
Vibrational Coherence in the Metal–Metal-Bonded Excited State of Pt(II) Complexes

1
School of Microelectronics, Shanghai University, Shanghai 201800, China
2
Department of Chemistry, State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
3
HKU-CAS Joint Laboratory on New Materials, The University of Hong Kong, Pokfulam Road, Hong Kong, China
*
Authors to whom correspondence should be addressed.
Inorganics 2023, 11(11), 441; https://doi.org/10.3390/inorganics11110441
Submission received: 10 October 2023
/
Revised: 6 November 2023
/
Accepted: 15 November 2023
/
Published: 18 November 2023
(This article belongs to the Special Issue Phosphorescent Materials Based on Organometallic Complexes)
Abstract
:In the past decade, there have been significant advancements in the investigation of coherence-related phenomena in organic systems such as biological photosynthetic reaction centers. The d8 Pt(II) dinuclear complex or molecular aggregate with a metal–metal-to-ligand charge transfer (MMLCT) or metal-centered (MC) excited state was reported to show the vibrational coherence phenomenon in the intersystem crossing (ISC) process, due to the Metal–metal (M-M) interaction at excited state. In this study, we review the coherence effect in the Pt(II)-Pt(II) complexes which are speculated to be a coherent energy conversion system. The impacts of coherence on the photo-physics of Pt(II) dinuclear complexes have been discussed and reviewed, including the intersystem crossing process and vibrational wavepacket dynamics.
1. Introduction
Plants and algae exhibit remarkable efficiency in converting solar energy into stored fuel, even in challenging environments that involve oxygen and water. They are also able to withstand extreme physiological temperatures and extremely low levels of light [1]. This stands in stark contrast to conventional materials used for solar energy conversion, which typically rely on long-range periodicity [2]. Despite their strong disorder, the photosynthetic machinery in biological systems demonstrates remarkable efficiency in converting solar energy. The coherence effect is believed to play a crucial role in facilitating energy transfer during photosynthesis in plants. Coherence refers to the phase relationships among the components of a superposition of waves. When the dynamics are coherent, these phase relationships are maintained for a sufficient duration to have a significant mechanistic and functional impact [3].
The coherence effect has significant implications for the design of light-harvesting materials, particularly in organic systems. There is speculation that coherence effects could be utilized to create more efficient light-harvesting materials, both in the context of organic photovoltaics and in the development of artificial photosynthetic systems [4,5]. By leveraging coherence effects, it may be possible to overcome the challenges presented by disorder and electron-vibration couplings, and to create materials that are more resilient to energy loss and decay [6].
Dinuclear platinum complexes have recently been the subject of intensive study in the context of coherent energy conversion systems [7,8,9,10,11,12,13,14,15,16]. The Pt(II) complexes, which possess a d8 electronic configuration, can generate significant intermolecular orbital overlap between two metal atoms, leading to the formation of a metal–metal-bonded excited state [17]. These dinuclear Pt(II) complex systems have exhibited abundant coherent phenomena [7,8,9,10,11,12,13,14,15]. This review summarizes recent advancements in the vibrational coherence effects of Pt(II) dinuclear complexes. The review begins with an overview of the metal–metal-to-ligand charge transfer (MMLCT) and metal-centered (MC) excited state and coherence effect, followed by a discussion of the spectroscopic evidence of vibrational coherence behavior in several Pt(II) dinuclear complexes or aggregation systems.
Several ultrafast spectroscopy techniques are discussed in this review, which utilize sequences of ultrashort laser pulses to study photoinduced dynamical processes in atoms, molecules and solids and have been proved to be fruitful in chemical reaction investigation [18,19]. Among the vast sea of studies, the coherent vibrational wavepacket (CVWP) formed by an impulsive excitation of laser pulse provide both the electronic and structural dynamics at the initial several picoseconds with ultrahigh time resolution. By detecting the signal related to CVWP, insights to the coherence-related phenomenon in molecular structural changes at excited states could be obtained. And the electronic excited state evolving on the potential surface could be mapped out in real time.
2. 1/3MMLCT and 1/3MC Excited States of d8 Metal Complexes
Metal complexes with d8 electronic configurations, such as Rh(I), Pt(II), and Pd(II) complexes, can form a planar geometry with an open axial site (Scheme 1). When these complexes form dimers or aggregates with close metal–metal (M-M) contacts, the strong M-M Pauli repulsion and orbital overlap between two metal dz2 orbitals can lead to the formation of an M-M bonding orbital of dσ and an antibonding orbital of dσ* (Figure 1a) [20]. Upon photo-excitation, an electron is promoted from the M-M antibonding orbital to the LUMO; an M-M bonding interaction is formed, leading to a contraction of the M-M distance at the excited state (Figure 1c). When the LUMO is a ligand-based π* orbital, this transition is known as the singlet or triplet metal–metal-to-ligand charge transfer (1/3MMLCT) excited state transition with an M-M bond order of 0.5. When the LUMO is a metal-based pσ bonding orbital, this transition is known as the singlet or triplet metal-centered (1/3MC) excited state transition with an M-M bond order of 1. In 1975, Gray and his colleagues reported that Rh(I) aggregates are capable of forming the MMLCT excited state when undergoing self-assembly in a solution [17]. In 1990 and 1991, Gliemann, Miskowski, and their colleagues were the first to identify and report the MMLCT excited state in a series of Pt(II) complexes in the solid state [21,22]. MMLCT emission of a dinuclear Pt(II) system in solution was first observed in a guanidine-bridged binuclear Pt(II) complex by Che and co-workers in 1992 [23]. A series of butterfly-like dinuclear Pt(II) complex was synthesized and reported by Ma and Thompson in 2005 [24]. By altering the bulkiness of the bridging ligand, these dinuclear Pt(II) complexes exhibit tunable metal-to-ligand charge transfer (MLCT) and MMLCT excited states. In 2002, Yam and his colleagues observed and reported the formation of the MMLCT excited state of Pt(II) assemblies through the aggregation process in solution [25]. Despite extensive research in the Pt(II) and Rh(I) systems, there have been limited studies on the emissive excited states of Pd(II) and Au(III) d8 isoelectronic complexes. This scarcity can be attributed to the prevailing belief in the academic community that Pd(II)-Pd(II) or Au(III)-Au(III) bonded excited states are unlikely to exist due to the electrophilic nature of the Pd(II) and Au(III) atom. The first phosphorescent MMLCT emission in Pd(II) molecular aggregates was reported in 2018, where the tridentate Pd(II) isocyanide complexes form aggregates with the MMLCT emission energy at 540 nm [26]. The LMMCT (ligand-to-metal–metal charge transfer) excited state was assigned to a series of Au(III) aggregates in 2019, where the M-M bonding orbital is located at the Au-6pz orbital at the excited state for Au(III) complex [27].
Compared to the MLCT excited state, the MMLCT or MC excited states have lower transition energy and exhibit a larger contribution from metal-ndz2 orbital for d8 metal complexes. Therefore, they can be utilized to make red or near-infrared (NIR) organic light-emitting diodes (OLEDs) with high efficiency. The solid-state thin film based on Pt(II) aggregates with the MMLCT excited state exhibits intense NIR emission with an emission peak maximum at ~740 nm and high emission quantum yield [28]. The MMLCT or MC excited state can also be used in bio-imaging [29] and inner-sphere photo-catalysis reactions [30].
3. Ultrafast Spectroscopy and CVWP
Extensive research has been dedicated to dinuclear d8-d8 metal complexes due to their remarkable photophysical and photochemical properties originating from the lowest singlet and triplet excited states. The dynamics of these excited states can be partially controlled by factors such as (1) solvent effects, (2) moiety rigidity, (3) the type of metal center, and (4) molecular structure, including ligand engineering.
Ultrafast spectroscopy stands as a potent tool for investigating the dynamics of excited states (Table 1). The advancements in laser technology have facilitated the emergence of various ultrafast spectroscopy techniques. The pump/probe wavelength range has been expanded into the extreme ultraviolet and terahertz regions, and temporal resolutions have reached sub-femtosecond scales at specific wavelengths [18,31,32]. The application of methodologies such as luminescence up-conversion, transient absorption anisotropy and multi-dimensional spectroscopies has made it possible to comprehensively track the evolution of excited states across various systems, encompassing the electronic, vibrational and spin states [33,34]. The utilization of white light (or wide range) probe is achieved with the assistance of non-collinear optical parametric amplification and super-continuum generation [35]. This enables the comprehensive monitoring of the entire energy transfer process following the initial photoexcitation, and thus provides an easily accessible but powerful tool for coherent effect study.
The exceptional temporal resolution of ultrafast spectroscopy enables the unraveling of relaxation pathways during photochemical reactions, including the identification of intermediate states and their respective lifetimes. An in-depth understanding and tunability of the excited state dynamics can help optimize the energy transfer properties within or across molecules. Various efforts have been made to study the excited state dynamics of Pt complexes and other inorganic compounds. Here, several examples are listed to show the ability of ultrafast spectroscopies, to name a few. The time-resolved infrared spectroscopy is used to monitor the electron transfer in a Pt(II)-trans-acetylide motif. The electron transfer rate is altered by mode-specific infrared excitation of vibrations that are coupled to the transfer pathway [36]. Two-dimensional infrared spectroscopy is used to track energy transfer across metal centers in platinum complexes featuring a triazole-terminated alkyne ligand of two and six carbons, a perfluorophenyl ligand, and two tri(p-tolyl)phosphine ligands, respectively. Vibrational modes and their coupling even hidden under a complicated linear spectrum of the compound are identified. The transfer is efficient if the high-frequency modes involved are delocalized over both ligands’ high-frequency modes, showing an oscillation of the cross peak along the waiting time [37]. Femtosecond transient absorption is also used to clarify the excited state decay pathway. With the aid of global fitting, the ISC time is measured in a N^N Pt(II) complex with two heteroleptic ligands sample [38].
The vibrational wave packets are commonly detected during the ISC events in transition metal complex dimers and trimers. For complex [FeII(bpy)3]2+ (bpy=2,2′-bipyridine), vibrational wave packets appear in the high spin excited quintet 5T2 state shown as a beating signal over the excited-state absorption region in the first picosecond in transient absorption spectra [39,40]. In the case of [Au(CN)2−] dimer or trimer oligomer, oscillation of the transient absorption in the first picosecond region accompanied with a drastic shortening of the Au-Au bonds is observed [41,42]. Similar metal–metal bond contraction was also observed in Pt(II) dimers [43,44]. The vibrational wavepacket is able to coherently transfer from an initially excited state to another state of different spin after ISC, which is observed in Pt(pop), Pt(ppy) and Pt(bzq) [9,14].
The temporal evolution of vibrational wavepacket provides a maker to inspect, if not able to act as a handle to control the energy relaxation trajectory of chemical reaction [45,46,47,48,49]. As depicted in Figure 2a, a molecule is excited by a laser pulse with a time duration so short that the excitation process is absent of a nuclear motion. Born–Oppenheimer approximation is used here to write product state wavefunctions to designate vibronic levels though it is usually not applicable near conical intersections, where energy transfer between states happens. The resulting excited state encompasses multiple vibrational states as a short-duration laser pulse typically covers a wide frequency domain. The vibrational states move in phase, forming a localized vibrational wavepacket. This vibrational wavepacket resides in a non-equilibrium position, which can be described by the Huang–Rhys factor and evolves over time. It essentially represents the molecule oscillating about its equilibrium position on the potential energy surface (PES), moving back and forth along the PES in relation to the potential coordinate. The coherence is gradually destroyed due to the coupling between the states with thermal bath.
The CVWP could be detected by various laser techniques, which usually behaves as one or several beating signals superimposed on the detected signal depending on the spectroscopy used [47,50,51,52,53]. Here, we limit our scope within the visible range white light pump–probe technique and diplatinum complex, in which an instantaneous light excitation ensures the vibration wavepacket by vertical transition. The evolution of the wavepacket is probed by a second laser pulse with its frequencies satisfying the Franck–Condon condition for vertical transitions, either on the S1 excited state or on other excited states. In contrary to the vibrational spectroscopy based on infrared lasers and X-ray spectroscopies, traditional pump–probe with supercontinuum white light gives an easy access for researchers to the structure and excited state dynamics of molecules. The readers could also refer to reviews for two-dimensional vibrational spectroscopy and X-ray spectroscopies [31,33,54].
Figure 2.
(a) Scheme of vibrational wavepacket induced by laser pulse in the excited state. The wavepacket is a linear combination of the oscillator eigenfunctions with coefficients cn [55]. The vertical arrow indicates an optical excitation of laser pulses. (b) Top: experimental stimulated emission spectra at different delay times for Pt-pop in ethylene glycol, excited at 370 nm. Bottom: simulated emission spectra for different delay times expressed as a function of the vibrational period T = 220 fs for the average vibrational quantum number of 6. The vertical grid lines facilitate the observation of the quantum interference fringes. Reprinted with permission from [43]. Copyright 2011 American Chemical Society.
Figure 2.
(a) Scheme of vibrational wavepacket induced by laser pulse in the excited state. The wavepacket is a linear combination of the oscillator eigenfunctions with coefficients cn [55]. The vertical arrow indicates an optical excitation of laser pulses. (b) Top: experimental stimulated emission spectra at different delay times for Pt-pop in ethylene glycol, excited at 370 nm. Bottom: simulated emission spectra for different delay times expressed as a function of the vibrational period T = 220 fs for the average vibrational quantum number of 6. The vertical grid lines facilitate the observation of the quantum interference fringes. Reprinted with permission from [43]. Copyright 2011 American Chemical Society.

The vibrational coherence revealed by ultrafast spectroscopy offers insights into the states involved in energy transfer and the molecular structural evolution. Consequently, it serves as a metric to differentiate between active and spectator relaxation channels. Nevertheless, some experiments yield conflicting results, highlighting the intricate interplay between electronic and vibrational states [11,15,43]. As a result, the full comprehension of vibrational wavepacket behavior is yet to be achieved to elucidate the dynamics of excited states in chemical reactions.
4. Molecular Structural Dynamics and Vibrational Wavepacket
The vibrational wavepacket reflects the spatial localization of the nuclei at a given instant in a specific mode. The oscillations of vibrational wavepacket over time could be extracted from the time-resolved spectra, providing insights into the molecular structural dynamics.
In Pt(II) complex, the vibrational wavepacket is observed in the time-resolved fluorescence and transient absorption (TA) measurement. At a certain time instant, the Franck–Condon overlap of the vibrational wave functions between the excited state and the ground state (in the case of time-resolved fluorescence) yields a spectrum in the probe wavelength axis, which could be mapped to the axis of the reaction coordinate. One example is the Pt-Pt bond length change for the d8-d8 binuclear complex [Pt2(P2O5H2)4]4− (Pt-pop) complex at excited state, as illustrated in Figure 3b [43]. In the time delay axis, an oscillating signal is overlayed over the transient spectrum signal, as seen in Figure 3a. The vibrational wave function spans a wide range at the initial excitation and gradually localizes, indicating the process of vibrational cooling. A nodal point near the maximum of S1 emission wavelength is observed [43]. Similar results are also observed in broad-band transient absorption spectroscopy of Pt dinuclear complex of [Pt(tBu2Pz)(N^C)]2 [tBu2Pz is 3,5-di-tert-butylpyrazole; N^C is 7,8-benzoquinoline] (Pt-bzq) and Pt-piq (N^C is 1-phenylisoquinoline) [10]. The oscillation signal exhibits a phase difference on either side of the nodal point. The spectral position of the node strongly indicates the origin of the CVWP motion. Ground-state coherence produces a nodal point around the absorption maximum, while excited-state coherence generates a node around the stimulated emission or excited state absorption maximum. Thus, the excited state and the ground state structural information is deduced by the probe wavelength resolved oscillating signal.
By mapping the energy difference between the displaced potential curves (emission energy) onto the Pt-Pt bond length axis, the vibrational wave packet becomes spectrally visualized. The spatial distribution of the wave packet, over time, is attributed to Pt-Pt bond length axis, displaying a harmonic oscillator-like behavior with vibrational cooling. The results reveal that in the case of Pt-pop, the excited S1 state undergoes a 0.191 Å shrinkage in Pt-Pt bond length compared to the S0 ground state, which is illustrated in Figure 3b [43]. Through Fourier analysis, TW Kim et al. were able to discern an upshifted beating frequency of vibrations, signifying the contraction motion of the Pt-Pt bond [10]. The frequencies measured at the ground state bleaching region and the stimulated emission/excited state absorption region in TA data differ for a Pt(II) dimer, for example, 100 and 150 . These distinct frequencies indicate the contraction of the Pt-Pt bond upon excitation, such as 157 at the excited state. The Pt-Pt bond shortening occurs due to the promotion of an electron from the antibonding orbital to a metal-based orbital, i.e., the 3MC transition. Such M-M atom distance shortening at the excited state is also confirmed in other closed-shell d8 and d10 metal complexes by ultrafast spectroscopy and/or DFT/TDDFT calculations [56]. Other experimental strategies, such as X-ray scattering/absorption spectra, further corroborate the bond contraction [44,57,58]. Notably, a Pt-Pt contraction of around 0.31 Å was probed by time-resolved EXAFS (Extended X-ray Absorption Fine Structure) spectroscopy at the triplet excited state [59]. DFT calculations predicted a Pt-Pt bond shortening (0.17–0.5 Å) in the triplet state (T1) using different functionals [59,60]. The coherent vibrational wavepacket thus empowers researchers to investigate structural details through visible-light ultrafast spectroscopy.
5. Excited State Dynamics and Coherent Time
When the electronic state onto which the wave packet is evolving is coupled to another electronic state, the vibrational coherence may be retained during population transfer. Thus, the amplitude change in the wavepacket on the initial and product state gives hint of the population relaxation pathway and timescale.
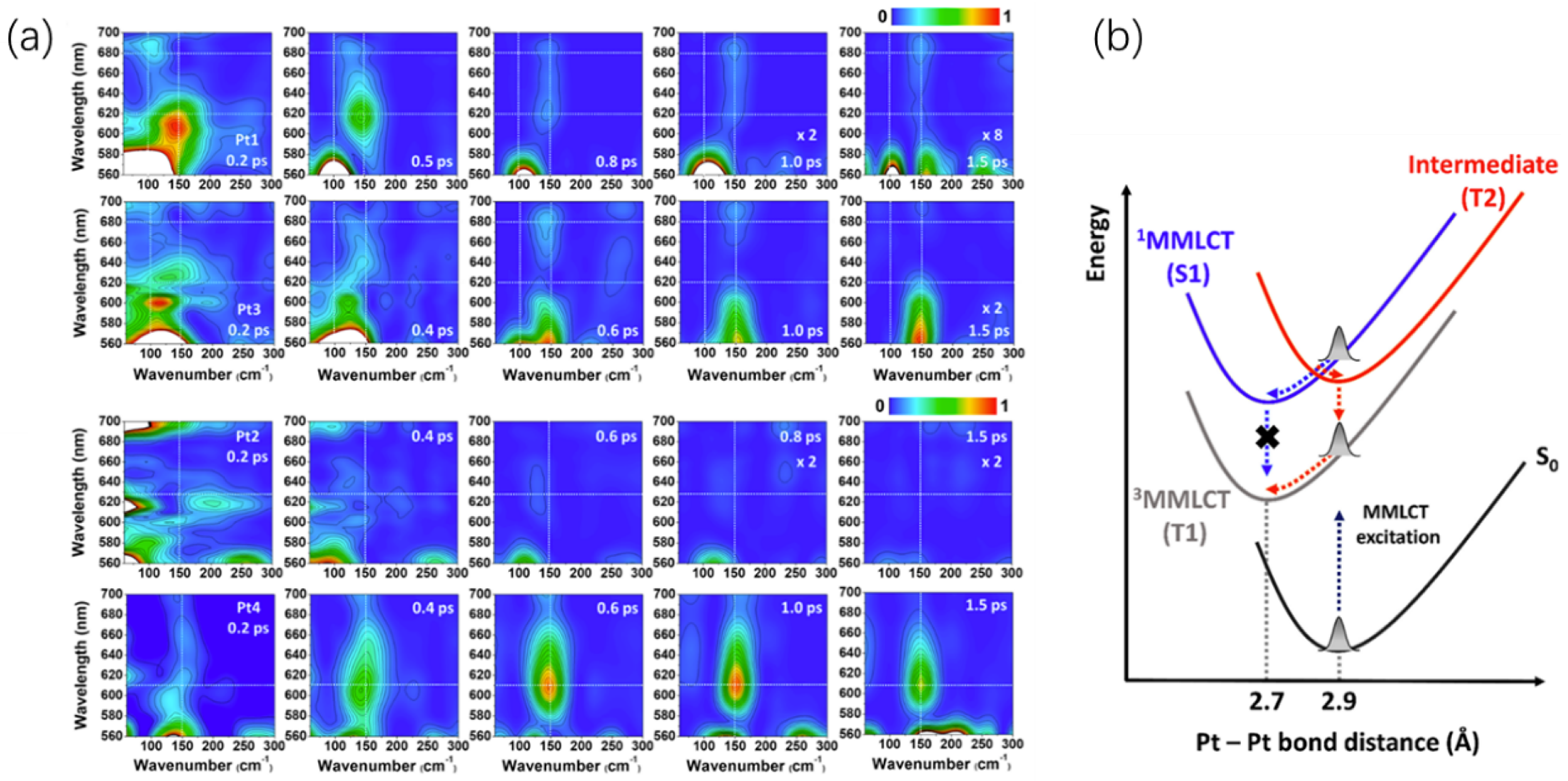
It is usually difficult to determine the relaxation path of the excited state due to the broadening and overlap of spectral features. The white light TA allows us to figure out the vibrational wavepacket evolution along the potential curves which is the probe wavelength in TA data. P. Kim et al. show a routine for excited state trajectory determination [9]. The short-time Fourier transform is utilized to portray the wavepacket evolving on certain potential surfaces. As seen in Figure 4, the amplitude of short-time Fourier transfer (STFT), as functions of time, probe wavelength and oscillation frequency, reveals the evolution of the wavepacket that is either cooling down on one potential curve indicated by a redshift of the oscillation amplitude as time or transferring to another orbital evidenced by a jump of oscillation frequency which is related to a different potential curvature. The results disclose the initial excited-state pathway of intersystem crossing. In Pt(II) dimer complexes with different ligands, there exist two distinct intersystem pathways, as are illustrated in Figure 4b, the vibrational wavepacket either experiences a cooling then transit to the triplet state, or transit to an intermediate triplet state without an observable cooling process then a relax to the final triplet state without spin flip.
Insights into the ISC trajectories from the initially populated Franck–Condon state to the final triplet state are achieved. In Figure 4a, the short-time Fourier transformation amplitude of the TA data for four Pt(II) dimers with different ligands are displayed. Pt1 with the cyclometalating ligand of 2-phenylpyridine (ppy) and bridging ligand of 2-hydroxy-6-methylpyridine (MePyO), and Pt3 with ppy and 2-hydroxy-6-phenylpyridine (PhPyO) ligand, show a matchable CVWP dephasing time to the TA rising time, together with an amplitude transfer from 100 (the ground state Pt-Pt stretching motion) to 150 (the excited state Pt-Pt stretching motion). The STFT map for sample Pt1 reveals a red shift of the 150 peak, which is followed by the CVWP dephasing with a time constant quantitatively matching the ISC. This indicates that the vibrational cooling happens on the singlet excited state potential surface after the excitation, followed by the ISC, which is the trajectory indicated by the blue arrows in Figure 4b. On the contrary, Pt4 with 7,8-benzoquinoline (bzq) and PhPyO shows a long dephasing time beyond the TA rising time, suggesting the retention of the Pt-Pt stretching CVWP motions during the ISC. With the aid of the calculated PESs, an alternative route, that is, S1 () to T2 then T1 excited state is proposed, is illustrated by the red arrows in Figure 4b. The conical intersection of S1 and T2 states resides near the Franck–Condon region, enabling the transition time from S1 to T1 via T2 much shorter than the Pt-Pt stretching period. Thus, most of the CVWP in the final T1 state is retained.
The results show that the ligands could fine-tune the potential energy surface to influence the proximity of the conical intersections of the excited states with the Franck–Condon state and thus to control the branching ratio of the dual ISC pathways. The STFT analysis of the CVWP data provides a visualizable method to study the ultrafast ISC in Pt(II) complex.
The dephasing time is usually complicated, which is related to the coupling between the vibronic states with other states. Thus, the dephasing time may conceive useful information of the excited state dynamics. In Pt-bzq and Pt-piq, two main oscillation frequencies are observed: the lower frequency mode related to the torsional motion of the bzq or piq ligand, and the higher frequency mode related to the Pt-Pt stretching mode. For the ligand torsional mode, the oscillation dephases in a sub-hundred femtosecond range. For the Pt-Pt stretching mode, the dephasing time constants are ~250 fs and 102 fs of Pt-bzq and Pt-piq, respectively, which are comparable or precedent to the ISC time. This suggests that the vibrational coherences associated with the initial formed singlet excited state undergoes dephasing during the ultrafast transition from state (or intermediate state) S1 to S3 [10], while, in contrast, the coherence is partially conserved after the initial ISC in Pt-ppy, with a dephasing time of between 200 and 500 fs [61]. The dephasing time difference leads to a speculation that the stiffness of the ligand structure affects the CVWP decoherence. A rigid ligand scaffold helps to sustain the CVWP motion. Through the comparison between Pt-bzq and Pt-piq complex, it was observed that a more rigid bzq ligand, compared to the piq ligand, leads to a longer CVWP motion in Pt-bzq than in Pt-piq. The large and floppy piq ligand induces a greater degree of structural constraint on the molecular motion, the Pt-Pt interaction. The ligand adopted structural motions circumventing the imposed steric hindrance like torsional motions. Thus, efficient redistribution of the vibrational energy along the torsional motion coordinates will be favored and ultimately bring about accelerated dephasing dynamics [10]. This makes the CVWP oscillating signal difficult to be directly mapped to the energy transfer path, especially when the dephasing time is shorter than the electronical transition time constant. However, the ligand structure engineering provides a tool to coherently control of the photoinduced charge-transfer processes in transition metal complexes (TMCs).
In Pt(II) complex, the pure CVWP dephasing time is not related to the solvent but the ISC rate is largely solvent-dependent [8]. For example, in Pt-pop, the dephasing time is ~2 ps, while the ISC time spans from several hundred femtoseconds to tens of picoseconds. The triplet state forms in a near unity quantum efficiency, despite its direct spin–orbital coupling between singlet and triplet states being symmetrically forbidden. The significant solvent effects on the ISC rate, as well as additional simulations, indicate that there exists an intermediate state assisting the ISC process from S1 to T1 state. In MeCN, an intermediate state (or a batch of states) mixes and solvates the high-lying LMMCT (ligand-to-metal–metal charge transfer) state, making it degenerate with the S1 state. Thus, the proper solvent accelerates the ISC. The CVWP signal reveals the energy transfer process by showing the oscillating amplitude on the ESA signal transfer from state S1 to state T1.
6. Prospective Future Directions
The metal–metal-bonded excited state is not only limited to Pt metal complexes. Other closed-shell d8 or d10 metal complexes, such as Rh(I), Pd(II), Au(I), Cu(I) and Ag(I) dinuclear complexes or molecular aggregates are well-known to form the 3MMLCT or 3MC excited state with a contraction of M-M distance upon photo-excitation. Could these metal complexes also generate the vibrational coherence at excited state as Pt(II) do? If so, what’s the difference of such a coherence phenomenon among various metal complexes? Besides the 3MMLCT and 3MC excited state, d8 Au(III) complexes can also show a Au-Au distance contraction at the T1 state due to the formation of 3LMMCT (ligand-to-metal–metal charge transfer) excited state [27]. When the electron jumps from the ligand to the metal orbital in the LMMCT state, instead of a metal-to-ligand orbital in the MMLCT state, how will the vibrational coherence evolve at the excited state for Au(III) complex?
Author Contributions
Conceptualization, T.Y. and Q.W.; writing—original draft preparation, T.Y. and Q.W.; writing—review and editing, T.Y. and Q.W.; supervision, T.Y. and Q.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant number 22090013).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no known competing financial interest or personal relationship that could have appeared to influence the work reported in this paper.
References
- Brédas, J.-L.; Sargent, E.H.; Scholes, G.D. Photovoltaic concepts inspired by coherence effects in photosynthetic systems. Nat. Mater. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Collini, E.; Wong, C.Y.; Wilk, K.E.; Curmi, P.M.; Brumer, P.; Scholes, G.D. Coherently wired light-harvesting in photosynthetic marine algae at ambient temperature. Nature 2010, 463, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.R.; Ondarse-Alvarez, D.; Oldani, N.; Rodriguez-Hernandez, B.; Alfonso-Hernandez, L.; Galindo, J.F.; Kleiman, V.D.; Fernandez-Alberti, S.; Roitberg, A.E.; Tretiak, S. Coherent exciton-vibrational dynamics and energy transfer in conjugated organics. Nat. Commun. 2018, 9, 2316. [Google Scholar] [CrossRef]
- Haedler, A.T.; Kreger, K.; Issac, A.; Wittmann, B.; Kivala, M.; Hammer, N.; Köhler, J.; Schmidt, H.-W.; Hildner, R. Long-range energy transport in single supramolecular nanofibres at room temperature. Nature 2015, 523, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Scholes, G.D.; Fleming, G.R.; Olaya-Castro, A.; Van Grondelle, R. Lessons from nature about solar light harvesting. Nat. Chem. 2011, 3, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Kim, P.; Fimmel, B.; Würthner, F.; Kim, D. Direct observation of ultrafast coherent exciton dynamics in helical π-stacks of self-assembled perylene bisimides. Nat. Commun. 2015, 6, 8646. [Google Scholar] [CrossRef] [PubMed]
- Leshchev, D.; Valentine, A.J.S.; Kim, P.; Mills, A.W.; Roy, S.; Chakraborty, A.; Biasin, E.; Haldrup, K.; Hsu, D.J.; Kirschner, M.S. Revealing Excited-State Trajectories on Potential Energy Surfaces with Atomic Resolution in Real Time. Angew. Chem. Int. Ed. 2023, 62, e202304615. [Google Scholar] [CrossRef]
- Monni, R.; Capano, G.; Auböck, G.; Gray, H.B.; Vlček, A.; Tavernelli, I.; Chergui, M. Vibrational coherence transfer in the ultrafast intersystem crossing of a diplatinum complex in solution. Proc. Natl. Acad. Sci. USA 2018, 115, E6396–E6403. [Google Scholar] [CrossRef]
- Kim, P.; Valentine, A.J.S.; Roy, S.; Mills, A.W.; Chakraborty, A.; Castellano, F.N.; Li, X.; Chen, L.X. Ultrafast Excited-State Dynamics of Photoluminescent Pt(II) Dimers Probed by a Coherent Vibrational Wavepacket. J. Phys. Chem. C 2021, 12, 6794–6803. [Google Scholar] [CrossRef]
- Kim, T.W.; Kim, P.; Mills, A.W.; Chakraborty, A.; Kromer, S.; Valentine, A.J.S.; Castellano, F.N.; Li, X.; Chen, L.X. Ligand-Structure-Dependent Coherent Vibrational Wavepacket Dynamics in Pyrazolate-Bridged Pt(II) Dimers. J. Phys. Chem. C 2022, 126, 11487–11497. [Google Scholar] [CrossRef]
- Kim, P.; Kelley, M.S.; Chakraborty, A.; Wong, N.L.; Van Duyne, R.P.; Schatz, G.C.; Castellano, F.N.; Chen, L.X. Coherent vibrational wavepacket dynamics in platinum (II) dimers and their implications. J. Phys. Chem. C 2018, 122, 14195–14204. [Google Scholar] [CrossRef]
- Iwamura, M.; Fukui, A.; Nozaki, K.; Kuramochi, H.; Takeuchi, S.; Tahara, T. Coherent Vibration and Femtosecond Dynamics of the Platinum Complex Oligomers upon Intermolecular Bond Formation in the Excited State. Angew. Chem. Int. Ed. 2020, 59, 23154–23161. [Google Scholar] [CrossRef] [PubMed]
- Radler, J.J.; Lingerfelt, D.B.; Castellano, F.N.; Chen, L.X.; Li, X. Role of vibrational dynamics on excited-state electronic coherence in a binuclear platinum complex. J. Phys. Chem. A 2018, 122, 5071–5077. [Google Scholar] [CrossRef]
- Monni, R.; Auböck, G.; Kinschel, D.; Aziz-Lange, K.M.; Gray, H.B.; Vlček, A.; Chergui, M. Conservation of vibrational coherence in ultrafast electronic relaxation: The case of diplatinum complexes in solution. Chem. Phys. Lett. 2017, 683, 112–120. [Google Scholar] [CrossRef]
- Cho, S.; Mara, M.W.; Wang, X.; Lockard, J.V.; Rachford, A.A.; Castellano, F.N.; Chen, L.X. Coherence in metal-metal-to-ligand-charge-transfer excited states of a dimetallic complex investigated by ultrafast transient absorption anisotropy. J. Phys. Chem. A 2011, 115, 3990–3996. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tong, G.S.M.; Wan, Q.; Cheng, G.; Tong, W.-Y.; Ang, W.-H.; Kwong, W.-L.; Che, C.-M. Highly phosphorescent platinum(II) emitters: Photophysics, materials and biological applications. Chem. Sci. 2016, 7, 1653–1673. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.R.; Gordon, J.; Gray, H.B. Characterization of oligomers of tetrakis (phenyl isocyanide) rhodium (I) in acetonitrile solution. J. Am. Chem. Soc. 1975, 97, 3553–3555. [Google Scholar] [CrossRef]
- Maiuri, M.; Garavelli, M.; Cerullo, G. Ultrafast Spectroscopy: State of the Art and Open Challenges. J. Am. Chem. Soc. 2020, 142, 3–15. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond. J. Phys. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Wan, Q.; Yang, J.; To, W.-P.; Che, C.-M. Strong metal-metal Pauli repulsion leads to repulsive metallophilicity in closed-shell d8 and d10 organometallic complexes. Proc. Natl. Acad. Sci. USA 2021, 118, e2019265118. [Google Scholar] [CrossRef]
- Biedermann, J.; Gliemann, G.; Klement, U.; Range, K.-J.; Zabel, M. Spectroscopic studies of cyclo-metallated Pt (lI) complexes: Optical absorption and emission and the structure of single crystal [Pt (bpm)(CN)2]·H2O (bpm = 2, 2′-bipyrimidine). Inorganica Chim. Acta 1990, 169, 63–70. [Google Scholar] [CrossRef]
- Miskowski, V.M.; Houlding, V.H. Electronic spectra and photophysics of platinum(II) complexes with a-diimine ligands solid-state effects. 2. Metal-metal interaction in double salts and linear chains. Inorg. Chem. 1991, 30, 4446–4452. [Google Scholar] [CrossRef]
- Yip, H.-K.; Che, C.-M.; Zhou, Z.-Y.; Mak, T. Photophysical Properties and X-ray Crystal Structure of a Luminescent Platinum (II) Dimer [Pt2(2,2-6,2-terpyridine)2(Gua)](CIO4)3.H2O (Gua = guanidine anion). Chem. Commun. 1992, 1369, 1369–1371. [Google Scholar] [CrossRef]
- Ma, B.; Li, J.; Djurovich, P.I.; Yousufuddin, M.; Bau, R.; Thompson, M.E. Synthetic control of pt…pt separation and photophysics of binuclear platinum complexes. J. Am. Chem. Soc. 2005, 127, 28–29. [Google Scholar] [CrossRef]
- Yam, V.W.-W.; Wong, K.M.-C.; Zhu, N. Solvent-Induced Aggregation through Metal…Metal/π…π Interactions: Large Solvatochromism of Luminescent Organoplatinum (II) Terpyridyl Complexes. J. Am. Chem. Soc. 2002, 124, 6506–6507. [Google Scholar] [CrossRef]
- Wan, Q.; To, W.-P.; Yang, C.; Che, C.-M. The metal-metal-to-ligand charge transfer excited state and supramolecular polymerization of luminescent pincer PdII-isocyanide complexes. Angew. Chem. Int. Ed. 2018, 57, 3089–3093. [Google Scholar] [CrossRef]
- Wan, Q.; Xia, J.; Lu, W.; Yang, J.; Che, C.-M. Kinetically controlled self-assembly of phosphorescent AuIII aggregates and ligand-to-metal–metal charge transfer excited state: A combined spectroscopic and DFT/TDDFT study. J. Am. Chem. Soc. 2019, 141, 11572–11582. [Google Scholar] [CrossRef]
- Ly, K.T.; Chen-Cheng, R.-W.; Lin, H.-W.; Shiau, Y.-J.; Liu, S.-H.; Chou, P.-T.; Tsao, C.-S.; Huang, Y.-C.; Chi, Y. Near-infrared organic light-emitting diodes with very high external quantum efficiency and radiance. Nat. Photonics 2017, 11, 63–68. [Google Scholar]
- Mauro, M.; Aliprandi, A.; Septiadi, D.; Kehr, N.S.; De Cola, L. When self-assembly meets biology: Luminescent platinum complexes for imaging applications. Chem. Soc. Rev. 2014, 43, 4144–4166. [Google Scholar] [CrossRef]
- Roundhill, D.M.; Gray, H.B.; Che, C.-M. Pyrophosphito-bridged diplatinum chemistry. Acc. Chem. Res. 1989, 22, 55–61. [Google Scholar] [CrossRef]
- Chergui, M.; Collet, E. Photoinduced Structural Dynamics of Molecular Systems Mapped by Time-Resolved X-ray Methods. Chem. Rev. 2017, 117, 11025–11065. [Google Scholar] [CrossRef] [PubMed]
- Fayer, M.D. Dynamics of Liquids, Molecules, and Proteins Measured with Ultrafast 2D IR Vibrational Echo Chemical Exchange Spectroscopy. Annu. Rev. Phys. Chem. 2009, 60, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Kraack, J.P.; Hamm, P. Surface-Sensitive and Surface-Specific Ultrafast Two-Dimensional Vibrational Spectroscopy. Chem. Rev. 2017, 117, 10623–10664. [Google Scholar] [CrossRef]
- Brixner, T.; Mančal, T.; Stiopkin, I.V.; Fleming, G.R. Phase-stabilized two-dimensional electronic spectroscopy. J. Chem. Phys. 2004, 121, 4221–4236. [Google Scholar] [CrossRef] [PubMed]
- Dubietis, A.; Couairon, A. Governing Physical Effects. In Ultrafast Supercontinuum Generation in Transparent Solid-State Media; Springer International Publishing: Cham, Switzerland, 2019; pp. 9–26. [Google Scholar]
- Delor, M.; Scattergood, P.A.; Sazanovich, I.V.; Parker, A.W.; Greetham, G.M.; Meijer, A.J.H.M.; Towrie, M.; Weinstein, J.A. Toward control of electron transfer in donor-acceptor molecules by bond-specific infrared excitation. Science 2014, 346, 1492–1495. [Google Scholar] [CrossRef]
- Leong, T.X.; Collins, B.K.; Dey Baksi, S.; Mackin, R.T.; Sribnyi, A.; Burin, A.L.; Gladysz, J.A.; Rubtsov, I.V. Tracking Energy Transfer across a Platinum Center. J. Phys. Chem. A 2022, 126, 4915–4930. [Google Scholar] [CrossRef]
- Wang, P.; Koo, Y.H.; Kim, W.; Yang, W.; Cui, X.; Ji, W.; Zhao, J.; Kim, D. Broadband Visible Light Harvesting N^N Pt(II) Bisacetylide Complex with Bodipy and Naphthalene Diimide Ligands: Förster Resonance Energy Transfer and Intersystem Crossing. J. Phys. Chem. C 2017, 121, 11117–11128. [Google Scholar] [CrossRef]
- Consani, C.; Prémont-Schwarz, M.; ElNahhas, A.; Bressler, C.; van Mourik, F.; Cannizzo, A.; Chergui, M. Vibrational Coherences and Relaxation in the High-Spin State of Aqueous [FeII(bpy)3]2+ Angew. Chem. Int. Ed. Engl. 2009, 48, 7184–7187. [Google Scholar] [CrossRef]
- Auböck, G.; Chergui, M. Sub-50-fs photoinduced spin crossover in [Fe(bpy)3]2+. Nat. Chem. 2015, 7, 629–633. [Google Scholar] [CrossRef]
- Iwamura, M.; Nozaki, K.; Takeuchi, S.; Tahara, T. Real-Time Observation of Tight Au–Au Bond Formation and Relevant Coherent Motion upon Photoexcitation of [Au(CN)2–] Oligomers. J. Am. Chem. Soc. 2013, 135, 538–541. [Google Scholar] [CrossRef]
- Iwamura, M.; Wakabayashi, R.; Maeba, J.; Nozaki, K.; Takeuchi, S.; Tahara, T. Coherent vibration and ultrafast dynamics upon bond formation in excited dimers of an Au(I) complex. Phys. Chem. Chem. Phys. 2016, 18, 5103–5107. [Google Scholar] [CrossRef]
- van der Veen, R.M.; Cannizzo, A.; van Mourik, F.; Vlček, A., Jr.; Chergui, M. Vibrational Relaxation and Intersystem Crossing of Binuclear Metal Complexes in Solution. J. Am. Chem. Soc. 2011, 133, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Haldrup, K.; Dohn, A.O.; Shelby, M.L.; Mara, M.W.; Stickrath, A.B.; Harpham, M.R.; Huang, J.; Zhang, X.; Møller, K.B.; Chakraborty, A.; et al. Butterfly Deformation Modes in a Photoexcited Pyrazolate-Bridged Pt Complex Measured by Time-Resolved X-ray Scattering in Solution. J. Phys. Chem. A 2016, 120, 7475–7483. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.S.; Rosker, M.J.; Zewail, A.H. Femtosecond real-time probing of reactions. IV. The reactions of alkali halides. J. Chem. Phys. 1989, 91, 7415–7436. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Cina, J.A. What can short-pulse pump-probe spectroscopy tell us about Franck-Condon dynamics? J. Chem. Phys. 1999, 110, 9793–9806. [Google Scholar] [CrossRef]
- Vos, M.H.; Rappaport, F.; Lambry, J.-C.; Breton, J.; Martin, J.-L. Visualization of coherent nuclear motion in a membrane protein by femtosecond spectroscopy. Nature 1993, 363, 320–325. [Google Scholar] [CrossRef]
- Jonas, D.M.; Bradforth, S.E.; Passino, S.A.; Fleming, G.R. Femtosecond Wavepacket Spectroscopy: Influence of Temperature, Wavelength, and Pulse Duration. J. Phys. Chem. 1995, 99, 2594–2608. [Google Scholar] [CrossRef]
- Dantus, M.; Lozovoy, V.V. Experimental Coherent Laser Control of Physicochemical Processes. Chem. Rev. 2004, 104, 1813–1860. [Google Scholar] [CrossRef]
- Letokhov, V.S.; Tyakht, V.V. Coherent Vibrational Wave Packet Dynamics in Femtosecond Laser Excitation of Diatomic Molecules. Isr. J. Chem. 1990, 30, 189–195. [Google Scholar] [CrossRef]
- Song, Y.; Clafton, S.N.; Pensack, R.D.; Kee, T.W.; Scholes, G.D. Vibrational coherence probes the mechanism of ultrafast electron transfer in polymer–fullerene blends. Nat. Commun. 2014, 5, 4933. [Google Scholar] [CrossRef]
- Cina, J.A.; Fleming, G.R. Vibrational Coherence Transfer and Trapping as Sources for Long-Lived Quantum Beats in Polarized Emission from Energy Transfer Complexes. J. Phys. Chem. A 2004, 108, 11196–11208. [Google Scholar] [CrossRef]
- Duan, H.G.; Jha, A.; Li, X.; Tiwari, V.; Ye, H.; Nayak, P.K.; Zhu, X.L.; Li, Z.; Martinez, T.J.; Thorwart, M.; et al. Intermolecular vibrations mediate ultrafast singlet fission. Sci. Adv. 2020, 6, eabb0052. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.B.; Wilk, K.E.; Curmi, P.M.G.; Scholes, G.D. Comparison of Electronic and Vibrational Coherence Measured by Two-Dimensional Electronic Spectroscopy. J. Phys. Chem. Lett. 2011, 2, 1904–1911. [Google Scholar] [CrossRef]
- Lanzani, G.; Cerullo, G.; De Silvestri, S. Coherent Vibrational Dynamics; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Schmidbaur, H.; Raubenheimer, H.G. Excimer and exciplex formation in gold (I) complexes preconditioned by aurophilic interactions. Angew. Chem. Int. Ed. 2020, 59, 14748–14771. [Google Scholar] [CrossRef] [PubMed]
- Haldrup, K.; Levi, G.; Biasin, E.; Vester, P.; Laursen, M.G.; Beyer, F.; Kjær, K.S.; Brandt van Driel, T.; Harlang, T.; Dohn, A.O.; et al. Ultrafast X-Ray Scattering Measurements of Coherent Structural Dynamics on the Ground-State Potential Energy Surface of a Diplatinum Molecule. Phys. Rev. Lett. 2019, 122, 063001. [Google Scholar] [CrossRef]
- Lockard, J.V.; Rachford, A.A.; Smolentsev, G.; Stickrath, A.B.; Wang, X.; Zhang, X.; Atenkoffer, K.; Jennings, G.; Soldatov, A.; Rheingold, A.L.; et al. Triplet Excited State Distortions in a Pyrazolate Bridged Platinum Dimer Measured by X-ray Transient Absorption Spectroscopy. J. Phys. Chem. A 2010, 114, 12780–12787. [Google Scholar] [CrossRef]
- van der Veen, R.M.; Milne, C.J.; El Nahhas, A.; Lima, F.A.; Pham, V.-T.; Best, J.; Weinstein, J.A.; Borca, C.N.; Abela, R.; Bressler, C.; et al. Structural determination of a photochemically active diplatinum molecule by time-resolved EXAFS spectroscopy. Angew. Chem. Int. Ed. 2009, 48, 2711–2714. [Google Scholar] [CrossRef]
- Novozhilova, I.V.; Volkov, A.V.; Coppens, P. Theoretical analysis of the triplet excited state of the [Pt2(H2P2O5)4]4- ion and comparison with time-resolved X-ray and spectroscopic results. J. Am. Chem. Soc. 2003, 125, 1079–1087. [Google Scholar] [CrossRef]
- Mewes, L.; Ingle, R.A.; Megow, S.; Böhnke, H.; Baranoff, E.; Temps, F.; Chergui, M. Ultrafast Intersystem Crossing and Structural Dynamics of [Pt(ppy)(μ-tBu2pz)]2. lnorg. Chem. 2020, 59, 14643–14653. [Google Scholar] [CrossRef]
Scheme 1.
Selected reported d8 metal complexes that can form the M-M bonding interaction at the excited state.
Scheme 1.
Selected reported d8 metal complexes that can form the M-M bonding interaction at the excited state.
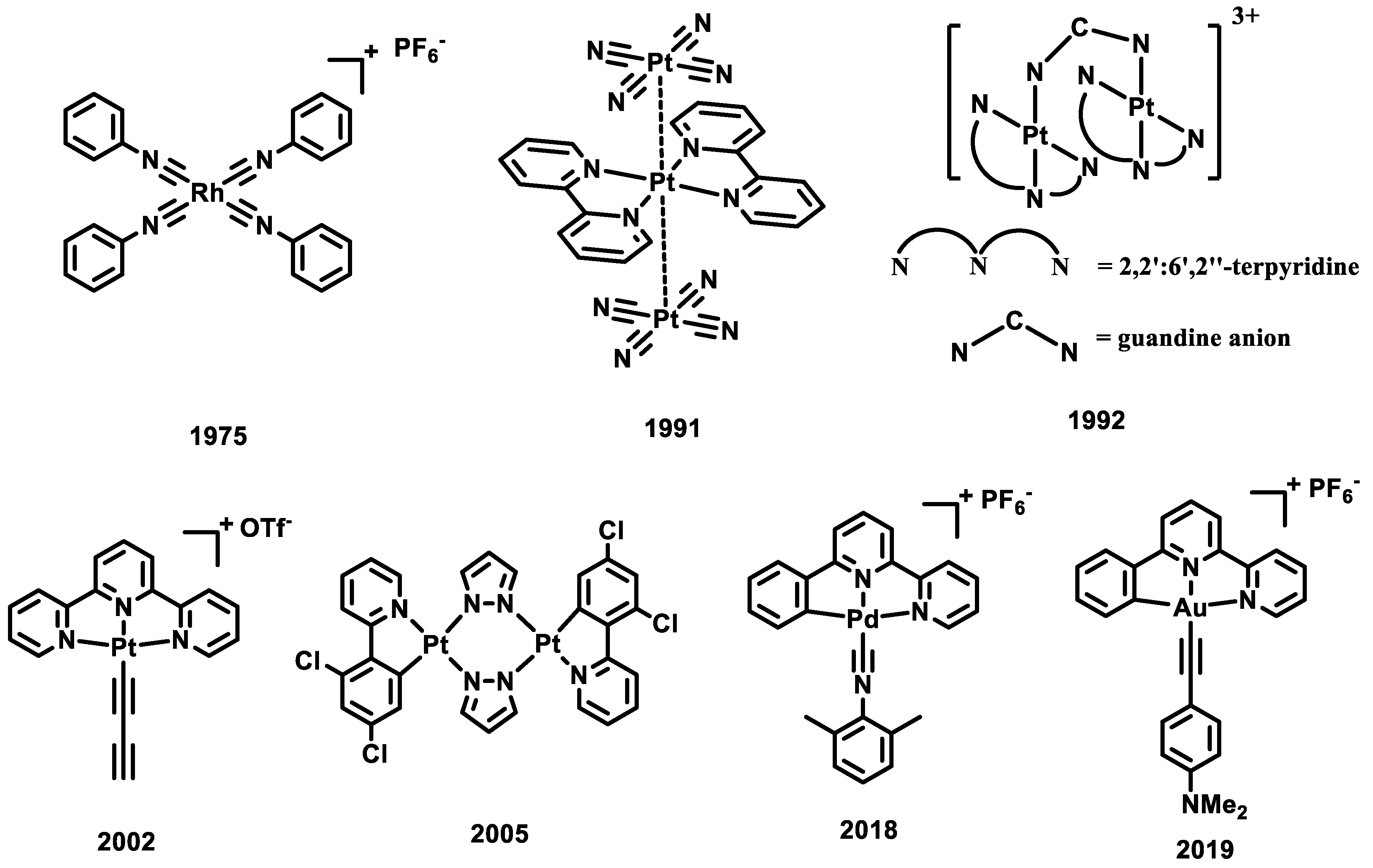
Figure 1.
Formation of the (a) MC and (b) MMLCT excited state in a d8-d8 metal complex system. (c) M-M bond contraction upon photoexcitation. 3MMLCT: triplet metal–metal-to-ligand charge transfer excited state. 3MC: triplet metal-centered excited state.
Figure 1.
Formation of the (a) MC and (b) MMLCT excited state in a d8-d8 metal complex system. (c) M-M bond contraction upon photoexcitation. 3MMLCT: triplet metal–metal-to-ligand charge transfer excited state. 3MC: triplet metal-centered excited state.

Figure 3.
(a) A 2D time–wavelength plot of the transient absorption spectra for Pt-pop in ethylene glycol excited with a ~100 fs laser pulse at 370 nm. The plotted area is the stimulated emission (SE) region. The false colored map shows of the sample where red indicate positive signal. Reprinted with permission from [43]. Copyright 2011 American Chemical Society. (b) Illustration for the metal–metal bond contraction of the Pt-pop molecule upon photoexcitation.
Figure 3.
(a) A 2D time–wavelength plot of the transient absorption spectra for Pt-pop in ethylene glycol excited with a ~100 fs laser pulse at 370 nm. The plotted area is the stimulated emission (SE) region. The false colored map shows of the sample where red indicate positive signal. Reprinted with permission from [43]. Copyright 2011 American Chemical Society. (b) Illustration for the metal–metal bond contraction of the Pt-pop molecule upon photoexcitation.
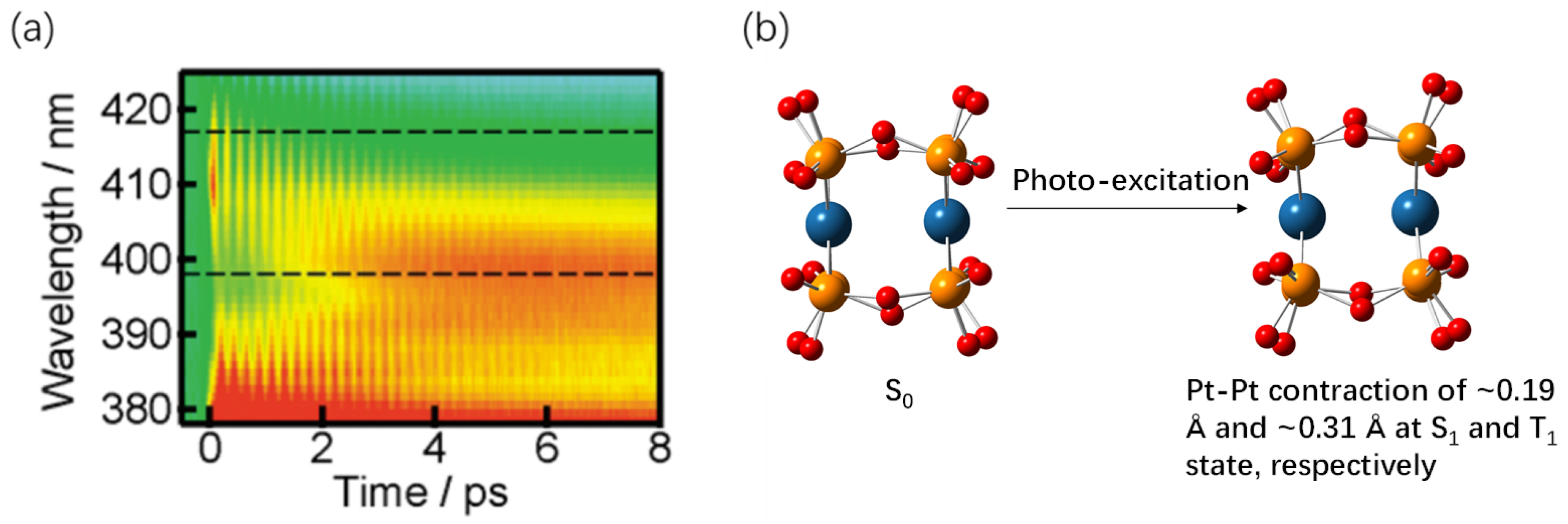
Figure 4.
(a) STFT maps for Pt(II) samples. From top to the bottom, the Pt1 (ppy and MePyO), Pt2 (bzq and PhPyO), Pt3 (ppy and PhPyO) and Pt4 (bzq and PhPyO). The data are acquired from the TA measurement of the samples in THF. (b) The two trajectories proposed for Pt−Pt stretching CVWP dynamics during ISC process. Blue arrows indicate the ISC pathway [S1(ν = n; n > 0) → S1(ν = 0) → T1], while the red arrows show another ISC path via the intermediate state T2 [S1 (ν = n; n > 0) → T2 → T1]. Reprinted with permission from [9]. Copyright 2011 American Chemical Society.
Figure 4.
(a) STFT maps for Pt(II) samples. From top to the bottom, the Pt1 (ppy and MePyO), Pt2 (bzq and PhPyO), Pt3 (ppy and PhPyO) and Pt4 (bzq and PhPyO). The data are acquired from the TA measurement of the samples in THF. (b) The two trajectories proposed for Pt−Pt stretching CVWP dynamics during ISC process. Blue arrows indicate the ISC pathway [S1(ν = n; n > 0) → S1(ν = 0) → T1], while the red arrows show another ISC path via the intermediate state T2 [S1 (ν = n; n > 0) → T2 → T1]. Reprinted with permission from [9]. Copyright 2011 American Chemical Society.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Commonly used ultrafast spectroscopy techniques.
| Technique | Advantages | Disadvantages |
|---|---|---|
| Visible transient absorption (TA) | Information about excited state dynamics such as internal conversion, ISC and so on. Simple setup. | Blurred signals due to broadening. Mandatory requirement of optically allowed electronic transition. |
| Time-resolved vibrational spectroscopy (IR) | Information about molecular vibrations at excited state. | Mandatory requirement of symmetry allowed vibrational modes. Challenging to set up. Costly lasers and detectors. |
| Time-resolved X-ray diffraction | Information about molecular structural changes at excited state. High temporal and spatial resolution. | Requirement of specially prepared samples. Potentially sample damage by radiation. Complexity and synchrotron radiation facilities are usually required. |
| 2D spectroscopy | Enhanced resolution for signals mixed due to inhomogeneous broadening, compared to TA technique. Direct inspection of correlated state. | Long data acquisition time, stable samples required. Challenging to set up. |
| Transient Raman | Information about molecular vibrations and conformational changes. | Weak signal. Interfered by fluorescence signal. |
| Time-resolved photoluminescence | Wide range of detection time delay. Simple setup. | Mandatory requirement of detectable photoluminescence of the sample. Inferior time resolution. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yan, T.; Wan, Q. Vibrational Coherence in the Metal–Metal-Bonded Excited State of Pt(II) Complexes. Inorganics 2023, 11, 441. https://doi.org/10.3390/inorganics11110441
AMA Style
Yan T, Wan Q. Vibrational Coherence in the Metal–Metal-Bonded Excited State of Pt(II) Complexes. Inorganics. 2023; 11(11):441. https://doi.org/10.3390/inorganics11110441
Chicago/Turabian StyleYan, Tengfei, and Qingyun Wan. 2023. "Vibrational Coherence in the Metal–Metal-Bonded Excited State of Pt(II) Complexes" Inorganics 11, no. 11: 441. https://doi.org/10.3390/inorganics11110441
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.