
Exploring the Use of Intracellular Chelation and Non-Iron Metals to Program Ferroptosis for Anticancer Application
 , and
, and
Abstract
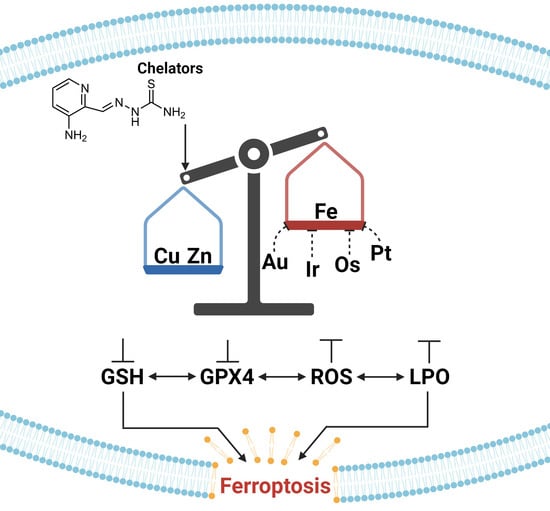
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
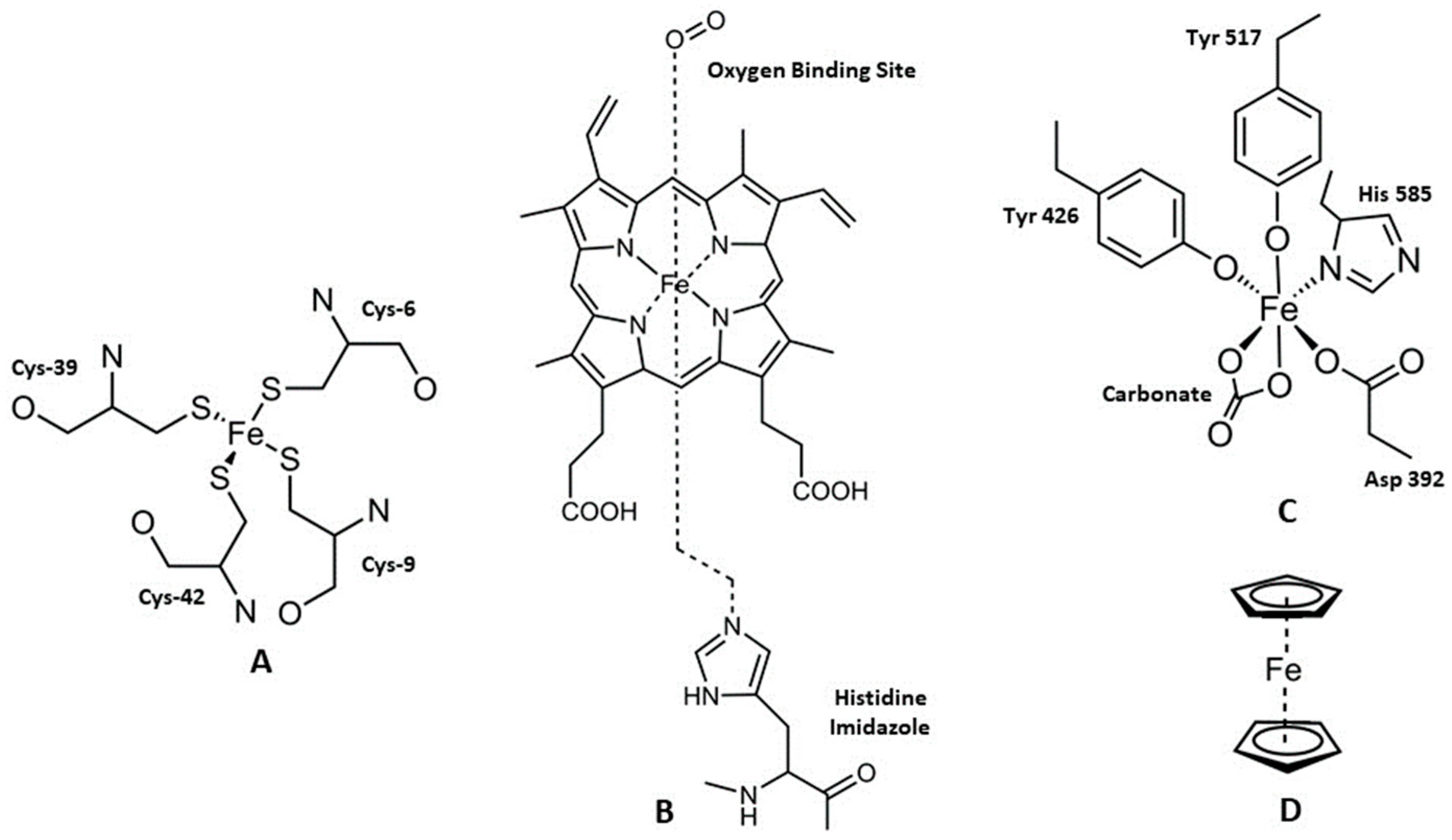
2. Iron and Cancer
2.1. Iron Chelation and Cancer Therapy
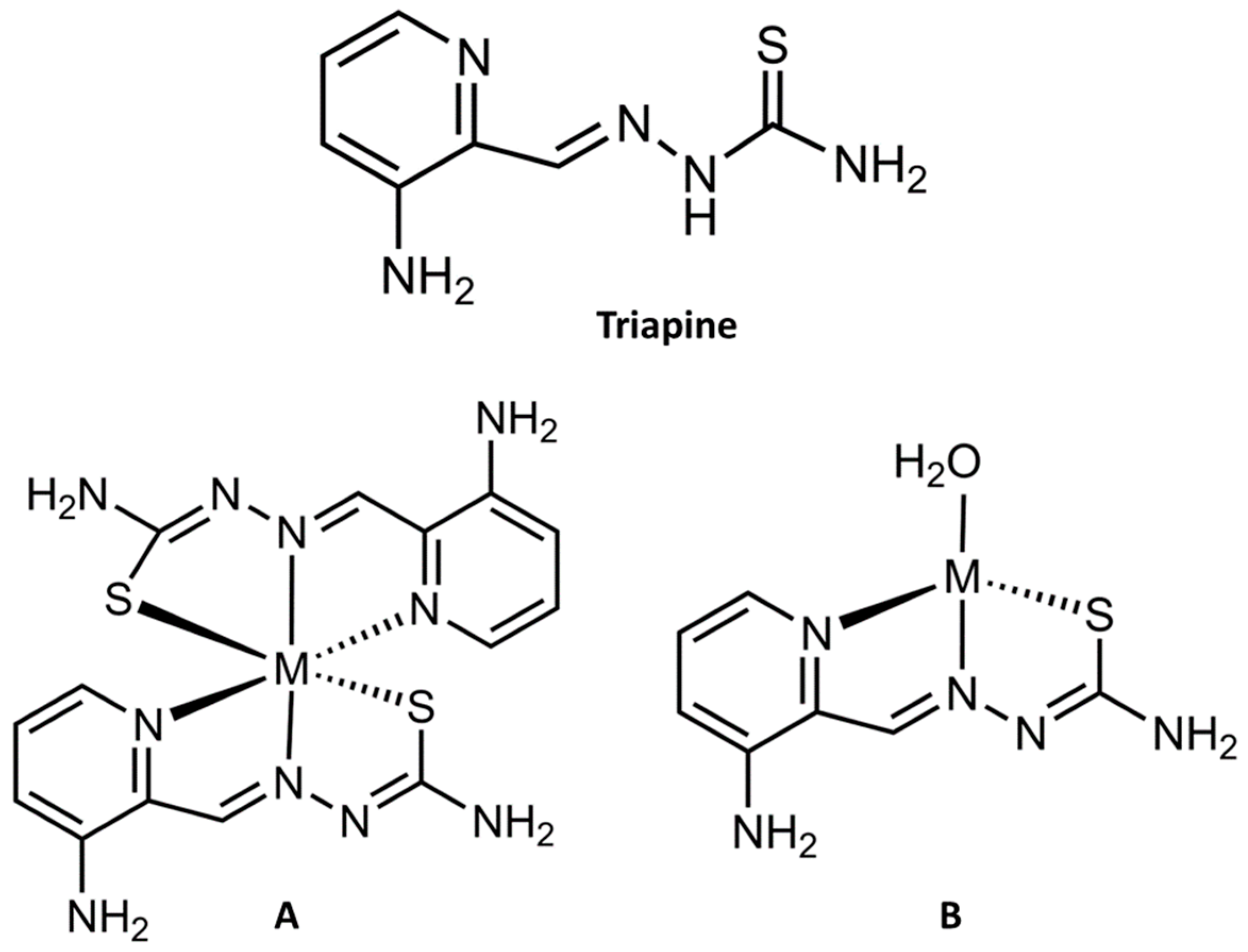
2.2. Fe Chelation by Triapine Induces Ferroptosis
3. Cu and Zn Contribute to Regulating Fe Homeostasis
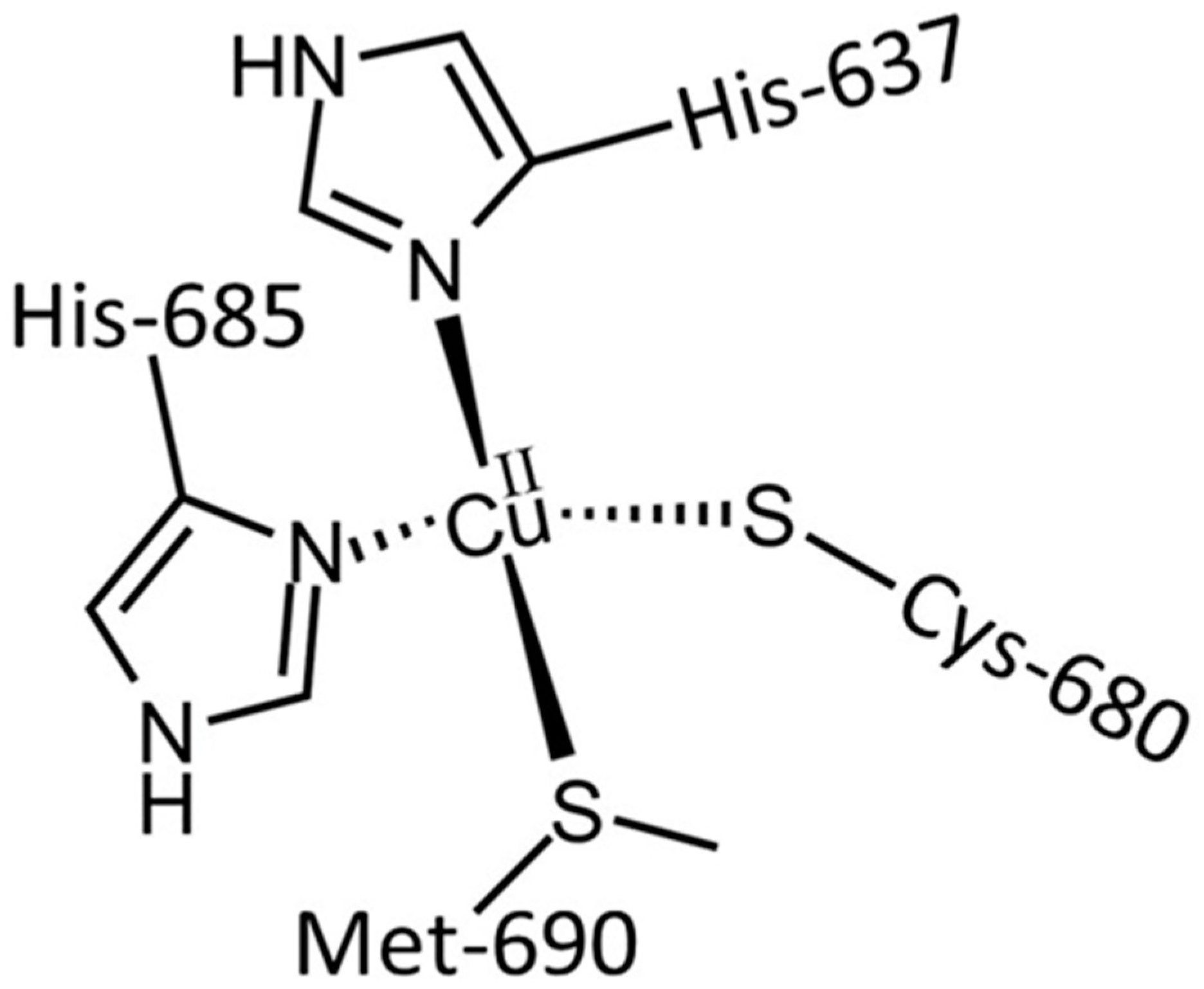
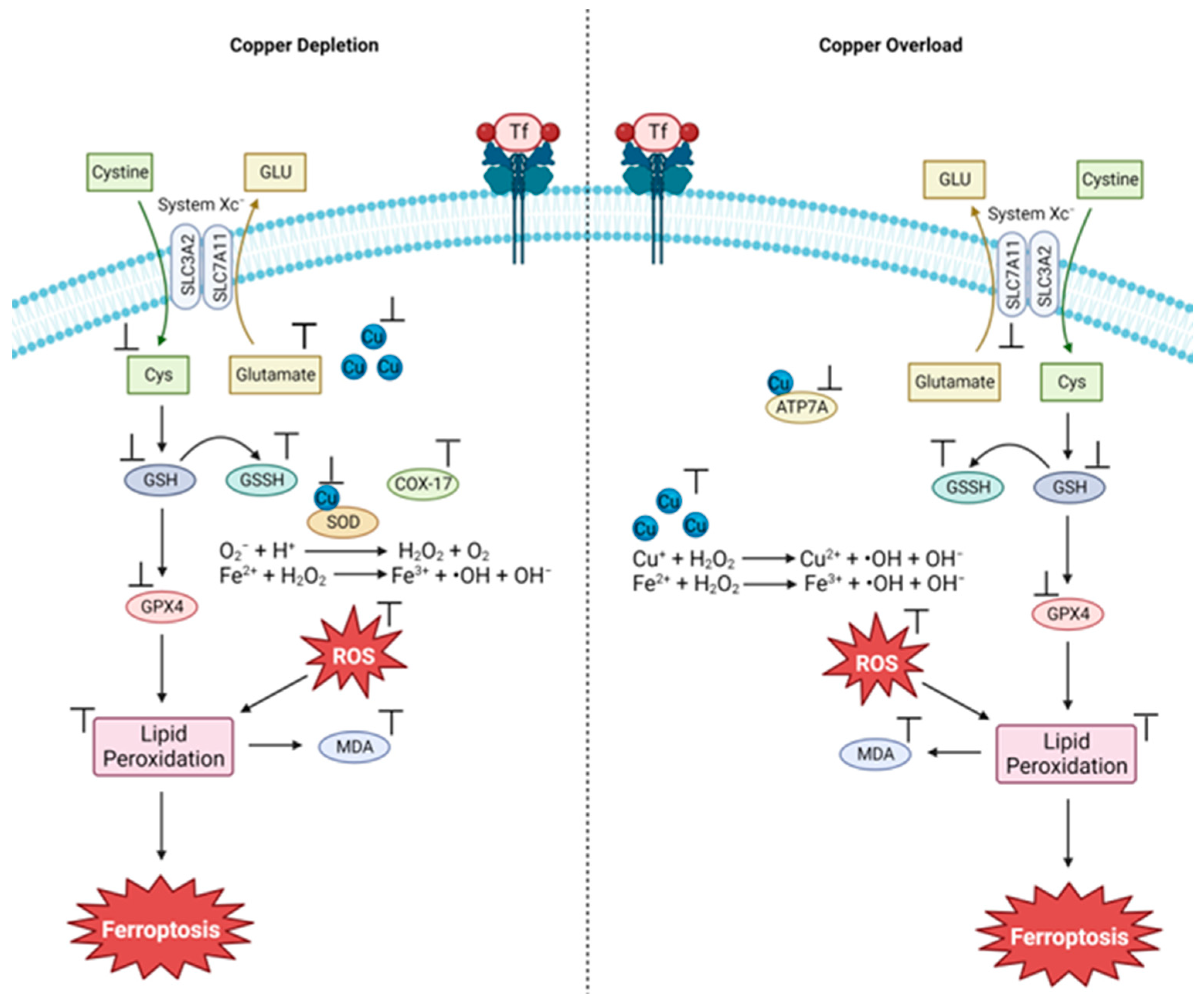
3.1. The Cu and Fe Interplay
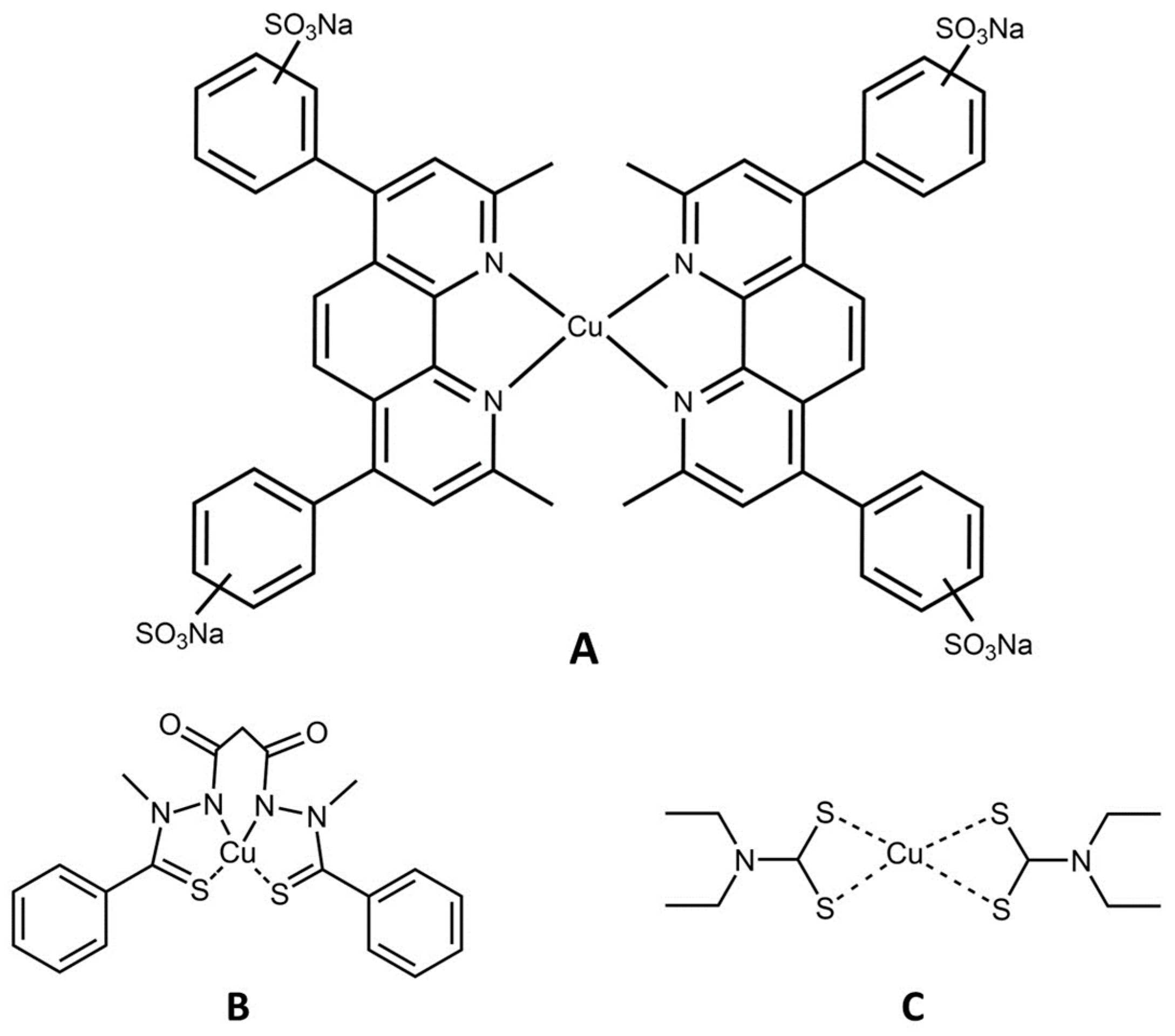
3.2. Cu(II) Chelation/Complexation Can Induce Ferroptosis
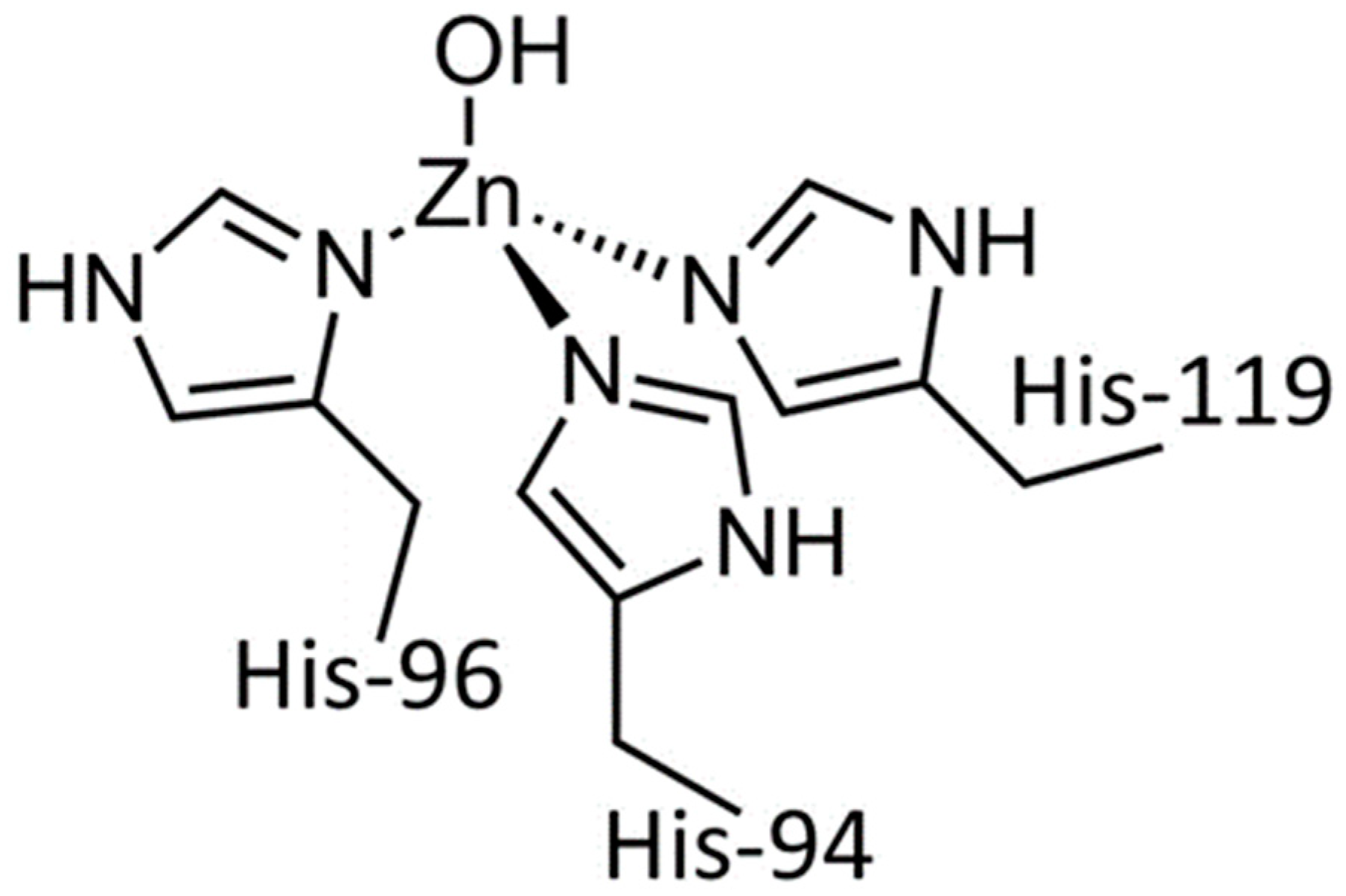
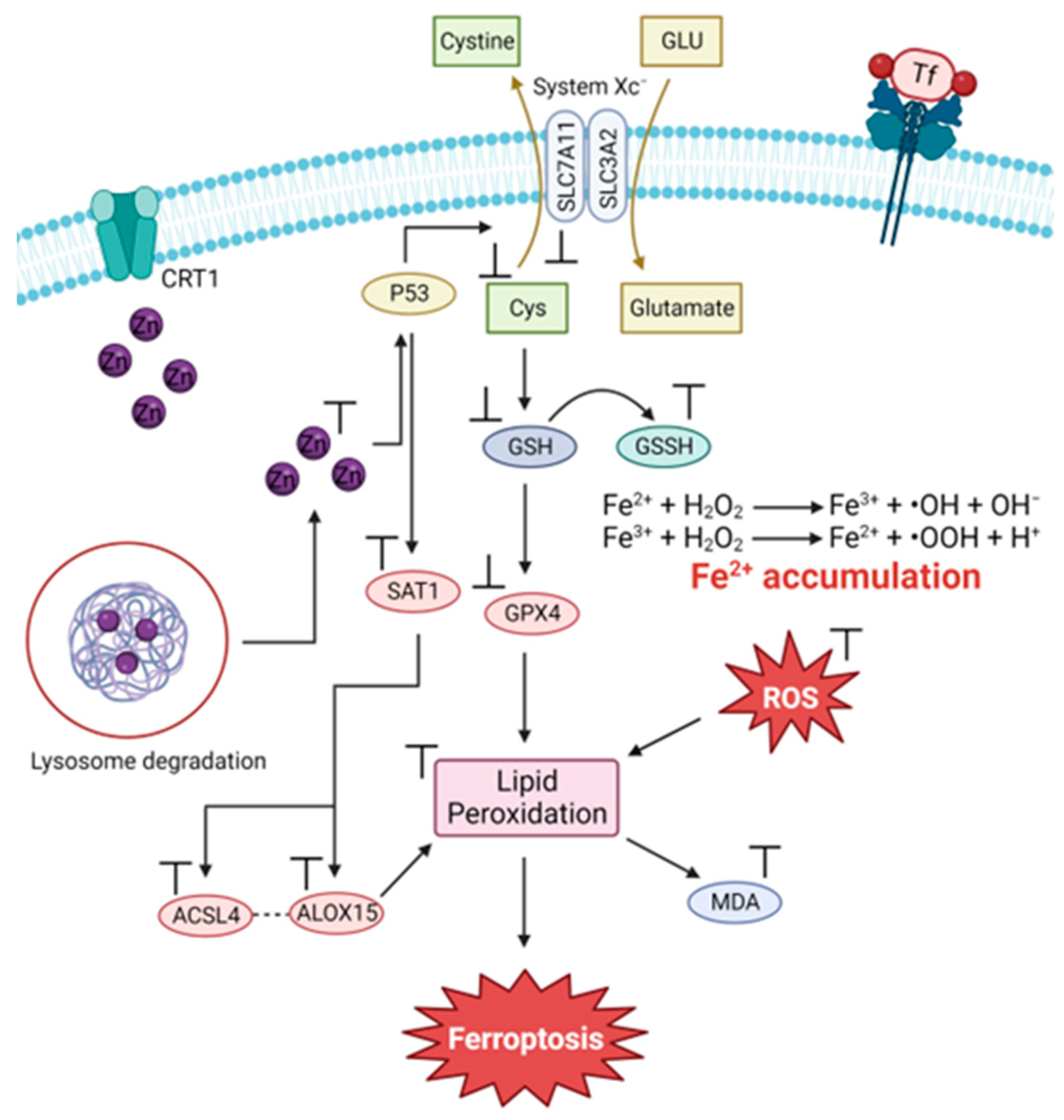
3.3. The Zn and Fe Interplay
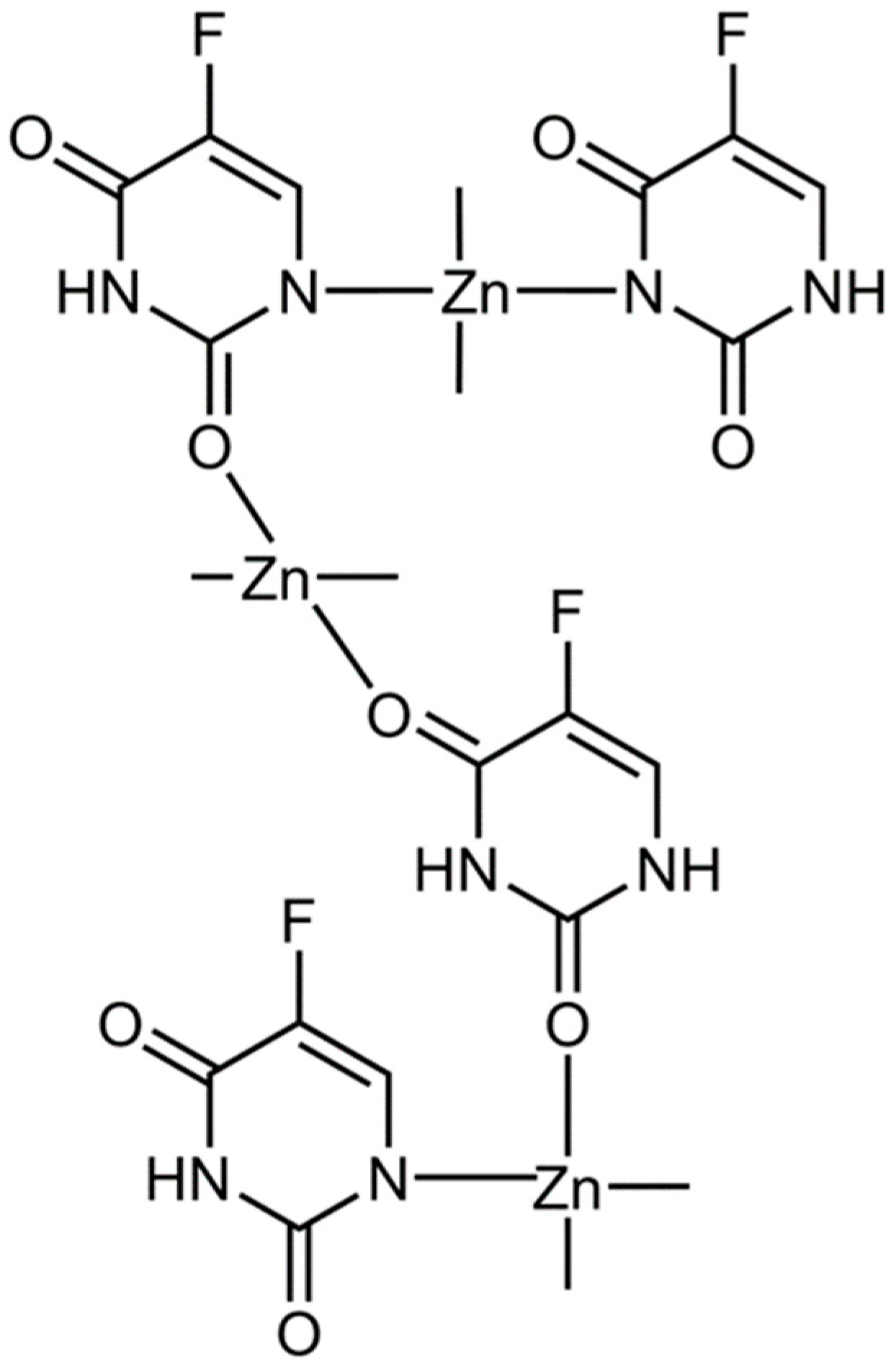
3.4. Modulating the Ability of Zn(II) to Induce Ferroptosis
3.5. Considering the Cu(II) and Zn(II) Coordination of Triapine and Derivatives
3.6. Ferroptotic Capacity of Zn and Cu Nanoparticles (NPs)
4. Metal Complexes That Induce Ferroptosis for Anticancer Application
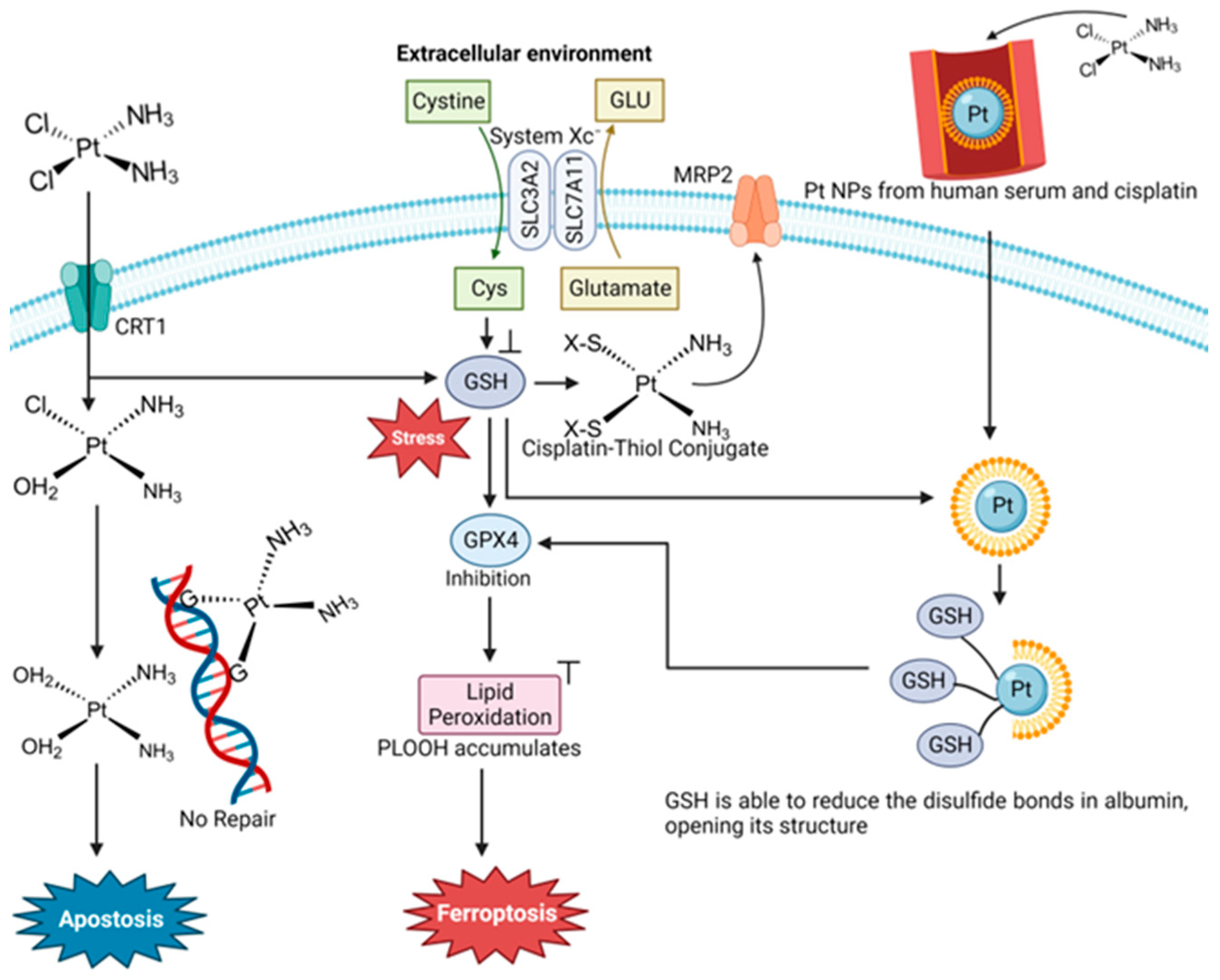
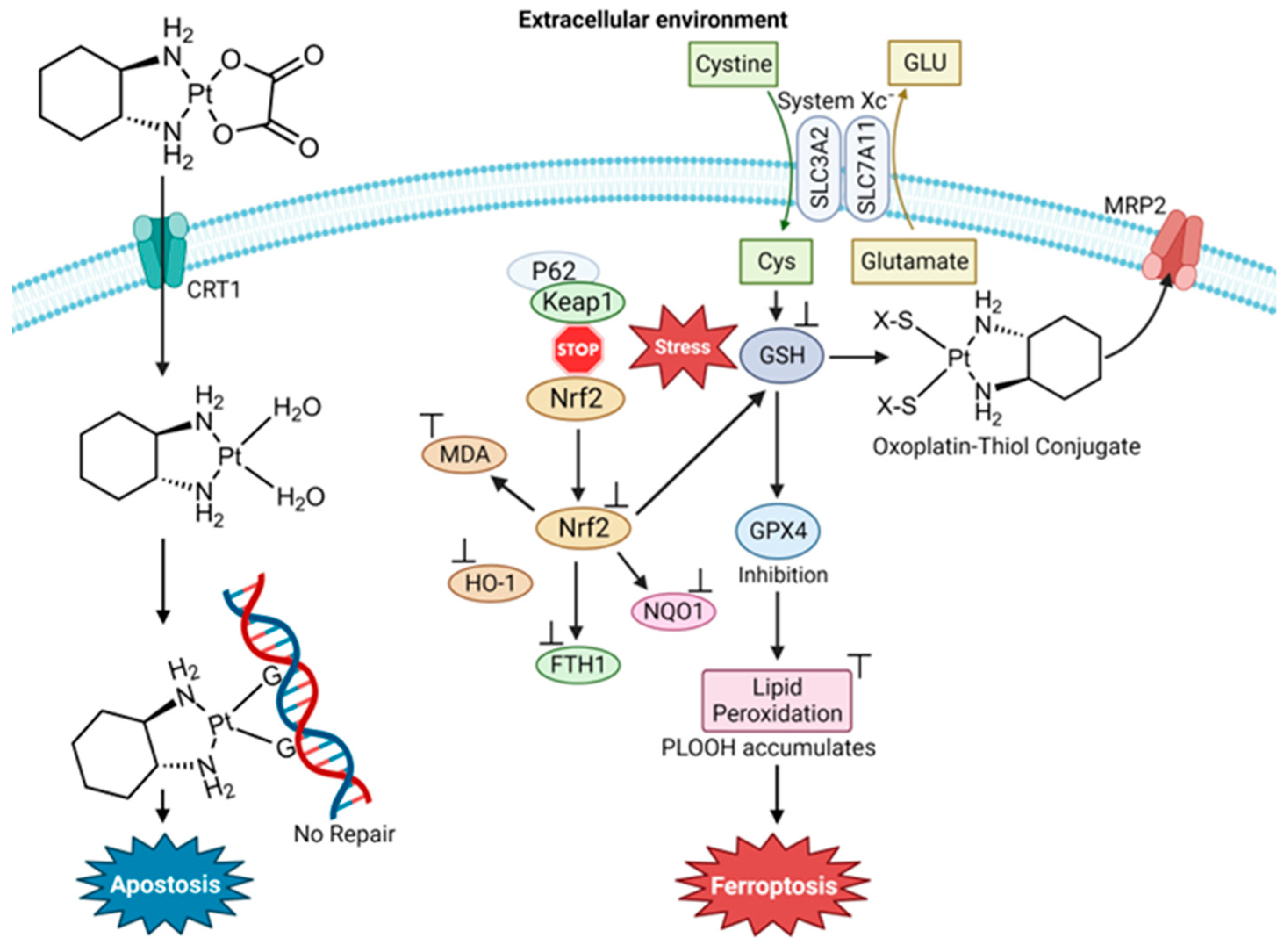
4.1. Cisplatin and Oxaliplatin
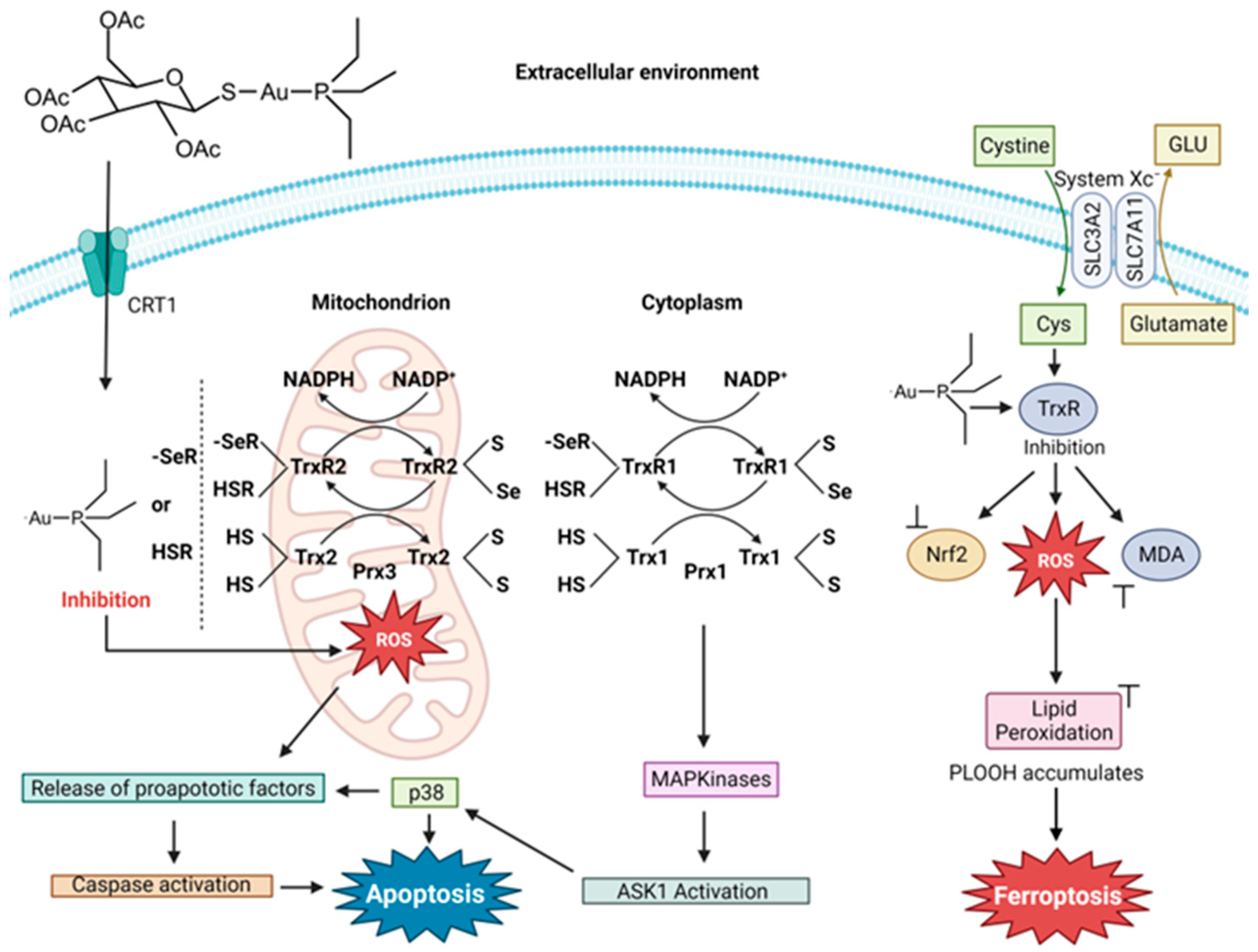
4.2. Auranofin
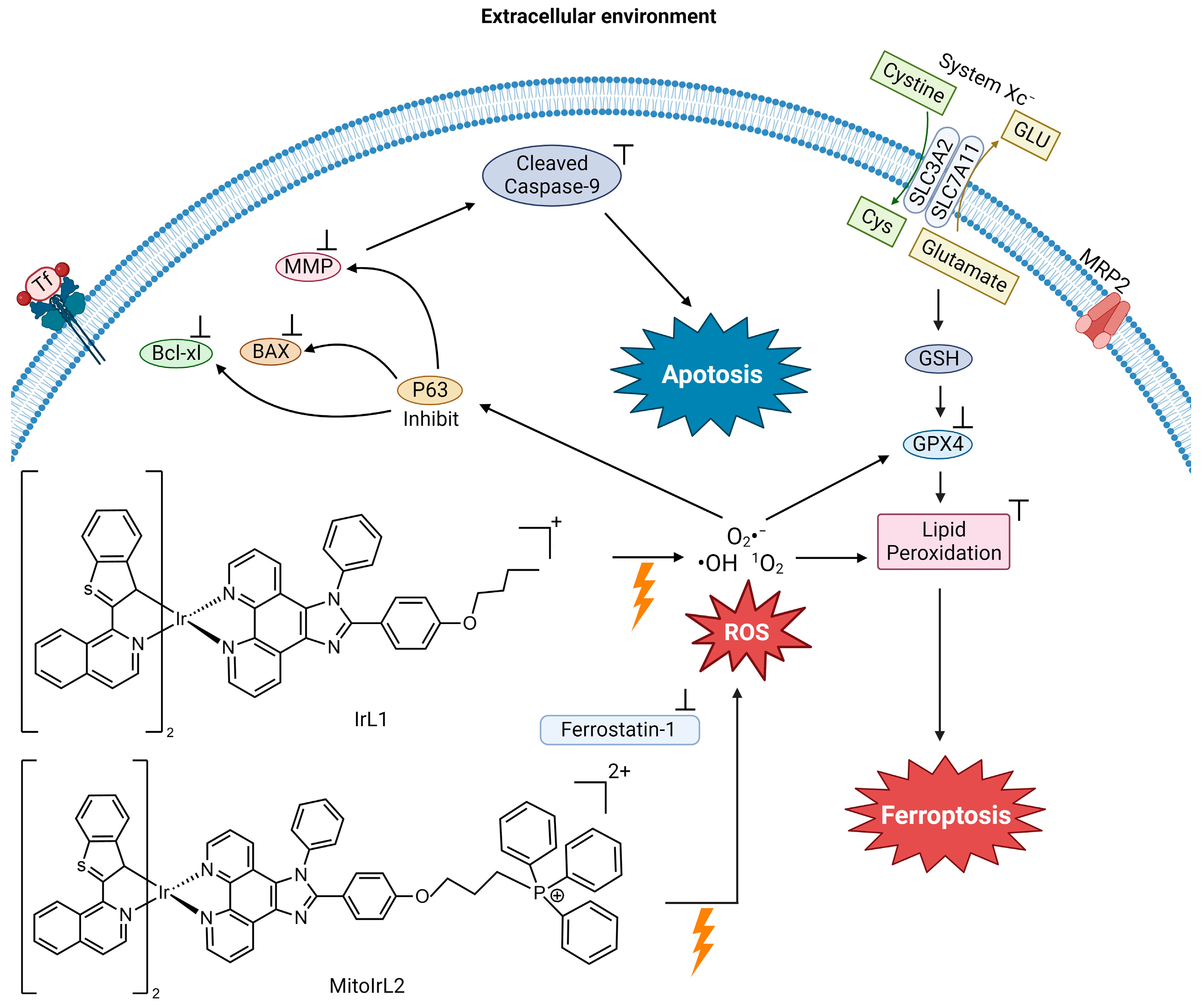
4.3. Photochemotherapeutic Metallodrugs
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target Ther. 2022, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Ferrari, D.; Los, M.; Wesselborg, S.; Peter, M.E. Apoptosis signaling by death receptors. Eur. J. Biochem. 1998, 254, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S.; Kitamura, E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 1980, 255, 2372–2376. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Palmer, L.D.; Jordan, A.T.; Maloney, K.N.; Farrow, M.A.; Gutierrez, D.B.; Gant-Branum, R.; Burns, W.J.; Romer, C.E.; Tsui, T.; Allen, J.L.; et al. Zinc intoxication induces ferroptosis in A549 human lung cells. Metallomics 2019, 11, 982–993. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Yang, X.; Wu, Q.; An, P.; Jin, X.; Liu, W.; Huang, X.; Li, Y.; Yan, S.; et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target Ther. 2020, 5, 138. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, C.; Wu, W.; Mao, Y.; Qin, Y.; Hu, J.; Hu, J.; Fu, J.; Hua, D.; Yin, J. Triapine/Ce6-loaded and lactose-decorated nanomicelles provide an effective chemo-photodynamic therapy for hepatocellular carcinoma through a reactive oxygen species-boosting and ferroptosis-inducing mechanism. Chem. Eng. J. 2021, 425, 131543. [Google Scholar] [CrossRef]
- Gao, W.; Huang, Z.; Duan, J.; Nice, E.C.; Lin, J.; Huang, C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol. Oncol. 2021, 15, 3527–3544. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Huang, Q.; He, B.; Liu, Y.; Huang, S.; Xiao, J. Regulation of ferroptosis by non-coding RNAs in the development and treatment of cancer (Review). Oncol. Rep. 2021, 45, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Geng, Z.; Bai, H.; Liu, T.; Zhang, B. Ammonium Ferric Citrate induced Ferroptosis in Non-Small-Cell Lung Carcinoma through the inhibition of GPX4-GSS/GSR-GGT axis activity. Int. J. Med. Sci. 2021, 18, 1899–1909. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Ann. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Shaghaghi, Z.; Motieian, S.; Alvandi, M.; Yazdi, A.; Asadzadeh, B.; Farzipour, S.; Abbasi, S. Ferroptosis Inhibitors as Potential New Therapeutic Targets for Cardiovascular Disease. Mini Rev. Med. Chem. 2022, 22, 2271–2286. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Liang, X.; Chen, M.; Bhattarai, P.; Hameed, S.; Tang, Y.; Dai, Z. Complementing Cancer Photodynamic Therapy with Ferroptosis through Iron Oxide Loaded Porphyrin-Grafted Lipid Nanoparticles. ACS Nano 2021, 15, 20164–20180. [Google Scholar] [CrossRef] [PubMed]
- Casas Fernandez, J.S.; Moreno Martinez, V.; Sanchez Gonzalez, A.; Sanchez Lopez, J.L.; Sordo Rodriguez, J. Quimica Bioinorganica; Editorial Sintesis: Madrid, Spain, 2002. [Google Scholar]
- Mannargudi, M.B.; Deb, S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: Is it a viable cancer therapy? J. Cancer Res. Clin. Oncol. 2017, 143, 1499–1529. [Google Scholar] [CrossRef] [PubMed]
- Greene, B.L.; Kang, G.; Cui, C.; Bennati, M.; Nocera, D.G.; Drennan, C.L.; Stubbe, J. Ribonucleotide reductases: Structure, chemistry, and metabolism suggest new therapeutic targets. Annu. Rev. Biochem. 2020, 89, 45–75. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Goodison, S.; Nicholson, B.; Tarin, D.; Urquidi, V. Expression of matrix metalloproteinase 8 (MMP-8) and tyrosinase-related protein-1 (TYRP-1) correlates with the absence of metastasis in an isogenic human breast cancer model. Differentiation 2003, 71, 114–125. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Chen, Y.; Fan, Z.; Yang, Y.; Gu, C. Iron metabolism and its contribution to cancer (Review). Int. J. Oncol. 2019, 54, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, L.M.; Rivella, S. Cancer cells with irons in the fire. Free Radic. Biol. Med. 2015, 79, 337–342. [Google Scholar] [CrossRef]
- Lane, D.J.; Merlot, A.M.; Huang, M.L.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef]
- Bertini, I.; Gray, H.B.; Stiefel, E.I.; Valentine, J.S. Biological Inorganic Chemistry: Structure and Reactivity; University Science Books: New York, NY, USA, 2007. [Google Scholar]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Grignano, E.; Birsen, R.; Chapuis, N.; Bouscary, D. From iron chelation to overload as a therapeutic strategy to induce ferroptosis in leukemic cells. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W. From iron chelation to overload as a therapeutic strategy to induce ferroptosis in hematologic malignancies. Hematology 2022, 27, 1163–1170. [Google Scholar] [CrossRef]
- Wu, L.Y.; Song, Z.Y.; Li, Q.H.; Mou, L.J.; Yu, Y.Y.; Shen, S.S.; Song, X.X. Iron chelators reverse organ damage in type 4B hereditary hemochromatosis. Medicine 2021, 100, e25258. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.T. The Effects of Treatment with Blood Transfusion, Iron Chelation and Hydroxyurea on Puberty, Growth and Spermatogenesis in Sickle Cell Disease (SCD): A short update. Acta Biomed. 2021, 92, 1–8. [Google Scholar] [CrossRef]
- Rayatpour, A.; Foolad, F.; Heibatollahi, M.; Khajeh, K.; Javan, M. Ferroptosis inhibition by deferiprone, attenuates myelin damage and promotes neuroprotection in demyelinated optic nerve. Sci. Rep. 2022, 12, 19630. [Google Scholar] [CrossRef] [PubMed]
- Yao, X. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen. Res. 2019, 14, 532–541. [Google Scholar]
- Jomen, W. Iron chelator deferasirox inhibits NF-κB activity in hepatoma cells and changes sorafenib-induced programmed cell deaths. Biomed. Pharmacother. 2022, 153, 1–12. [Google Scholar] [CrossRef]
- Ryan, S.K.; Ugalde, C.L.; Rolland, A.S.; Skidmore, J.; Devos, D.; Hammond, T.R. Therapeutic inhibition of ferroptosis in neurodegenerative disease. Trends Pharmacol. Sci. 2023, 44, 674–688. [Google Scholar] [CrossRef]
- Fratta Pasini, A.M.; Stranieri, C.; Busti, F.; Di Leo, E.G.; Girelli, D.; Cominacini, L. New Insights into the Role of Ferroptosis in Cardiovascular Diseases. Cells 2023, 12, 867. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, X.; Zeng, Y.; Mo, X.; Hong, S.; He, H.; Li, J.; Fatima, S.; Liu, Q. Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci. Rep. 2023, 13, 15515. [Google Scholar] [CrossRef]
- Enyedy, É.A. Complex formation and cytotoxicity of Triapine derivatives: A comparative solution study on the effect of the chalcogen atom and NH-methylation. Dalton Trans. 2020, 49, 16887–16902. [Google Scholar] [CrossRef]
- Enyedy, E.A.; Primik, M.F.; Kowol, C.R.; Arion, V.B.; Kiss, T.; Keppler, B.K. Interaction of triapine and related thiosemicarbazones with iron(III)/(II) and gallium(III): A comparative solution equilibrium study. Dalton Trans. 2011, 40, 5895–5905. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Long, M.J.C.; Stubbe, J. Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase in Vitro and in Mammalian Cells. J. Biol. Chem. 2012, 287, 35768–35778. [Google Scholar] [CrossRef] [PubMed]
- Plamthottam, S.; Sun, D.; Van Valkenburgh, J.; Valenzuela, J.; Ruehle, B.; Steele, D.; Poddar, S.; Marshalik, M.; Hernandez, S.; Radu, C.G.; et al. Activity and electrochemical properties: Iron complexes of the anticancer drug triapine and its analogs. J. Biol. Inorg. Chem. 2019, 24, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Schweigel-Röntgen, M. Chapter Nine—The Families of Zinc (SLC30 and SLC39) and Copper (SLC31) Transporters. In Current Topics in Membranes; Bevensee, M.O., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 73, pp. 321–355. [Google Scholar]
- Shi, W.; Zhang, H.; Zhang, Y.; Lu, L.; Zhou, Q.; Wang, Y.; Pu, Y.; Yin, L. Co-exposure to Fe, Zn, and Cu induced neuronal ferroptosis with associated lipid metabolism disorder via the ERK/cPLA2/AA pathway. Environ. Pollut. 2023, 336, 122438. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, R.; Chen, M.; Zheng, J.; Chen, M.; Braidy, N.; Liu, S.; Liu, G.; Maimaitiming, Z.; Shen, T.; et al. Multi-copper ferroxidase deficiency leads to iron accumulation and oxidative damage in astrocytes and oligodendrocytes. Sci Rep 2019, 9, 9437. [Google Scholar] [CrossRef] [PubMed]
- Kondaiah, P.; Yaduvanshi, P.S.; Sharp, P.A.; Pullakhandam, R. Iron and Zinc Homeostasis and Interactions: Does Enteric Zinc Excretion Cross-Talk with Intestinal Iron Absorption? Nutrients 2019, 11, 1885. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, X.; Liu, H.; Liu, M.; Yue, Z.; Wu, Z.; Liu, L.; Li, F. Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms. Antioxidants 2022, 11, 2084. [Google Scholar] [CrossRef]
- Collins, J.F.; Prohaska, J.R.; Knutson, M.D. Metabolic crossroads of iron and copper. Nutr. Rev. 2010, 68, 133–147. [Google Scholar] [CrossRef]
- La Fontaine, S.; Ackland, M.L.; Mercer, J.F. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: Emerging roles. Int. J. Biochem. Cell Biol. 2010, 42, 206–209. [Google Scholar] [CrossRef]
- Myint, Z.W.; Oo, T.H.; Thein, K.Z.; Tun, A.M.; Saeed, H. Copper deficiency anemia: Review article. Ann. Hematol. 2018, 97, 1527–1534. [Google Scholar] [CrossRef]
- Chen, H.; Attieh, Z.; Su, T.; Syed, B.; Gao, H.; Alaeddine, R.; Fox, T.; Usta, J.; Naylor, C.; Evans, R.; et al. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood 2004, 103, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Yan, D.; Chen, X.; Li, X.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Chen, X.; Tang, D.; Liu, J. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 2023, 19, 1982–1996. [Google Scholar] [CrossRef] [PubMed]
- Kannappan, V.; Ali, M.; Small, B.; Rajendran, G.; Elzhenni, S.; Taj, H.; Wang, W.; Dou, Q.P. Recent Advances in Repurposing Disulfiram and Disulfiram Derivatives as Copper-Dependent Anticancer Agents. Front. Mol. Biosci. 2021, 8, 741316. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, Y.; Zhou, Y.; Hu, W.; Yang, C.; Jing, Q.; Zhou, C.; Wang, X.; Hu, J.; Wang, L.; et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox. Biol. 2021, 46, 102122. [Google Scholar] [CrossRef] [PubMed]
- Bin, B.-H.; Seo, J.; Kim, S.T. Function, Structure, and Transport Aspects of ZIP and ZnT Zinc Transporters in Immune Cells. J. Immunol. Res. 2018, 2018, 9365747. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.P. Iron, Zinc, and Physical Performance. Biol. Trace Elem. Res. 2019, 188, 135–139. [Google Scholar] [CrossRef]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K.; et al. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef]
- Abdelhaleim, A.F.; Abdo Soliman, J.S.; Amer, A.Y.; Abdo Soliman, J.S. Association of Zinc Deficiency with Iron Deficiency Anemia and its Symptoms: Results from a Case-control Study. Cureus 2019, 11, e3811. [Google Scholar] [CrossRef]
- Sanna, A.; Firinu, D.; Zavattari, P.; Valera, P. Zinc Status and Autoimmunity: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 68. [Google Scholar] [CrossRef]
- Eide, D.J. Zinc transporters and the cellular trafficking of zinc. Biochim. Biophys. Acta 2006, 1763, 711–722. [Google Scholar] [CrossRef]
- Lei, L.; Dong, Z.; Xu, L.; Yang, F.; Yin, B.; Wang, Y.; Yue, R.; Guan, G.; Xu, J.; Song, G.; et al. Metal-fluorouracil networks with disruption of mitochondrion enhanced ferroptosis for synergistic immune activation. Theranostics 2022, 12, 6207–6222. [Google Scholar] [CrossRef] [PubMed]
- Ohui, K.; Stepanenko, I.; Besleaga, I.; Babak, M.V.; Stafi, R.; Darvasiova, D.; Giester, G.; Pósa, V.; Enyedy, E.A.; Vegh, D.; et al. Triapine Derivatives Act as Copper Delivery Vehicles to Induce Deadly Metal Overload in Cancer Cells. Biomolecules 2020, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Enyedy, É.A.; Nagy, N.V.; Zsigó, É.; Kowol, C.R.; Arion, V.B.; Keppler, B.K.; Kiss, T. Comparative solution equilibrium study of the interactions of copper(II), iron(II) and zinc(II) with triapine (3-aminopyridine-2-carbaldehyde thiosemicarbazone) and related ligands. Eur. J. Inorg. Chem. 2010, 2010, 1717–1728. [Google Scholar] [CrossRef]
- Hager, S.; Pape, V.F.S.; Pósa, V.; Montsch, B.; Uhlik, L.; Szakács, G.; Tóth, S.; Jabronka, N.; Keppler, B.K.; Kowol, C.R.; et al. High Copper Complex Stability and Slow Reduction Kinetics as Key Parameters for Improved Activity, Paraptosis Induction, and Impact on Drug-Resistant Cells of Anticancer Thiosemicarbazones. Antioxid. Redox Signal. 2020, 33, 395–414. [Google Scholar] [CrossRef] [PubMed]
- Popovic-Bijelic, A.; Kowol, C.R.; Lind, M.E.; Luo, J.; Himo, F.; Enyedy, E.A.; Arion, V.B.; Graslund, A. Ribonucleotide reductase inhibition by metal complexes of triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): A combined experimental and theoretical study. J. Inorg. Biochem. 2011, 105, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Ngamchuea, K.; Batchelor-McAuley, C.; Compton, R.G. The Copper(II)-Catalyzed Oxidation of Glutathione. Chem. Eur. J. 2016, 22, 15937–15944. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Niu, C.; Yi, J.; Sun, L.; Cao, H.; Fang, Y.; Jin, T.; Li, Y.; Lou, C.; Kang, J.; et al. Novel Triapine Derivative Induces Copper-Dependent Cell Death in Hematopoietic Cancers. J. Med. Chem. 2019, 62, 3107–3121. [Google Scholar] [CrossRef]
- Gilleran, J.A.; Yu, X.; Blayney, A.J.; Bencivenga, A.F.; Na, B.; Augeri, D.J.; Blanden, A.R.; Kimball, S.D.; Loh, S.N.; Roberge, J.Y.; et al. Benzothiazolyl and Benzoxazolyl Hydrazones Function as Zinc Metallochaperones to Reactivate Mutant p53. J. Med. Chem. 2021, 64, 2024–2045. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. In Cancer Nanotechnology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 25–37. [Google Scholar]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef]
- Wang, S.; Luo, J.; Zhang, Z.; Dong, D.; Shen, Y.; Fang, Y.; Hu, L.; Liu, M.; Dai, C.; Peng, S.; et al. Iron and magnetic: New research direction of the ferroptosis-based cancer therapy. Am. J. Cancer Res. 2018, 8, 1933–1946. [Google Scholar] [PubMed]
- Shubhra, Q.T.H. Iron oxide nanoparticles in magnetic drug targeting and ferroptosis-based cancer therapy. Med. Rev. 2023, 3, 444–447. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Zhang, Y.; Ma, L.; Song, E.; Song, Y. “Iron free” zinc oxide nanoparticles with ion-leaking properties disrupt intracellular ROS and iron homeostasis to induce ferroptosis. Cell Death Dis. 2020, 11, 183. [Google Scholar] [CrossRef]
- Yao, T.; Lu, R.; Zhang, Y.; Zhang, Y.; Zhao, C.; Lin, R.; Lin, Z. Cervical cancer stem cells. Cell Prolif. 2015, 48, 611–625. [Google Scholar] [CrossRef]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef]
- Lei, J.-Y.; Li, S.-X.; Li, F.; Li, H.; Lei, Y.-S. Zinc oxide nanoparticle regulates the ferroptosis, proliferation, invasion and steaminess of cervical cancer by miR-506-3p/CD164 signaling. Cancer Nanotechnol. 2022, 13, 33. [Google Scholar] [CrossRef]
- Tu, M.; Cai, L.; Zheng, W.; Su, Z.; Chen, Y.; Qi, S. CD164 regulates proliferation and apoptosis by targeting PTEN in human glioma. Mol. Med. Rep. 2017, 15, 1713–1721. [Google Scholar] [CrossRef]
- Pandey, N.K.; Chudal, L.; Phan, J.; Lin, L.; Johnson, O.; Xing, M.; Liu, J.P.; Li, H.; Huang, X.; Shu, Y.; et al. A facile method for the synthesis of copper–cysteamine nanoparticles and study of ROS production for cancer treatment. J. Mater. Chem. B 2019, 7, 6630–6642. [Google Scholar] [CrossRef]
- Yuan, H. Photothermal Nanozymatic Nanoparticles Induce Ferroptosis and Apoptosis through Tumor Microenvironment Manipulation for Cancer Therapy. Small 2022, 18, 2202161. [Google Scholar] [CrossRef]
- Tong, Z.; Gao, Y.; Huang, Y.; Wang, W.; Mao, Z. Nanomaterials for cascade promoted catalytic cancer therapy. View 2021, 2, 20200133. [Google Scholar] [CrossRef]
- Dabrowiak, J.C. Metals in Medicine, 2nd ed.; Wiley: Hoboken, NJ, USA, 2017; pp. 91–146. [Google Scholar]
- Wang, X.; Guo, Z. The role of sulfur in platinum anticancer chemotherapy. Anticancer Agents Med. Chem. 2007, 7, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Mao, C.Q.; Chen, S.; Ma, G.; Wang, J.; Liu, Y. Combating the drug resistance of cisplatin using a platinum prodrug based delivery system. Angew. Chem. 2012, 51, 6742–6747. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Liu, B.; Wang, H. Oxaliplatin induces ferroptosis and oxidative stress in HT29 colorectal cancer cells by inhibiting the Nrf2 signaling pathway. Exp. Ther. Med. 2022, 23, 394. [Google Scholar] [CrossRef] [PubMed]
- Imberti, C.; Zhang, P.; Huang, H.; Sadler, P.J. New Designs for Phototherapeutic Transition Metal Complexes. Angew. Chem. 2020, 59, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yang, Y.; Xu, Q.; Ling, M.; Lin, H.; Ma, W.; Sun, R.; Xu, Y.; Liu, X.; Li, N.; et al. Self-Amplification of Tumor Oxidative Stress with Degradable Metallic Complexes for Synergistic Cascade Tumor Therapy. Nano Lett. 2020, 20, 8141–8150. [Google Scholar] [CrossRef] [PubMed]
- Freeman, E.C.; Weiland, L.M.; Meng, W.S. Modeling the proton sponge hypothesis: Examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 2013, 24, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Sutton, B.M.; McGusty, E.; Walz, D.T.; DiMartino, M.J. Oral gold. Antiarthritic properties of alkylphosphinegold coordination complexes. J. Med. Chem. 1972, 15, 1095–1098. [Google Scholar] [CrossRef]
- Schattenkirchner, M.; Bröll, H.; Kaik, B.; Müller-Fassbender, H.; Rau, R.; Zeidler, H. Auranofin and gold sodium thiomalate in the treatment of rheumatoid arthritis: A one-year, double-blind, comparative multicenter study. Klin. Wochenschr. 1988, 66, 167–174. [Google Scholar] [CrossRef]
- Simon, T.M.; Kunishima, D.H.; Vibert, G.J.; Lorber, A. Cellular antiproliferative action exerted by auranofin. J. Rheumatol. Suppl. 1979, 5, 91–97. [Google Scholar]
- Allardyce, C.S.; Dyson, P.J. Metal-based drugs that break the rules. Dalton Trans. 2016, 45, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Vega, L.; Ruiz Silva, V.A.; Domínguez-González, T.M.; Claudio-Betancourt, S.; Toro-Maldonado, R.E.; Capre Maso, L.C.; Sanabria Ortiz, K.; Pérez-Verdejo, J.A.; Román González, J.; Rosado-Fraticelli, G.T.; et al. Evaluating ligand modifications of the titanocene and auranofin moieties for the development of more potent anticancer drugs. Inorganics 2020, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, J. Redox Modulation and Induction of Ferroptosis as a New Therapeutic Strategy in Hepatocellular Carcinoma. Transl. Oncol. 2020, 13, 100785. [Google Scholar] [CrossRef]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Scutari, G.; Folda, A.; Bindoli, A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem. Pharmacol. 2004, 67, 689–696. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef]
- Ye, S.; Chen, X.; Yao, Y.; Li, Y.; Sun, R.; Zeng, H.; Shu, Y.; Yin, H. Thioredoxin Reductase as a Novel and Efficient Plasma Biomarker for the Detection of Non-Small Cell Lung Cancer: A Large-scale, Multicenter study. Sci. Rep. 2019, 9, 2652. [Google Scholar] [CrossRef]
- Karlenius, T.C.; Tonissen, K.F. Thioredoxin and Cancer: A Role for Thioredoxin in all States of Tumor Oxygenation. Cancers 2010, 2, 209–232. [Google Scholar] [CrossRef]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J., 3rd; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Tian, J.; Huang, B.; Nawaz, M.H.; Zhang, W. Recent advances of multi-dimensional porphyrin-based functional materials in photodynamic therapy. Coord. Chem. Rev. 2020, 420, 213410. [Google Scholar] [CrossRef]
- Yuan, H.; Han, Z.; Chen, Y.; Qi, F.; Fang, H.; Guo, Z.; Zhang, S.; He, W. Ferroptosis Photoinduced by New Cyclometalated Iridium(III) Complexes and Its Synergism with Apoptosis in Tumor Cell Inhibition. Angew. Chem. 2021, 60, 8174–8181. [Google Scholar] [CrossRef] [PubMed]
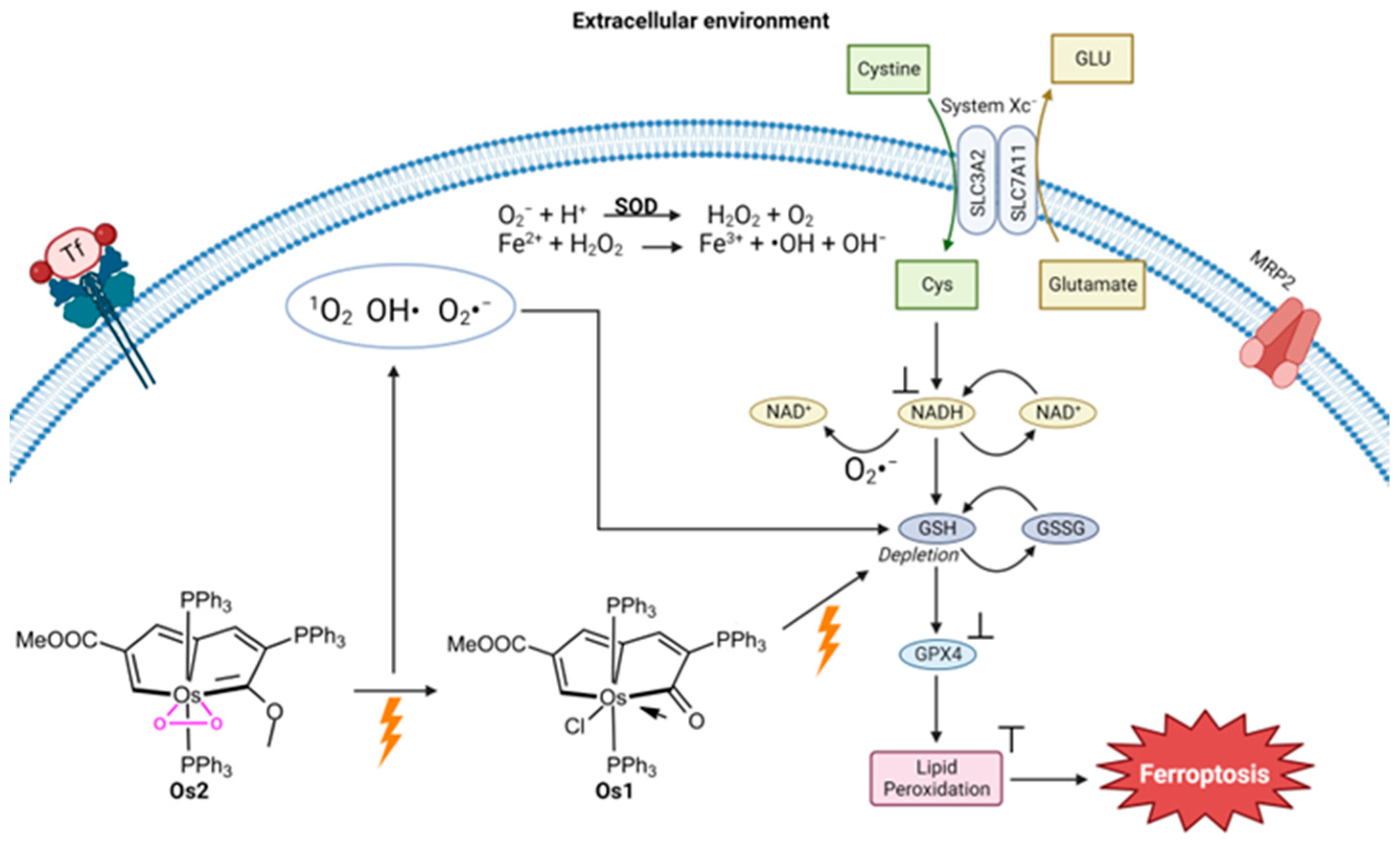
- Lu, N.; Deng, Z.; Gao, J.; Liang, C.; Xia, H.; Zhang, P. An osmium-peroxo complex for photoactive therapy of hypoxic tumors. Nat. Commun. 2022, 13, 2245. [Google Scholar] [CrossRef] [PubMed]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claudio-Ares, O.; Luciano-Rodríguez, J.; Del Valle-González, Y.L.; Schiavone-Chamorro, S.L.; Pastor, A.J.; Rivera-Reyes, J.O.; Metzler, C.L.; Domínguez-Orona, L.M.; Vargas-Pérez, B.L.; Skouta, R.; et al. Exploring the Use of Intracellular Chelation and Non-Iron Metals to Program Ferroptosis for Anticancer Application. Inorganics 2024, 12, 26. https://doi.org/10.3390/inorganics12010026
Claudio-Ares O, Luciano-Rodríguez J, Del Valle-González YL, Schiavone-Chamorro SL, Pastor AJ, Rivera-Reyes JO, Metzler CL, Domínguez-Orona LM, Vargas-Pérez BL, Skouta R, et al. Exploring the Use of Intracellular Chelation and Non-Iron Metals to Program Ferroptosis for Anticancer Application. Inorganics. 2024; 12(1):26. https://doi.org/10.3390/inorganics12010026
Chicago/Turabian StyleClaudio-Ares, Oscar, Jeileen Luciano-Rodríguez, Yolmarie L. Del Valle-González, Selene L. Schiavone-Chamorro, Alex J. Pastor, Javier O. Rivera-Reyes, Carmen L. Metzler, Lizandra M. Domínguez-Orona, Brenda Lee Vargas-Pérez, Rachid Skouta, and et al. 2024. "Exploring the Use of Intracellular Chelation and Non-Iron Metals to Program Ferroptosis for Anticancer Application" Inorganics 12, no. 1: 26. https://doi.org/10.3390/inorganics12010026