.11 × 10−3 |
| EDTA vs. SST | Secondary alcohol | HMDB | 44 | 1.68 × 10−4 | 4.75 × 10−3 |
| EDTA vs. SST | Primary alcohol | HMDB | 32 | 3.61 × 10−8 | 1.09 × 10−5 |
| EDTA vs. SST | Glycerophospholipids | Lipid Maps | 24 | 4.36 × 10−4 | 3.23 × 10−2 |
| EDTA vs. SST | 1,2-Diol | HMDB | 21 | 3.54 × 10−3 | 2.97 × 10−2 |
| EDTA vs. SST | Cyclohexane | HMDB | 18 | 1.04 × 10−3 | 1.31 × 10−2 |
| EDTA vs. SST | Secondary carboxylic acid amide | HMDB | 17 | 1.42 × 10−5 | 7.15 × 10−4 |
| EDTA vs. SST | Carboxamide group | HMDB | 17 | 9.65 × 10−5 | 3.24 × 10−3 |
| EDTA vs. SST | Prenol Lipids | HMDB | 17 | 1.61 × 10−3 | 1.58 × 10−2 |
| EDTA vs. SST | Saccharide | HMDB | 16 | 3.11 × 10−3 | 2.68 × 10−2 |
| EDTA vs. SST | Bicyclohexane | HMDB | 11 | 1.31 × 10−3 | 1.41 × 10−2 |
| EDTA vs. SST | Allyl alcohol | HMDB | 10 | 1.73 × 10−4 | 4.75 × 10−3 |
| EDTA vs. SST | Sesterterpene | HMDB | 10 | 1.99 × 10−4 | 5.01 × 10−3 |
| EDTA vs. SST | Decaline | HMDB | 10 | 4.44 × 10−3 | 3.12 × 10−2 |
| EDTA vs. SST | Choline | HMDB | 9 | 2.13 × 10−3 | 1.95 × 10−2 |
| EDTA vs. SST | Quaternary ammonium salt | HMDB | 9 | 4.11 × 10−3 | 3.12 × 10−2 |
| P100 vs. SST | Secondary alcohol | HMDB | 37 | 1.51 × 10−3 | 1.84 × 10−2 |
| P100 vs. SST | Primary alcohol | HMDB | 27 | 1.04 × 10−6 | 3.17 × 10−4 |
| P100 vs. SST | Glycerophospholipids | Lipid Maps | 20 | 1.56 × 10−3 | 2.03 × 10−2 |
| P100 vs. SST | 1,2-Diol | HMDB | 19 | 3.84 × 10−3 | 3.45 × 10−2 |
| P100 vs. SST | Prenol Lipids | HMDB | 17 | 3.49 × 10−4 | 8.42 × 10−3 |
| P100 vs. SST | Cyclohexane | HMDB | 17 | 6.05 × 10−4 | 1.04 × 10−2 |
| P100 vs. SST | Cyclic alcohol | HMDB | 15 | 2.91 × 10−4 | 8.42 × 10−3 |
| P100 vs. SST | Secondary carboxylic acid amide | HMDB | 14 | 1.64 × 10−4 | 7.15 × 10−3 |
| P100 vs. SST | Carboxamide group | HMDB | 14 | 7.69 × 10−4 | 1.12 × 10−2 |
| P100 vs. SST | Saccharide | HMDB | 14 | 5.72 × 10−3 | 4.09 × 10−2 |
| P100 vs. SST | Bicyclohexane | HMDB | 11 | 4.36 × 10−4 | 9.50 × 10−3 |
| P100 vs. SST | Sesterterpene | HMDB | 10 | 6.68 × 10−5 | 5.67 × 10−3 |
| P100 vs. SST | Decaline | HMDB | 10 | 1.71 × 10−3 | 2.01 × 10−2 |
| P100 vs. SST | Drimane-skeleton | HMDB | 8 | 6.15 × 10−4 | 1.04 × 10−2 |
| P100 vs. SST | Allyl alcohol | HMDB | 8 | 1.41 × 10−3 | 1.79 × 10−2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruickshank-Quinn, C.; Zheng, L.K.; Quinn, K.; Bowler, R.; Reisdorph, R.; Reisdorph, N. Impact of Blood Collection Tubes and Sample Handling Time on Serum and Plasma Metabolome and Lipidome. Metabolites 2018, 8, 88. https://doi.org/10.3390/metabo8040088
Cruickshank-Quinn C, Zheng LK, Quinn K, Bowler R, Reisdorph R, Reisdorph N. Impact of Blood Collection Tubes and Sample Handling Time on Serum and Plasma Metabolome and Lipidome. Metabolites. 2018; 8(4):88. https://doi.org/10.3390/metabo8040088
Chicago/Turabian StyleCruickshank-Quinn, Charmion, Laura K. Zheng, Kevin Quinn, Russell Bowler, Richard Reisdorph, and Nichole Reisdorph. 2018. "Impact of Blood Collection Tubes and Sample Handling Time on Serum and Plasma Metabolome and Lipidome" Metabolites 8, no. 4: 88. https://doi.org/10.3390/metabo8040088