Quantitative 1H NMR Metabolomics Reveal Distinct Metabolic Adaptations in Human Macrophages Following Differential Activation

 , ,
, , 
Abstract
:1. Introduction
2. Results
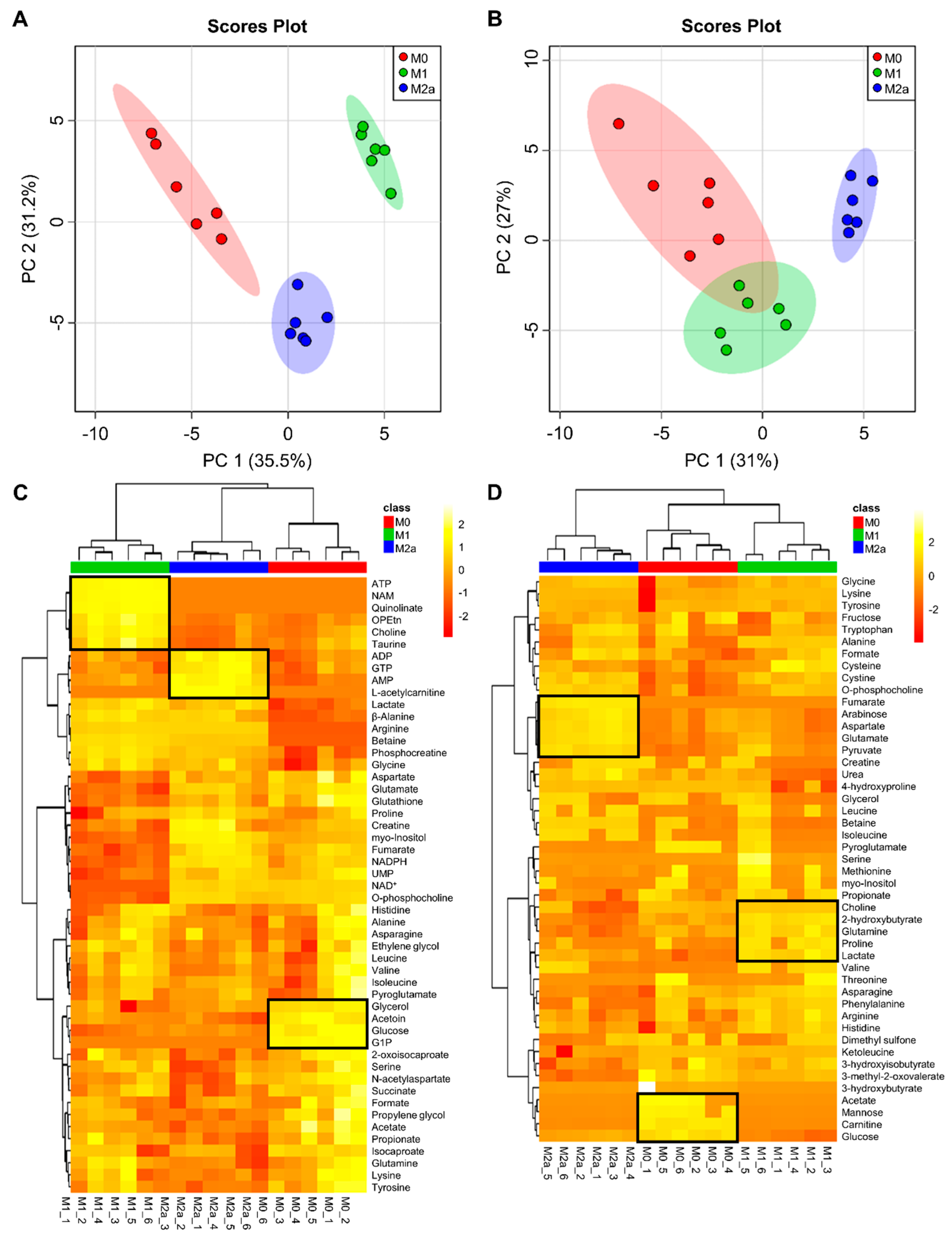
2.1. Quantitative Metabolic Profiles Differentiate between MΦ Activation States
2.2. Glycolytic Activity and Lactic Acid Fermentation Are Universal Markers of Activation
2.3. M1 and M2a MΦs Exhibit Distinct Anaplerotic Trends Corresponding to the TCA Cycle
2.4. Activated MΦs Undergo Significant Oxidative Stress
2.5. M1 MΦs Manipulate the Kennedy Pathway
3. Discussion
4. Materials and Methods
4.1. Primary Human Monocyte Isolation
4.2. Culture of Primary Human Monocyte-Derived MΦs
4.3. Activation of Primary Human Monocyte-Derived MΦs
4.4. Antibodies and Flow Cytometry
4.5. Intra- and Extracellular Metabolite Extraction
4.6. Determination of Protein Content
4.7. NMR Sample Preparation
4.8. NMR Spectra Acquisition and Preprocessing
4.9. NMR Data Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic instruction of immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C. Systems biology: Metabonomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—the link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Galván-Peña, S.; O’Neill, L.A. Metabolic reprogramming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewshi, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016, 17, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Woollard, K.J.; Geissmann, F. Monocytes in atherosclerosis: Subsets and functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Platt, A.M.; Bain, C.C.; Bordon, Y.; Sester, D.P.; Mowat, A.M. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 2010, 184, 6843–6854. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J. Clin. Invest. 2008, 118, 2269–2280. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyoc, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Diskin, C.; Pålsson-McDermott, E.M. Metabolic modulation in macrophage effector function. Front. Immunol. 2018, 9, 270. [Google Scholar] [CrossRef]
- Pavlou, S.; Wang, L.; Xu, H.; Chen, M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J. Inflamm. (Lond.) 2017, 14, 4. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Michl, J.; Ohlbaum, D.J.; Silverstein, S.C. 2-Deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages II. Dissociation of the inhibitory effects of 2-deoxyglucose on phagocytosis and ATP generation. J. Exp. Med. 1976, 144, 1484–1493. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.C.; Nathan, C.F. Man is not a mouse: Reply. J. Leukoc. Biol. 2007, 81, 580. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Mattila, J.T. “Of mice and men”: Arginine metabolism in macrophages. Front. Immunol. 2014, 5, 479. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Milde, R.; Varin, A.; Melgert, B.N.; Draijer, C.; Thomas, B.; Fabbri, M.; Crawshaw, A.; Ho, L.P.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood 2013, 121, e57–e69. [Google Scholar] [CrossRef]
- León, Z.; García-Cañaveras, J.C.; Donato, M.T.; Lahoz, A. Mammalian cell metabolomics: Experimental design and sample preparation. Electrophoresis 2013, 34, 2762–2775. [Google Scholar] [CrossRef]
- Wishart, D.S. Quantitative metabolomics using NMR. Trends Analyt. Chem. 2008, 27, 228–237. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; de Winther, M.P. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J. Vis. Exp. 2015, 105, e53424. [Google Scholar] [CrossRef]
- Feng, J.; Li, L.; Ou, Z.; Li, Q.; Gong, B.; Zhao, Z.; Qi, W.; Zhou, T.; Zhong, J.; Cai, W.; et al. IL-25 stimulates M2 macrophage polarization and thereby promotes mitochondrial respiratory capacity and lipolysis in adipose tissues against obesity. Cell Mol. Immunol. 2018, 15, 493–505. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Longo, D.M.; Louie, B.; Wang, E.; Pos, Z.; Marincola, F.M.; Hawtin, R.E.; Cesano, A. Inter-donor variation in cell subset specific immune signaling responses in healthy individuals. Am. J. Clin. Exp. Immunol. 2012, 1, 1–11. [Google Scholar] [PubMed]
- Carpenter, D.J.; Granot, T.; Matsuoka, N.; Senda, T.; Kumar, B.V.; Thome, J.J.C.; Gordon, C.L.; Miron, M.; Weiner, J.; Connors, T.; et al. Human immunology studies using organ donors: Impact of clinical variations on immune parameters in tissues and circulation. Am. J. Transplant. 2018, 18, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, S.; De Majo, F.; Kim, J.; Trenti, A.; Trevisi, L.; Paolo Fadini, G.; Bolego, C.; Zandstra, P.W.; Cignarella, A.; Vitiello, L. Convenience versus biological significance: Are PMA-differentiated THP-1 cells a reliable substitute for blood-derived macrophages when studying in vitro polarization? Front. Pharmacol. 2018, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy pathway--de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef]
- Tavakoli, S.; Short, J.D.; Downs, K.; Nguyen, H.N.; Lai, Y.; Zhang, W.; Jerabek, P.; Goins, B.; Sadeghi, M.M.; Asmis, R. Differential regulation of macrophage glucose metabolism by macrophage colony-stimulating factor and granulocyte-macrophage colony-stimulating factor: Implications for 18F FDG PET imaging of vessel wall inflammation. Radiology 2017, 283, 87–97. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, C.C.; Batra, S.; McKerrow, J.H.; Loke, P. Delineation of diverse macrophage activation programs in response to intracellular parasites and cytokines. PLoS Negl. Trop. Dis. 2010, 4, e648. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006. [Google Scholar] [CrossRef] [Green Version]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-in-dependent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of SdhB, but not SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Brück, J.; Kellerer, C.; Deng, C.; Peng, H.; Rothfuss, O.; Hussain, R.Z.; Gocke, A.R.; Respa, A.; Glocova, I.; et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 2011, 208, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The reactive oxygen species in macrophage polarization: Reflecting its dual role in progression and treatment of human diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Cha, Y.N.; Kim, C. Taurine chloramine protects RAW 264.7 macrophages against hydrogen peroxide-induced apoptosis by increasing antioxidants. J. Clin. Biochem. Nutr. 2011, 49, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Ryan, A.J.; Murthy, S.; Carter, A.B. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J. Biol. Chem. 2013, 288, 20745–20757. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Kontny, E. Taurine and inflammatory diseases. Amino Acids 2014, 46, 7–20. [Google Scholar] [CrossRef]
- Snider, S.A.; Margison, K.D.; Ghorbani, P.; LeBlond, N.D.; O’Dwyer, C.; Nunes, J.R.C.; Nguyen, T.; Xu, H.; Bennett, S.A.L.; Fullerton, M.D. Choline transport links macrophage phospholipid metabolism and inflammation. J. Biol. Chem. 2018, 293, 11600–11611. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.K.; Singh, B.P.; Arora, N.; Gaur, S.N. Choline attenuates inflammation and suppresses oxidative stress in patients with asthma. Immunobiology 2010, 215, 527–534. [Google Scholar] [CrossRef]
- Richter, K.; Mathes, V.; Fronius, M.; Althaus, M.; Hecker, A.; Krasteva-Christ, G.; Padberg, W.; Hone, A.J.; McIntosh, J.M.; Zakrzewicz, A.; et al. Phosphocholine—An agonist of metabotropic but not of ionotropic functions of α9-containing nicotinic acetylcholine receptors. Sci. Rep. 2016, 6, 28660. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Mamedova, L.K.; Barton, B.; Bradford, B.J. Choline regulates the function of bovine immune cells and alters the mRNA abundance of enzymes and receptors involved in its metabolism in vitro. Front. Immunol. 2018, 9, 2448. [Google Scholar] [CrossRef]
- Gohil, V.M.; Zhu, L.; Baker, C.D.; Cracan, V.; Yaseen, A.; Jain, M.; Clish, C.B.; Brookes, P.S.; Bakovic, M.; Mootha, V.K. Meclizine inhibits mitochondrial respiration through direct targeting of cytosolic phosphoethanolamine metabolism. J. Biol. Chem. 2013, 288, 35387–35395. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Sävman, K.; Heyes, M.P.; Svedin, P.; Karlsson, A. Microglia/macrophage-derived inflammatory mediators galectin-3 and quinolinic acid are elevated in cerebrospinal fluid from newborn infants after birth asphyxia. Transl. Stroke Res. 2013, 4, 228–235. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef]
- Pláteník, J.; Stopka, P.; Vejrazka, M.; Stípek, S. Quinolinic acid-iron(II) complexes: Slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radic. Res. 2001, 34, 445–459. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; López, S.; Castell, J.V.; Donato, M.T.; Lahoz, A. Extending metabolome coverage for untargeted metabolite profiling of adherent cultured hepatic cells. Anal. Bioanal. Chem. 2016, 408, 1217–1230. [Google Scholar] [CrossRef]
- Ammons, M.C.; Tripet, B.P.; Carlson, R.P.; Kirker, K.R.; Gross, M.A.; Stanisich, J.J.; Copié, V. Quantitative NMR metabolite profiling of methicillin-resistant and methicillin-susceptible Staphylococcus aureus discriminates between biofilm and planktonic phenotypes. J. Proteome Res. 2014, 13, 2973–2985. [Google Scholar] [CrossRef]
- Fathi, F.; Brun, A.; Rott, K.H.; Falco Cobra, P.; Tonelli, M.; Eghbalnia, H.R.; Caviedes-Vidal, E.; Karasov, W.H.; Markley, J.L. NMR-based identification of metabolites in polar and non-polar extracts of avian liver. Metabolites 2017, 7, 61. [Google Scholar] [CrossRef]
- Ramm Sander, P.; Peer, M.; Grandl, M.; Bogdahn, U.; Schmitz, G.; Robert Kalbitzer, H. NMR spectroscopy of macrophages loaded with native, oxidized or enzymatically degraded lipoproteins. PLoS ONE 2013, 8, e56360. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.J.; Peters, T.R.; Tripet, B.; Van Vuren, A.; Rakesh; Lee, R.E.; Copié, V.; Teintze, M. Exposure of methicillin-resistant Staphylococcus aureus to low levels of the antibacterial THAM-3ΦG generates a small colony drug-resistant phenotype. Sci. Rep. 2018, 8, 9850. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.L.; Weaver, A.J., Jr.; Tripet, B.P.; Ammons, M.C.B.; Teintze, M.; Copié, V. Characterization of the antibacterial activity of Bald’s eyesalve against drug resistant Staphylococcus aureus and Pseudomonas aeruginosa. PLoS ONE 2018, 13, e0208108. [Google Scholar] [CrossRef] [PubMed]
- Mercier, P.; Lewis, M.J.; Chang, D.; Baker, D.; Wishart, D.S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton NMR spectra. J. Biomol. NMR 2011, 49, 307–323. [Google Scholar] [CrossRef]
- Emwas, A.H.; Saccenti, E.; Gao, X.; McKay, R.T.; Dos Santos, V.A.P.M.; Roy, R.; Wishart, D.S. Recommended strategies for spectral processing and post-processing of 1D 1H-NMR data of biofluids with a particular focus on urine. Metabolomics 2018, 14, 31. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinformatics 2011, 34, 14.10.1–14.10.48. [Google Scholar] [CrossRef]
- Araníbar, N.; Ott, K.H.; Roongta, V.; Mueller, L. Metabolomic analysis using optimized NMR and statistical methods. Anal. Biochem. 2006, 355, 62–70. [Google Scholar] [CrossRef]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourgue, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | M1 MΦs | M2a MΦs | ||
|---|---|---|---|---|
| FC | p-Value | FC | p-Value | |
| 2-Oxoisocaproate | –1.28 | NS | –1.88 | ** |
| Acetate | –1.66 | * | –2.11 | ** |
| ADP | 1.05 | NS | 1.42 | *** |
| AMP | 1.33 | ** | 2.43 | **** |
| Arginine | 7.54 | **** | 7.44 | **** |
| Aspartate | –1.60 | ** | –1.21 | NS |
| ATP | 9.14 | **** | ND | N/A |
| β-Alanine | 3.01 | **** | 2.53 | *** |
| Betaine | 6.38 | **** | 2.95 | **** |
| Choline | 1.63 | *** | –1.18 | NS |
| Creatine | –1.24 | ** | 1.18 | NS |
| Creatine phosphate | 2.81 | **** | 2.13 | *** |
| Fumarate | –1.68 | *** | 1.11 | NS |
| Glucose | –9.85 | *** | –6.68 | ** |
| Glucose-1 phosphate | –5.10 | **** | –5.10 | **** |
| Glutamate | –2.67 | *** | –1.22 | NS |
| Glutamine | 1.02 | NS | –1.33 | * |
| Glutathione | –1.81 | ** | –1.34 | * |
| Glycerol | –3.10 | *** | –2.53 | *** |
| Glycine | 1.27 | ** | 1.21 | * |
| GTP | –1.01 | NS | 1.48 | *** |
| Lactate | 1.79 | **** | 1.66 | **** |
| Lysine | –1.11 | NS | –1.25 | * |
| Myo-Inositol | –2.43 | **** | 1.88 | *** |
| NAD+ | –3.79 | **** | 1.07 | NS |
| NADPH | –2.40 | ** | 1.34 | * |
| Niacinamide | 5.19 | **** | ND | N/A |
| O-phosphocholine | –9.23 | **** | 1.03 | NS |
| O-phosphoethanolamine | 2.00 | *** | –1.02 | NS |
| Propionate | –1.13 | NS | –1.57 | * |
| Quinolinate | 25.90 | **** | ND | N/A |
| Serine | –1.12 | NS | –1.67 | * |
| Succinate | –1.23 | NS | –1.82 | ** |
| Taurine | 1.23 | * | –1.18 | * |
| Tyrosine | –1.20 | NS | –1.50 | * |
| UMP | –1.67 | *** | –1.04 | NS |
| Metabolite | Concentration (Mean ± SD) | p-Value | ||||
|---|---|---|---|---|---|---|
| M0 MΦs | M1 MΦs | M2a MΦs | M1 vs. M0 | M2a vs. M0 | M1 vs. M2a | |
| 2-Hydroxybutyrate | 48.65 ± 13.52 | 132.89 ± 17.24 | 23.43 ± 7.29 | **** | ** | **** |
| 2-Oxoisocaproate | 60.64 ± 10.85 | 26.96 ± 5.31 | 2.81 ± 2.92 | *** | **** | **** |
| 3-Hydroxybutyrate | –20.75 ± 13.78 | –116.64 ± 8.81 | –96.50 ± 3.85 | **** | **** | ** |
| 3-Hydroxyisobutyrate | 18.01 ± 3.36 | 16.87 ± 2.38 | 12.49 ± 2.03 | NS | ** | ** |
| 3-Methyl-2-oxovalerate | 108.34 ± 49.08 | 61.13 ± 9.68 | 21.34 ± 3.60 | NS | ** | **** |
| Acetate | 39.73 ± 73.93 | –150.67 ± 13.89 | –213.14 ± 27.60 | ** | *** | ** |
| Alanine | –60.54 ± 73.40 | 98.11 ± 67.21 | 20.73 ± 35.65 | ** | * | * |
| Arabinose | –107.36 ± 12.28 | –109.08 ± 12.55 | –75.83 ± 4.50 | NS | *** | *** |
| Arginine | 852.67 ± 399.21 | 1349.61 ± 290.20 | 788.70 ± 198.71 | * | NS | ** |
| Aspartate | –791.19 ± 90.54 | –797.31 ± 93.74 | –573.21 ± 22.03 | NS | ** | ** |
| Carnitine | 37.60 ± 39.79 | –10.46 ± 1.09 | –9.89 ± 0.39 | * | * | NS |
| Choline | 13.83 ± 12.57 | 52.20 ± 9.38 | 6.41 ± 14.85 | *** | NS | *** |
| Creatine | 1.50 ± 4.85 | –5.73 ± 5.69 | 3.80 ± 3.65 | * | NS | ** |
| Cysteine | –147.54 ± 22.37 | –103.71 ± 24.21 | –125.00 ± 11.15 | ** | NS | NS |
| Cystine | 183.44 ± 50.04 | 419.86 ± 62.88 | 468.59 ± 62.40 | **** | **** | NS |
| Formate | 140.60 ± 46.15 | 195.50 ± 61.22 | 228.89 ± 66.43 | NS | * | NS |
| Fumarate | ND | ND | 9.51 ± 1.67 | N/A | **** | **** |
| Glucose | –21044.40 ± 1478.98 | –42208.15 ± 4621.45 | –30552.65 ± 1595.13 | **** | **** | ** |
| Glutamate | –674.75 ± 78.82 | –595.54 ± 116.43 | –363.96 ± 18.48 | NS | *** | ** |
| Glutamine | 1235.12 ± 211.97 | 1976.33 ± 320.59 | 731.62 ± 127.38 | ** | *** | **** |
| Glycine | 179.01 ± 122.04 | 404.88 ± 106.51 | 501.91 ± 115.32 | ** | *** | NS |
| Histidine | 41.76 ± 24.39 | 98.44 ± 23.25 | 56.36 ± 17.82 | ** | NS | ** |
| Isoleucine | –78.84 ± 63.44 | –13.97 ± 166.32 | 60.25 ± 54.71 | NS | ** | NS |
| Lactate | 29467.61 ± 2714.66 | 51046.16 ± 4806.45 | 38150.09 ± 2610.21 | **** | *** | *** |
| Leucine | –141.51 ± 88.78 | 28.65 ± 126.92 | 19.93 ± 35.22 | * | ** | NS |
| Lysine | 25.26 ± 51.14 | 325.04 ± 43.60 | 209.60 ± 20.31 | **** | *** | *** |
| Mannose | 9.84 ± 12.78 | –54.62 ± 7.85 | –43.79 ± 7.40 | **** | **** | * |
| O-phosphocholine | –81.15 ± 20.78 | –50.46 ± 7.86 | –42.20 ± 2.26 | * | ** | * |
| Phenylalanine | 2.21 ± 11.13 | 26.02 ± 18.03 | –9.66 ± 9.85 | * | NS | ** |
| Proline | –51.72 ± 53.94 | 60.38 ± 54.56 | –27.75 ± 21.10 | ** | NS | ** |
| Pyroglutamate | 121.33 ± 345.73 | –564.30 ± 551.92 | –386.27 ± 161.22 | * | * | NS |
| Pyruvate | –642.79 ± 67.35 | –489.93 ± 121.02 | –336.50 ± 17.62 | * | **** | * |
| Serine | –179.15 ± 49.58 | –45.09 ± 75.77 | –284.36 ± 89.47 | ** | * | *** |
| Urea | –1521.10 ± 205.87 | –2142.83 ± 454.07 | –1152.24 ± 159.89 | * | ** | ** |
| Valine | –25.75 ± 27.43 | 89.95 ± 97.95 | 10.00 ± 50.05 | * | NS | NS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, A.L.; Schiller, S.M.; Keegan, W.J.; Ammons, M.C.B.; Eilers, B.; Tripet, B.; Copié, V. Quantitative 1H NMR Metabolomics Reveal Distinct Metabolic Adaptations in Human Macrophages Following Differential Activation. Metabolites 2019, 9, 248. https://doi.org/10.3390/metabo9110248
Fuchs AL, Schiller SM, Keegan WJ, Ammons MCB, Eilers B, Tripet B, Copié V. Quantitative 1H NMR Metabolomics Reveal Distinct Metabolic Adaptations in Human Macrophages Following Differential Activation. Metabolites. 2019; 9(11):248. https://doi.org/10.3390/metabo9110248
Chicago/Turabian StyleFuchs, Amanda L., Sage M. Schiller, Wyatt J. Keegan, Mary Cloud B. Ammons, Brian Eilers, Brian Tripet, and Valérie Copié. 2019. "Quantitative 1H NMR Metabolomics Reveal Distinct Metabolic Adaptations in Human Macrophages Following Differential Activation" Metabolites 9, no. 11: 248. https://doi.org/10.3390/metabo9110248