
Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes


Department of Chemistry, Graduate School of Natural Science and Technology, Shimane University, 1060 Nishikawatsu, Matsue 690-8504, Shimane, Japan
*
Authors to whom correspondence should be addressed.
Inorganics 2024, 12(2), 41; https://doi.org/10.3390/inorganics12020041
Submission received: 10 January 2024
/
Revised: 23 January 2024
/
Accepted: 25 January 2024
/
Published: 26 January 2024
(This article belongs to the Special Issue Editorial Board Members’ Collection Series in “Featuring Ligands and Their Applications in Coordination Chemistry”)
Abstract
:Two ferrocenecarboxylate (fca)-bridged dirhodium (Rh2) complexes, [Rh2(fca)4] (1) and [Rh2(fca)(piv)3] (2; piv = pivalate), were prepared through the carboxylate-exchange reactions of [Rh2(O2CCH3)4(H2O)2] and [Rh2(piv)4], respectively, with fcaH and characterized by 1H NMR, ESI-TOF-MS, and elemental analyses. Single-crystal X-ray diffraction analyses of [Rh2(fca)4(MeOH)2] (1(MeOH)2) and [Rh2(fca)(piv)3(MeOH)2] (2(MeOH)2), which are recrystallized from MeOH-containing solutions of 1 and 2, revealed that (1) 1(MeOH)2 and 2(MeOH)2 possess homoleptic and heteroleptic paddlewheel-type dinuclear structures, respectively; (2) both complexes have a single Rh–Rh bond (2.3771(3) Å for 1(MeOH)2, 2.3712(3) Å for 2(MeOH)2); and (3) the cyclopentadienyl rings of the fca ligands in 1(MeOH)2 adopt an eclipsed conformation, whereas those in 2(MeOH)2 are approximately 12–14° rotated from the staggered conformation. Density functional theory (DFT) calculations revealed that (1) the electronic configurations of the Rh2 core in 1(MeOH)2 and 2(MeOH)2 are π4σ2δ2π*2δ*2π*2 and π4σ2δ2δ*2π*4, respectively; and (2) the occupied molecular orbitals (MOs) localized on the fca ligands are energetically degenerate and relatively more unstable than those on the Rh2 cores. Absorption features and electrochemical properties of 1 and 2 were investigated in a 9:1 CHCl3-MeOH solution and compared with those of fcaH and [Rh2(piv)4]. Through examining the obtained results in detail using time-dependent DFT (TDDFT) and unrestricted DFT, we found that 1 and 2 exhibit charge transfer excitations between the fca ligands and Rh2 cores, and 1 shows electronic interactions between ferrocene units through the Rh2 core in the electrochemical oxidation process.
1. Introduction
In modern coordination chemistry, the synthetic strategy utilizing “metalloligands” or “metal-organic ligands”, which possess substituents such as carboxylic acid that act as free coordination sites, are of great interest in the development of multinuclear complexes, supramolecular metal complexes, coordination polymers (CPs), and metal–organic frameworks (MOFs) [1,2,3,4,5,6]. Rational photocatalysts [7,8], sensors [9,10], and functional magnetic materials [11,12] can be developed by appropriately combining photo- or redox-active metalloligands with specific metal cations or clusters. Among the various metalloligands, ferrocenecarboxylate (fca; see Scheme 1a) and its derivatives [13], which are redox-active organometallic compounds, have been especially applied because of their structural accommodation, chemical stability, and reversible redox behavior [14,15,16]. Multinuclear complexes, in which multiple fca ligands are connected to multinuclear building blocks, are expected to exhibit redox interactions between (1) a few fca ligands through a multinuclear core, or (2) a multinuclear core and fca ligands. For example, two fca-bearing square tetraplatinum (Pt4) complexes exhibit weak interactions between two ferrocene units through a Pt4 core in the electrochemical oxidation of the fca ligands [17].
Paddlewheel-type dinuclear (M2) complexes, which have metal–metal bonds or interactions, have long been known as versatile building blocks that can rationally coordinate organic ligands or metalloligands at the equatorial positions of the M2 cores [18]. Fca-bearing paddlewheel-type M2 complexes have been investigated for several M2 cores, such as Cu, Zn, Mo, Ru, and Rh [19,20,21,22,23,24,25,26,27,28]. For example, Churchill reported the crystal structure of [Cu2(fca)4(THF)2], in which four fca and two THF ligands are coordinated to the equatorial and axial positions, respectively, of a Cu2 core, and four ferrocene units adopt both eclipsed and staggered conformations in a 2:2 ratio [19]. Similar eclipsed and staggered conformations of ferrocene units are observed in the crystal structure of [Cu2(fca)4 (MeOH)2] [20]. An initial attempt to isolate and determine the structure of an fca-bearing heteroleptic paddlewheel M2 complex was reported by Cotton et al., who prepared trans-[Mo2(fca)2(O2CCH3)2] as an intermediate complex in the synthesis of homoleptic [Mo2(fca)4] [22].
Fca-bearing paddlewheel-type Rh2 complexes are considered to be particularly interesting because this type of Rh2 complex shows excellent functional properties such as catalysis, sensing, and antitumor activities [29,30,31,32,33,34,35,36,37]. Homoleptic paddlewheel-type Rh2 complexes bridged with four fca derivative ligands, for example, [Rh2(fca)4], act as catalysts for carbene insertion, shape-selective alkane functionalization, and asymmetric intramolecular C–H insertion reactions [26,27,38]; however, the details of the synthesis, characterization, electrochemical and absorption properties, and X-ray crystal structures of the Rh2 complexes have not been previously reported. In addition, a heteroleptic paddlewheel-type Rh2 complex bridged with an fca ligand and three acetate ligands, [Rh2(fca)(O2CCH3)3], was recently reported as a catalyst for carbene C–H insertion reaction; however, its crystal structure has not yet been reported [28]. It is important to investigate fca-bearing homoleptic and heteroleptic paddlewheel-type Rh2 complexes in detail not only for further development of these complexes as catalysts but also for a deep and systematic understanding of paddlewheel-type M2 complexes with metalloligands. Hence, in this study, we closely investigated the synthesis, characterization, crystal structure, and absorption and electrochemical properties of fca-bearing homoleptic and heteroleptic paddlewheel-type Rh2 complexes, [Rh2(fca)4] (1; Scheme 1b) and [Rh2(fca)(piv)3] (2; piv = pivalate, Scheme 1c), using a combination of experimental and theoretical techniques.
2. Results and Discussion
2.1. Synthesis and Characterization
Homoleptic complex 1 was easily prepared in a high yield (94.7%) using a facile reflux reaction of [Rh2(O2CCH3)4(H2O)2] with eight equivalents of fcaH in chlorobenzene, whereas heteroleptic complex 2 was obtained in a 29.1% yield via the partial carboxylate-exchange reaction of [Rh2(piv)4] with one equivalent of fcaH in N,N-dimethylaniline at 433 K followed by purification with silica-gel column chromatography. The yield of 2 was higher than that of a 3:1-coordination arrangement of carboxylate-bridged Rh2 complexes such as [Rh2(O2CCH3)3(PABC)] (15.9% yield; PABC = p-aminobenzenecarboxylate) [39]. One of the reasons for the relatively high yield of 2 is thought to be the strong coordination ability of the electron-donating pivalate ligands to the Rh2 core, which suppresses excessive unfavorable carboxylate-exchange reactions. In fact, unreacted [Rh2(piv)4] (recover yield: 31.4%) and scarce amounts of [Rh2(fca)x(piv)4-x] (x = 2–4) mixture were found in different fractions of the column chromatography. Both Rh2 complexes are air-stable compounds with diamagnetic closed-shell electronic structures, where 1 is less soluble in most organic solvents, but dissolves slightly in a mixed 9:1 CHCl3-MeOH solution. In contrast, 2 exhibits excellent solubility in common organic solvents.
To characterize 1 and 2, 1H NMR, ESI-TOF-MS, and elemental analyses were performed. The 1H NMR spectrum of 1 in DMSO-d6 showed three broad signals at 4.62, 4.22, and 3.85 ppm with an integral ratio of 8:8:20. As shown in Figure S1, because no additional proton signal was observed in the spectrum, only the homoleptic Rh2 complex was confirmed to be isolated. In 2, proton signals were observed at 4.61 (2H), 4.29 (2H), and 3.97 (5H) ppm, which can be attributed to an fca ligand, and at 0.90 (9H) and 0.89 (18H) ppm, which can be attributed to pivalate ligands at the trans and cis positions, respectively, relative to the fca ligand (see Figure S2). The ESI-TOF-MS spectra of 1 and 2 show positive ion peaks at 1144.7777 and 760.9762 m/z, respectively, in good agreement with their simulated [M+Na]+ values (1144.7814 and 760.9762 m/z, respectively). As shown in Figures S3 and S4, the shapes of the isotope patterns of 1 and 2 fit the corresponding simulated ones well. Furthermore, elemental analyses confirmed the purities of the obtained Rh2 complexes; the observed CHN values of 1 and 2 were very close to the calculated ones of desolvated 1 and 2 (see the experimental section).
2.2. Crystal Structures
Single crystals of 1(MeOH)2 and 2(MeOH)2 suitable for SCXRD measurements were grown using the method described in the experimental section. Diffraction analysis revealed that 1(MeOH)2 crystallizes in a monoclinic system with the P 21/n space group, whereas 2(MeOH)2 crystallizes in a triclinic system with the P-1 space group. Final refined structures of 1(MeOH)2 {R1(I > 2σ(I)) = 2.48%} and 2(MeOH)2 {R1(I > 2σ(I)) = 4.03%} are shown in Figure 1 and Figure 2, respectively.
In the structure of 1(MeOH)2, the asymmetric unit consists of one-half of a molecule comprising a Rh2+ cation, two fca ligands, and a MeOH ligand. A crystallographic inversion center is located at the midpoint of the Rh–Rh bond, and the overall structure forms a paddlewheel unit, in which four fca and two MeOH ligands are coordinated to the equatorial and axial positions, respectively, of the Rh2 core, and is isostructural with the previously reported [Cu2(fca)4(MeOH)2] [20], of which the crystal system (triclinic, P-1) is different. In contrast, the asymmetric unit of 2(MeOH)2 contains two crystallographically independent molecules; both Rh2 molecules adopt a heteroleptic paddlewheel unit, in which the Rh2 cores are equatorially bridged with fca and three pivalate ligands and axially coordinated via two MeOH ligands.
The Rh2+ ions in 1(MeOH)2 and 2(MeOH)2 exist in distorted octahedral environments. The Rh–Rh bond lengths of 1(MeOH)2 and 2(MeOH)2 are 2.3771(3) and 2.3712(3)/2.3724(3) Å, respectively, which are in the typical range of single Rh–Rh bonds in [Rh2(O2CR)4(L)2] complexes (2.35–2.45 Å) including [Rh2(piv)4(H2O)2] (2.371(1) Å) [18,40]. This indicates that the ground-state charge transfer interactions between the Rh2 core and the fca ligands in 1(MeOH)2 and 2(MeOH)2 are negligible. The Rh–O(fca) bond lengths of 1(MeOH)2 are in the range of 2.0131(12)–2.0502(12) Å (average length: 2.033 Å), which are slightly longer than those of [Cu2(fca)4(MeOH)2] (average Cu–O(fca): 1.967 Å) [20] and slightly shorter than those of [Mo2(fca)4(CH3CN)(DMSO)] (average Mo–O(fca): 2.112 Å) [22]. In 2(MeOH)2, the Rh–O(fca) bond lengths are in the range of 2.015(3)–2.044(2) Å (average length: 2.031 Å), which are consistent with the Rh–O(fca) bond lengths of 1(MeOH)2. There was no significant difference in the length of Rh–O(piv) bonds located in the trans (average length: 2.030 Å) and cis positions (average length: 2.027 Å) relative to the Rh–O(fca) bonds. In both complexes, the Rh–Rh–O(fca) and Rh–Rh–O(piv) angles are nearly perpendicular, and the C–O bond lengths and O–C–O angles of the bridging carboxylate moieties are approximately 1.26–1.28 Å and 124–125°, respectively, which are approximately equal to those of paddlewheel-type Rh2 complexes.
In the ferrocene moieties, the average distances from the centroids of the cyclopentadienyl rings to the corresponding Fe atom in 1(MeOH)2 and 2(MeOH)2 are 1.649 Å (∆max = 0.009 Å) and 1.642 Å (∆max = 0.009 Å), respectively, which are nearly equal to those in ferrocenecarboxylic acid (1.638 Å) [13] and [Cu2(fca)4(MeOH)2] (1.651 Å) [20]. The cyclopentadienyl rings in 1(MeOH)2 adopt an eclipsed conformation (rotation angles: 2.20–2.85° and 6.01–6.68° for each asymmetric fca unit), whereas those in 2(MeOH)2 are rotated by approximately 12–14° from the staggered conformation (rotation angles: 21.83–23.84° and 22.62–24.33° for fca units belonging to crystallographically different Rh2 molecules).
2.3. Optimized Geometries and Electronic Structures
Spin-restricted DFT calculations (B3LYP method) of 1(MeOH)2 and 2(MeOH)2 were performed to clarify the molecular geometries and electronic structures of 1 and 2 in their crystal states and in the CHCl3-MeOH solution. As shown in Figure S5, the optimized geometries of 1(MeOH)2 and 2(MeOH)2 reproduced the experimentally observed (X-ray) geometries without significant structural changes (Tables S1 and S2). The Rh–Rh bond lengths of the optimized geometries of 1(MeOH)2 and 2(MeOH)2 were calculated to be 2.407 and 2.405 Å, respectively, which are almost identical (~0.034 Å difference) to those of observed geometries. In the primary coordination sphere of the Rh2 cores in 1(MeOH)2, although the averaged Rh–O(fca) bond length (2.066 Å) of the optimized geometry is close to that of the observed geometry, the averaged Rh–O(MeOH) bond length (2.389 Å) of the optimized geometry is 0.105 Å longer than that of the observed geometry. A similar tendency was found in 2; the averaged Rh–O(fca) (2.067 Å), Rh–O(trans-piv) (2.067 Å), and Rh–O(cis-piv) (2.066 Å) bond lengths agree well with each other and those of the observed geometry, whereas the averaged Rh–O(MeOH) bond length (2.389 Å) of the optimized geometry is 0.094 Å longer than that of the observed geometry. The longer Rh–O(MeOH) bond lengths in the optimized geometries are presumably due to crystal packing stress [41]. In the ferrocene moieties, the cyclopentadienyl rings in the optimized geometries of 1(MeOH)2 and 2(MeOH)2 adopt an eclipsed conformation. The average distances from the centroids of the cyclopentadienyl rings to the corresponding Fe atom in optimized geometries of 1(MeOH)2 and 2(MeOH)2 are 1.680 and 1.679 Å, respectively, which are also close to the distances in the observed geometries.
Figure 3 shows the electronic structure diagrams with selected molecular orbitals (MOs) of optimized geometries of 1(MeOH)2 and 2(MeOH)2. In the occupied orbital spaces, (1) electronic configurations of the Rh2 core in 1(MeOH)2 and 2(MeOH)2 are π4σ2δ2π*2δ*2π*2 and π4σ2δ2δ*2π*4, respectively, indicating that a single bond is formed between two Rh ions in 1(MeOH)2 and 2(MeOH)2, similarly to other paddlewheel-type Rh2 complexes [41,42,43], and (2) the occupied MOs localized on fca moieties are energetically degenerate and relatively more unstable than those on Rh2 moieties. For instance, the HOMO to HOMO-7 in 1(MeOH)2 and HOMO and HOMO-1 in 2(MeOH)2 are mainly localized on the fca ligands (where, HOMO is the highest occupied MO). Two π*2(Rh2) and one δ*2(Rh2) antibonding orbitals were also energetically degenerate and found in the HOMO-8 to HOMO-10 of 1(MeOH)2 and HOMO-2 to HOMO-4 of 2(MeOH)2. The HOMO-11 of 1(MeOH)2 and HOMO-6 of 2(MeOH)2, whose orbital energies are approximately 0.7 and 1.0 eV more stable than those of MOs with π*2(Rh2)/δ*2(Rh2) characters, are mainly localized on the δ2(Rh2) orbitals with minor contributions from d(Fe) orbitals. The σ2(Rh2) orbitals, which are found in the HOMO-16 (−6.82 eV) of 1(MeOH)2 and HOMO-7 (−6.81 eV) of 2(MeOH)2, strongly interact with the p(O) orbitals of the MeOH moieties, and their orbital energies are almost identical. The π4(Rh2) orbitals of 1(MeOH)2 and 2(MeOH)2 are remarkably stable and are observed as HOMO-24 and HOMO-25, and HOMO-9 and HOMO-11, respectively.
In the unoccupied orbital spaces of 1(MeOH)2 and 2(MeOH)2, the σ*2(Rh2) orbitals, which interact with the p(O) orbitals of the MeOH moieties, are the lowest unoccupied MOs (LUMOs). The HOMO-LUMO gap energies of 1(MeOH)2 and 2(MeOH)2 are estimated to be 3.93 and 3.97 eV, respectively. The LUMO+1 of 1(MeOH)2 and 2(MeOH)2 exhibit antibonding orbital interactions with dx2-y2(Rh2) character. The energies of LUMO and LUMO+1 of 1(MeOH)2 and 2(MeOH)2 are identical, respectively, between the complexes. The unoccupied MOs localized on the fca moieties are found in the LUMO+2 and LUMO+3 of 1(MeOH)2 and LUMO+2 of 2(MeOH)2. The LUMO+6 of 1(MeOH)2 and LUMO+3 of 2(MeOH)2 are localized on the bonding orbital interactions with the dx2-y2(Rh2) character.
2.4. Absorption Features
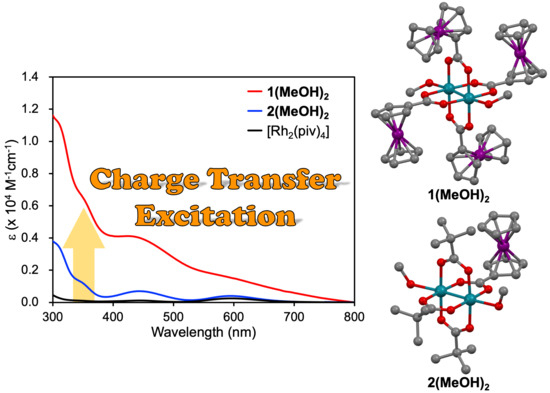
Figure 4 shows the absorption spectra of 1, 2, [Rh2(piv)4], and fcaH in a 9:1 CHCl3-MeOH solution. In the visible light region, [Rh2(piv)4] possesses two absorption bands at 595 and 444 nm due to π*(Rh2) → σ*(Rh2) and π*(Rh2) → antibonding dx2-y2(Rh2) excitations [41], respectively, whereas fcaH has an absorption band at 447 nm due to the d–d(Fe) excitation [44]. The absorption spectral shape of 2 behaves like a superposition of those of [Rh2(piv)4] and fcaH; the band maxima of 2 are observed at 597 (ε = 400) and 443 nm (ε = 695), whereas the absorption coefficients are slightly higher than those of the corresponding bands of [Rh2(piv)4] and fcaH. Multiple coordination of fca ligands to the Rh2 core further increases the absorption coefficients; 1 has an intense absorption maximum at 423 nm whose absorption coefficient (ε = 4122) is apparently higher than the sum of the absorption coefficients of one [Rh2(piv)4] and four fcaH at 423 nm. In the longer-wavelength regions, the low-lying absorption bands due to the d–d excitations of the Rh2 core cannot be confirmed because they are hidden by an intense band at 423 nm. On the other hand, the solid-state diffuse reflectance spectrum of 1 in the visible light region shows a shoulder band at approximately 610 nm in addition to a higher-energy band at 444 nm (see Figure S6).
In the ultraviolet region, 1 and 2 show unique absorption features similarly to fcaH, with a shoulder band around 350 nm, but their absorption coefficients are relatively higher than those of fcaH. These results indicate that the absorption features of 1 and 2 may possess new characteristics, such as charge transfer (CT) excitation, in addition to the respective absorption characteristics of the Rh2 cores and fca ligands.
TDDFT calculations of 1(MeOH)2 and 2(MeOH)2 were performed to elucidate the absorption features of 1 and 2 in CHCl3-MeOH solution. The computed excitation wavelengths, oscillator strengths, and excitation components of 1(MeOH)2 and 2(MeOH)2 are summarized in Tables S3 and S4, respectively. A comparison of the experimental spectra and calculated vertical excitations of 1(MeOH)2 and 2(MeOH)2 are shown in Figure S7. The excitation characteristics of 1(MeOH)2 and 2(MeOH)2 are very complicated, consisting of several excitation components because 1(MeOH)2 and 2(MeOH)2 form degenerated electronic structures. In general terms, the absorption bands observed at approximately 590–610 nm in 1(MeOH)2 and 2(MeOH)2 are comprised of d–d excitations of the fca ligands and Rh2 core (dπ*(Rh2) → dσ*(Rh2) or antibonding-dx2-y2(Rh2)) as the major contributions and CT excitations from fca ligands to the Rh2 core (dσ*(Rh2)) as the minor contributions. The dominant excitation characters of 1(MeOH)2 and 2(MeOH)2 at approximately 440 nm can be assigned to d–d excitations of the Rh2 core (dπ*(Rh2) → dσ*(Rh2) or antibonding-dx2-y2(Rh2)), and 1(MeOH)2 also includes a slight contribution of CT excitation from the fca ligands to the Rh2 core. The calculated oscillator strengths of 1(MeOH)2 in the visible light region are relatively higher than those of 2(MeOH)2, and this tendency is consistent with the experimental results. In the ultraviolet region, 1(MeOH)2 and 2(MeOH)2 possess CT excitation characteristics from the Rh2 core to the fca ligands in addition to d–d excitations of the fca ligands and the Rh2 core and CT excitations from the fca ligands to the Rh2 core. One of the reasons why the absorption coefficients of 1(MeOH)2 and 2(MeOH)2 are higher than those of fcaH and [Rh2(piv)4] may be the occurrence of CT excitations of 1(MeOH)2 and 2(MeOH)2.
2.5. Electrochemical Properties
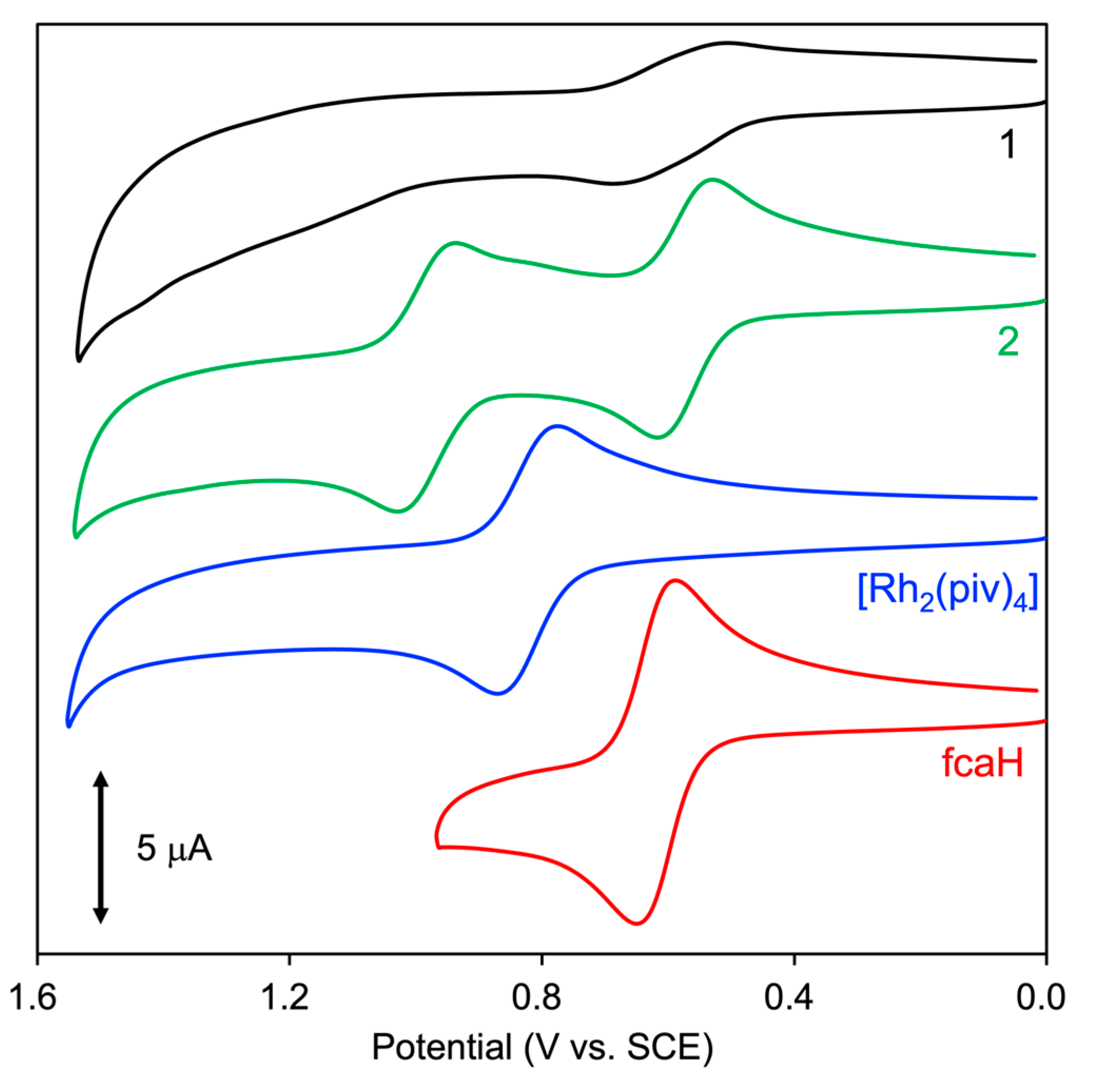
To investigate the electrochemical properties of 1 and 2, cyclic voltammetry (CV) analyses were performed in a degassed 9:1 CHCl3-MeOH solution. As shown in Figure 5, the fcaH and [Rh2(piv)4] exhibit reversible one-electron oxidation waves at redox potentials E1/2 = 0.620 and 0.822 V vs. SCE, respectively, in which the separation of the cathodic (Epc) and anodic (Epa) peak potentials (∆E) is estimated to be 59 and 94 mV, respectively. The CV diagram of 2 shows two reversible one-electron redox waves at 0.574 V (∆E = 90 mV) and 0.984 V vs. SCE (∆E = 92 mV), which are ascribed to oxidation processes of the fca ligand and Rh2 core, respectively. The reason for the positive shift of potential for Rh2 oxidation in 2 compared to [Rh2(piv)4] may be due to the electron-withdrawing effect of the oxidized fca ligand (fca+). In the CV diagram of 1, only one reversible and broad redox wave is observed at 0.597 V vs. SCE. The reason is inferred that (1) the observed redox wave is an aggregate of several waves of fca oxidation processes because the ∆E value was estimated to be 180 mV, which is obviously larger than those of fcaH (∆E = 59 mV) and 2 (∆E = 90 mV for the fca ligand) and (2) the potential for the Rh2 oxidation process was not found in the observed window because of the strong electron-withdrawing effects of the four fca (or fca+) ligands on the Rh2 core. The result of the differential pulse voltammetry (DPV) measurement of 1 supported the first explanation above; the DPV diagram of 1 took a shape that merged two consecutive potential peaks (See Figure S8). Furthermore, these explanations are consistent with the unrestricted DFT (uDFT) calculation results of one-electron oxidation species of 1(MeOH)2 and 2(MeOH)2; the spin density distributions of 1(MeOH)2+ and 2(MeOH)2+ are predominantly localized on the fca ligands (see Figure 6). Remarkably, this calculation result also clearly indicates that 1(MeOH)2+ exhibits redox interactions between four fca ligands through a Rh2 core.
3. Materials and Methods
3.1. Chemicals and Instruments
[Rh2(O2CCH3)4(H2O)2] and [Rh2(piv)4] were prepared according to previous studies [40,45,46]. The other chemicals used in this study were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan).
Proton nuclear magnetic resonance (1H NMR) was performed using a JEOL JNM-ECX500 spectrometer (500 MHz) (JEOL, Tokyo, Japan) in DMSO-d6. Chemical shifts (δ) are reported as parts per million (ppm) relative to residual DMSO (δ = 2.49 ppm). Electrospray time-of-flight ionization mass spectrometry (ESI-TOF-MS) was measured on a Bruker micrOTOFII instrument (Bruker, Billerica, MA, USA) in the positive ion mode. Mass calibration was performed using sodium formate. Elemental analyses (EA) were carried out with a Yanako CHN Corder MT-6 analyzer (Yanaco, Tokyo, Japan). Electrochemical analyses of 1 (0.10 mM), 2, [Rh2(piv)4], and fcaH (0.50 mM) were performed in a 9:1 CHCl3-MeOH solution containing 0.10 M tetrabutylammonium hexafluorophosphate (TBAPF6) using a HOKUTO DENKO HZ-7000 HAG1232m system (Meiden Hokuto Coorporation, Tokyo, Japan) with a glassy carbon working electrode, platinum counter wire, and saturated calomel reference electrode (SCE) under an argon atmosphere. Absorption spectra were recorded with a JASCO UV-670 spectrophotometer (JASCO, Tokyo, Japan).
3.2. Synthesis of [Rh2(fca)4] (1)
A mixture of [Rh2(O2CCH3)4(H2O)2] (47.8 mg, 0.10 mmol), ferrocenecarboxylic acid (fcaH; 184.0 mg, 0.80 mmol), and chlorobenzene (15.0 mL) was refluxed under nitrogen for 36 h. Resultant precipitate was filtrated and washed with methanol and acetone and dried at 393 K under vacuum for 3 h. A brown powder was obtained in a 94.7% yield (106.3 mg). 1H NMR (500 MHz, DMSO-d6, δ): 4.62 (br, 8H), 4.22 (br, 8H), 3.85 (br, 20H) ppm. EA calculated (%) for C44H36Fe4O8Rh2: C 47.10, H 3.23; found C 46.84, H 3.45. ESI-TOF-MS calculated for C44H36Fe4O8Rh2Na [M+Na]+: 1144.7814 m/z; found 1144.7777 m/z.
3.3. Synthesis of [Rh2(fca)(piv)3] (2)
[Rh2(piv)4] (122.1 mg, 0.20 mmol) and fcaH (46.0 mg, 0.20 mmol) were dissolved in N,N-dimethylaniline (10.0 mL) and heated at 433 K under nitrogen for 36 h. The solvent was then removed under vacuum. The residue was dissolved in CHCl3 and washed four times with a 2 M HCl solution and water using a separating funnel. The organic layer was evaporated and purified using silica-gel column chromatography (eluent: CH3CN-hexane-CH2Cl2 = 2: 48~0: 50~98, in volume). The yellowish-green fraction was separated and evaporated to dryness. The resultant solid was dried at 393 K under vacuum for 3 h. A yellowish-green powder was obtained in a 29.1% yield (42.9 mg). 1H NMR (500 MHz, DMSO-d6, δ): 4.61 (t, 2H), 4.29 (t, 2H), 3.97 (s, 5H), 0.90 (s, 9H), 0.89 (s, 18H) ppm. EA calculated (%) for C26H36FeO8Rh2: C 42.30, H 4.92; found C 42.53, H 4.74. ESI-TOF-MS calculated for C26H36FeO8Rh2Na [M+Na]+: 760.9762 m/z; found 760.9762 m/z.
3.4. Crystallography
Single crystals of [Rh2(fca)4(MeOH)2] (1(MeOH)2) suitable for SCXRD analysis were grown using a slow diffusion of hexane into MeOH/CH2Cl2 solution containing 1, whereas those of [Rh2(fca)(piv)3(MeOH)2] (2(MeOH)2) were obtained using a slow diffusion of water into MeOH solution containing 2. Diffraction data were collected on a Rigaku HyPix-6000 detector system (Mo Kα radiation; λ = 0.71073 Å) at 150 K. Data collection and reduction were performed with CrysAlisPro (version 1.171.39.43a) software [47]. The structures were solved with the SHELXT program [48], and the full-matrix least-square refinements on F2 were performed using the SHELXL program [49] via the Olex2 (version 1.5) software [50]. All nonhydrogen atoms were refined anisotropically, whereas hydrogen atoms were placed in calculated positions and refined as a riding model. In the refinement of 2(MeOH)2, the residual electron density of disorder solvents was removed using the solvent mask routine of the Olex2. Crystallographic data of final refined structures are summarized in Table 1, and selected bond lengths and angles are given in Tables S5 and S6 in the Supplementary Materials. These crystallographic data can be obtained free of charge from Cambridge Crystallographic Data Centre (CCDC); deposition numbers of 1(MeOH)2 and 2(MeOH)2 are CCDC-2324331 and 2324332, respectively.
3.5. Theoretical Calculation Method
All density functional theory (DFT) calculations were performed using the B3LYP functional [51] in conjunction with the LANL08f for Rh atom, SVP for Fe atom, aug-cc-pVDZ for O atom, and cc-pVDZ for C and H atoms on the Gaussian 16 program package [52]. The solvent effect of CHCl3 was taken into account using the self-consistent reaction field (SCRF) through the polarizable continuum model (PCM) theory [53]. The initial structures for geometry optimizations were produced from the CIF files, and obtained optimized structures were confirmed as the minima via frequency analyses. The singlet vertical excitations were calculated using the time-dependent density functional theory (TDDFT). Optimized geometries, molecular orbitals, and spin density distributions were drawn using a GaussView 5.0 [54].
4. Conclusions
This study described the synthesis, characterization, structural determination, and absorption and electrochemical properties of fca-bearing homoleptic and heteroleptic paddlewheel-type Rh2 complexes, 1 and 2. SCXRD analyses of 1(MeOH)2 and 2(MeOH)2, of which crystals were obtained using recrystallization from MeOH-containing solutions of 1 and 2, clearly proved that the fca ligands are connected to the equatorial positions of the Rh2 core. To the best of our knowledge, this study is the first result of the structural determination of fca-bearing paddlewheel-type Rh2 complexes. Optimized geometries of 1(MeOH)2 and 2(MeOH)2, which were obtained using DFT calculations, reproduced the experimentally observed geometries without significant structural changes, and their electronic structure analyses revealed that (1) a single bond is formed between two Rh ions in 1(MeOH)2 and 2(MeOH)2, similarly to other paddlewheel-type Rh2 complexes, and (2) the occupied MOs localized on fca moieties are energetically degenerate and relatively more unstable than those on Rh2 moieties. Complexes 1 and 2 showed unique absorption features in the 9:1 CHCl3-MeOH solution; absorption coefficients of 1 and 2 are apparently higher than those of [Rh2(piv)4] and fcaH. TDDFT calculations indicated that the higher absorption coefficients of 1 and 2 are due to the CT excitations between fca ligands and Rh2 cores in 1 and 2. CV results and uDFT calculations clarified that 2 undergoes stepwise oxidation in the order of a fca ligand and a Rh2 core, whereas 1 exhibited redox interactions between four fca ligands through a Rh2 core in the oxidation process. It is necessary to improve the solubilities of homoleptic Rh2 complexes coordinated by metalloligands with ferrocene units for further investigation of absorption and electrochemical properties as well as applications as the catalysts and building blocks for the CPs and MOFs. These studies are currently being conducted by introducing various substituents on Rh2 and fca units in our laboratory.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics12020041/s1, Table S1: Averaged bond lengths (Å) of observed and optimized geometries of [Rh2(fca)4(MeOH)2] (1(MeOH)2); Table S2: Averaged bond lengths (Å) of observed and optimized geometries of [Rh2(fca)(piv)3(MeOH)2] (2(MeOH)2); Table S3: TDDFT results (excitation wavelength, oscillator strength, and orbital contribution) of 1(MeOH)2; Table S4: TDDFT results (excitation wavelength, oscillator strength, and orbital contribution) of 2(MeOH)2; Table S5: Selected bond lengths (Å) and angles (°) of 1(MeOH)2; Table S6: Selected bond lengths (Å) and angles (°) of 2(MeOH)2; Figure S1: 1H NMR spectrum of 1 in DMSO-d6; Figure S2: 1H NMR spectrum of 2 in DMSO-d6; Figure S3: Observed and simulated ESI-TOF-MS spectra of 1; Figure S4: Observed and simulated ESI-TOF-MS spectra of 2; Figure S5: Optimized geometries of 1(MeOH)2 (left) and 2(MeOH)2 (right); Figure S6: Diffuse reflectance (DR) spectrum of 1; Figure S7. Observed spectra (black line) and calculated vertical excitations (red line) of (a) 1(MeOH)2 and (b) 2(MeOH)2; Figure S8. Differential pulse voltammetry (DPV) diagram of 1 (0.10 mM) in 9:1 CHCl3-MeOH solution containing 0.10 M TBAPF6.
Author Contributions
Conceptualization, Y.K. and M.H.; validation, Y.K., K.S., N.Y. and M.H.; formal analysis, Y.K., K.S. and N.Y.; investigation, K.S.; resources, Y.K. and N.Y.; data curation, N.Y.; writing—original draft preparation, Y.K.; writing—review and editing, K.S., N.Y. and M.H.; visualization, N.Y.; supervision, Y.K. and M.H.; project administration, Y.K.; funding acquisition, Y.K. and N.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI, Grant Numbers 22K14765 and 19K15588.
Data Availability Statement
Crystallographic data of 1(MeOH)2 and 2(MeOH)2 can be obtained free of charge from Cambridge Crystallographic Data Centre (CCDC); deposition numbers of 1(MeOH)2 and 2(MeOH)2 are CCDC-2324331 and 2324332, respectively.
Acknowledgments
A part of this work was conducted at the Institute for Molecular Science, supported by the Nanotechnology Platform Program <Molecule and Material Synthesis> of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jansze, S.M.; Severin, K. Clathrochelate Metalloligands in Supramolecular Chemistry and Materials Science. Acc. Chem. Res. 2018, 51, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, N.; Kuwamura, N.; Kojima, T.; Konno, T. Development of coordination chemistry with thiol-containing amino acids. Coord. Chem. Rev. 2023, 474, 214857. [Google Scholar] [CrossRef]
- Kumar, G.; Gupta, R. Molecularly designed architectures—The metalloligand way. Chem. Soc. Rev. 2013, 42, 9403–9453. [Google Scholar] [CrossRef] [PubMed]
- Garibay, S.J.; Stork, J.R.; Cohen, S.M. The Use of Metalloligands in Metal-Organic Frameworks. Prod. Inorg. Chem. 2009, 56, 335–378. [Google Scholar]
- Das, M.C.; Xiang, S.; Zhang, Z.; Chen, B. Functional Mixed Metal-Organic Frameworks with Metalloligands. Angew. Chem. Int. Ed. 2011, 50, 10510–10520. [Google Scholar] [CrossRef]
- Gao, W.X.; Zhang, H.N.; Jin, G.X. Supramolecular catalysis based on discrete heterometallic coordination-driven metallacycles and metallacages. Coord. Chem. Rev. 2019, 386, 69–84. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C-Photochem. Rev. 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane. Angew. Chem. Int. Ed. 2006, 45, 6215–6218. [Google Scholar] [CrossRef]
- Singh, A.K.; Pandey, D.S.; Xu, Q.; Braunstein, P. Recent advances in supramolecular and biological aspects of arene ruthenium(II) complexes. Coord. Chem. Rev. 2014, 270, 31–56. [Google Scholar] [CrossRef]
- Gil-Rubio, J.; Vicente, J. The Coordination and Supramolecular Chemistry of Gold Metalloligands. Chem. Eur. J. 2018, 24, 32–46. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Kong, X.J.; Zheng, Z.; Long, L.S.; Zheng, L.S. High-Nuclearity Lanthanide-Containing Clusters as Potential Molecular Magnetic Coolers. Acc. Chem. Res. 2018, 51, 517–525. [Google Scholar] [CrossRef]
- Chiu, C.C.; Cheng, M.C.; Lin, S.H.; Yan, C.W.; Lee, G.H.; Chang, M.C.; Lin, T.S.; Peng, S.M. Structure and magnetic properties of a novel heteroheptanuclear metal string complex [Ni3Ru2Ni2(μ7-teptra)4(NCS)2](PF6). Dalton Trans. 2020, 49, 6643–6653. [Google Scholar] [CrossRef]
- Cotton, F.A.; Reid, A.H. Solid-State structure of Ferrocenecarboxylic acid, [Fe(C5H4CO2H)(C5H5)]. Acta Cryst. 1985, C41, 686–688. [Google Scholar] [CrossRef]
- Prokopuk, N.; Shriver, D.F. A one-dimensional array of clusters: Na2Mo6Cl8(O2CC5H4FeCp)6·CH3OH. Inorg. Chem. 1997, 36, 5609–5613. [Google Scholar] [CrossRef]
- Fan, Y.; Cui, Y.; Zou, G.D.; Duan, R.H.; Zhang, X.; Dong, Y.X.; Lv, H.T.; Cao, J.T.; Jing, Q.S. A ferrocenecarboxylate-functionalized titanium-oxo-cluster: The ferrocene wheel as a sensitizer for photocurrent response. Dalton Trans. 2017, 46, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Li, G.; Li, L.; Zhu, Y.; Meng, X.; Fan, Y. Synthesis, crystal structures, and magnetic properties of three novel ferrocenecarboxylato-bridged lanthanide dimers. Inorg. Chem. 2003, 42, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Mashima, K. Interaction of Ferrocene Moieties Across a Square Pt4 Unit: Synthesis, Characterization, and Electrochemical Properties of Carboxylate-Bridged Bimetallic Pt4Fen (n = 2, 3, and 4) Complexes. Inorg. Chem. 2011, 50, 11384–11393. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer Science and Business Media: New York, NY, USA, 2005. [Google Scholar]
- Churchill, M.R.; Li, Y.J.; Nalewajek, D.; Schaber, P.M.; Dorfman, J. Preparation, Crystal and Molecular Structure, and Properties of Tetrakis(ferrocenecarboxylato)bis(tetrahydrofuran)dicopper(II). A Structure Containing both Eclipsed and Staggered Ferrocenyl Fragments. Inorg. Chem. 1985, 24, 2684–2687. [Google Scholar] [CrossRef]
- Artetxe, B.; Vitoria, P.; Pache, A.; Reinoso, S.; Gutiérrez-Zorrilla, J.M. Tetrakis(μ2-ferrocenecarboxylato-κ2O:O′)bis[(methanol-κO)copper(II)] methanol disolvate. Acta Cryst. 2011, E67, m1840–m1841. [Google Scholar] [CrossRef]
- Zhang, E.; Hou, H.; Meng, X.; Liu, Y.; Liu, Y.; Fan, Y. Ferrocenyl Functional Coordination Polymers Based on Mono-, Bi-, and Heterotrinuclear Organometallic Building Blocks: Syntheses, Structures, and Properties. Cryst. Growth Des. 2009, 9, 903–913. [Google Scholar] [CrossRef]
- Cotton, F.A.; Falvello, L.R.; Reid, A.H.; Tocher, J.H. Mixed-ligand systems containing quadruple bonds. Capture and structural characterization of an intermediate in the ligand exchange process leading to new carboxylates of the dimolybdenum(4+) unit. Synthesis and X-ray crystallographic and electrochemical studies of Mo2[(η5-C5H4CO2)Fe(η5-C5H5)]2(O2CCH3)2(C5H5N)2 and [Mo2](η5-C5H4CO2)Fe(η5-C5H5)]4(ax-CH3CN)(ax-DMSO)](DMSO)2. J. Organomet. Chem. 1987, 319, 87–97. [Google Scholar]
- Cai, X.M.; Meister, T.K.; Pöthig, A.; Kühn, F.E. Filling a Gap: Electrochemical Property Comparison of the Completed Compound Series [Mo2(DArF)n(O2C-Fc)4–n] (DArF = N,N′-Diarylformamidinate; O2C-Fc = Ferrocenecarboxylate). Inorg. Chem. 2016, 55, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.W.; Cameron, T.S.; Robertson, K.N.; Swarts, J.C.; Aquino, M.A.S. Structure and electrochemistry of various diruthenium(II,III) tetrametallocenecarboxylates. Organometallics 2002, 21, 5962–5971. [Google Scholar] [CrossRef]
- Boyd, D.A.; Cao, Z.; Song, Y.; Wang, T.W.; Fanwick, P.E.; Crutchley, R.J.; Ren, T. Diruthenium Compounds Bearing Equatorial Fc-containing Ligands: Synthesis and Electronic Structure. Inorg. Chem. 2010, 49, 11525–11531. [Google Scholar] [CrossRef] [PubMed]
- Noels, A.F.; Demonceau, A.; Petiniot, N.; Hubert, A.J.; Teyssie, P. Transition-metal-catalyzed reaction of diazocompounds, efficient synthesis of functionalized ethers by carbene insertion into the hydroxylic bond of alcohols. Tetrahedron 1982, 38, 2733–2739. [Google Scholar] [CrossRef]
- Demonceau, A.; Noels, A.F.; Teyssie, P.; Hubert, A.J. Shape selective alkane functionalisation by ethyl diazoacetate catalysed by rhodium carboxylates. J. Mol. Catal. 1988, 49, L13–L17. [Google Scholar] [CrossRef]
- Vosáhlo, P.; Harmach, P.; Císarová, I.; Stepnicka, P. Synthesis and characterisation of dirhodium(II) tetraacetates bearing axial ferrocene ligands. J. Organomet. Chem. 2021, 953, 122065. [Google Scholar] [CrossRef]
- Hansen, J.; Davies, H.M.L. High Symmetry Dirhodium(II) Paddlewheel Complexes as Chiral Catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef]
- Fiori, K.W.; Du Bois, J. Catalytic Intermolecular Amination of C−H Bonds: Method Development and Mechanistic Insights. J. Am. Chem. Soc. 2007, 129, 562–568. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Handa, M.; Kawamoto, T. Intrinsic Hydrogen Evolution Capability and Theoretically Supported Reaction Mechanism of Paddlewheel-type Dirhodium Complex. Dalton Trans. 2019, 48, 7302–7312. [Google Scholar] [CrossRef]
- Kataoka, Y.; Sato, K.; Yano, N. Hydroxypyridinate-bridged paddlewheel-type dirhodium complex as a catalyst for photochemical and electrochemical hydrogen evolution. J. Chem. Phys. 2023, 159, 204304. [Google Scholar] [CrossRef] [PubMed]
- Hilderbrand, S.A.; Lim, M.H.; Lippard, S.J. Dirhodium Tetracarboxylate Scaffolds as Reversible Fluorescence-Based Nitric Oxide Sensors. J. Am. Chem. Soc. 2004, 126, 4972–4978. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Kohara, Y.; Yano, N.; Kawamoto, T. Unique vapochromism of a paddlewheel-type dirhodium complex accompanied by dynamic structural and phase transitions. Dalton Trans. 2020, 49, 14373–14377. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Dunber, K.R. Interactions of Metal−Metal-Bonded Antitumor Active Complexes with DNA Fragments and DNA. Acc. Chem. Res. 2005, 38, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Yano, N.; Mikuriya, M.; Handa, M. Coordination polymers and metal–organic frameworks based on paddlewheel-type dirhodium(II) tetracarboxylates. Coord. Chem. Rev. 2022, 472, 214796. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Mikuriya, M.; Handa, M. Paddlewheel-type dirhodium complexes with N,N’-bridging ligands. Coord. Chem. Rev. 2023, 479, 214997. [Google Scholar] [CrossRef]
- Sawamura, M.; Sasaki, H.; Nakata, T.; Ito, Y. Synthesis of Optically Active Ferrocene Analogues of Salicylic Acid Derivatives and Rhodium(II)-Catalyzed Asymmetric Intramolecular C–H Insertion of α-Diazo β-Keto Esters Using New Chiral Carboxylato Ligands. Bull. Chem. Soc. Jpn. 1993, 66, 2725–2729. [Google Scholar] [CrossRef]
- Arakawa, K.; Yano, N.; Imasaki, N.; Kohara, Y.; Yatsushiro, D.; Atarashi, D.; Handa, M.; Kataoka, Y. Coordination-Induced Self-Assembly of a Heteroleptic Paddlewheel-Type Dirhodium Complex. Crystals 2020, 10, 85. [Google Scholar] [CrossRef]
- Cotton, F.A.; Felthouse, T.R. Structural studies of three tetrakis(carboxylato)dirhodium(II) adducts in which carboxylate groups and axial ligands are varied. Inorg. Chem. 1980, 19, 323–328. [Google Scholar] [CrossRef]
- Kataoka, Y.; Fukumoto, R.; Yano, N.; Atarashi, D.; Tanaka, H.; Kawamoto, T.; Handa, M. Synthesis, Characterization, Absorption Properties, and Electronic Structures of Paddlewheel-Type Dirhodium(II) Tetra-μ-(n-naphthoate) Complexes: An Experimental and Theoretical Study. Molecules 2019, 24, 447. [Google Scholar] [CrossRef]
- Sizova, O.V.; Skripnikov, L.V.; Sokolov, A.Y.; Ivanova, N.V. Rhodium and ruthenium tetracarboxylate nitrosyl complexes: Electronic structure and metal-metal bond. Russ. J. Inorg. Chem. 2007, 33, 588–593. [Google Scholar] [CrossRef]
- Norman, J.G.; Kolari, H.J. Strength and trans influence of Rh-Rh bond in rhodium(II) carboxylate dimers. J. Am. Chem. Soc. 1978, 100, 791–799. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, H.; Sun, M.; Liu, Y.; Pang, X.; Yu, X.; Liu, B.; Li, Z. Substitution effect on the geometry and electronic structure of the ferrocene. J. Comput. Chem. 2007, 28, 2260–2274. [Google Scholar] [CrossRef] [PubMed]
- Legzdins, P.; Mitchell, R.W.; Rempel, G.L.; Ruddick, J.D.; Wilkinsin, G. The protonation of ruthenium- and rhodium-bridged carboxylates and their use as homogeneous hydrogenation catalysts for unsaturated substances. J. Chem. Soc. A 1970, 3322–3326. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Kawamoto, T.; Handa, M. Isolation of the Intermediate in the Synthesis of Paddlewheel-type Dirhodium Tetraacetate. Eur. J. Inorg. Chem. 2015, 34, 5650–5655. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; Rigaku Oxford Diffraction: Tokyo, Japan, 2018.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- John, D. Roy, Keith, Todd, Millam, GaussView 5.0; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
Scheme 1.
Molecular structures of: (a) fcaH ligand, and homoleptic and heteroleptic paddlewheel-type Rh2 complexes; (b) [Rh2(fca)4] (1); (c) [Rh2(fca)(piv)3] (2).
Scheme 1.
Molecular structures of: (a) fcaH ligand, and homoleptic and heteroleptic paddlewheel-type Rh2 complexes; (b) [Rh2(fca)4] (1); (c) [Rh2(fca)(piv)3] (2).

Figure 1.
Crystal structure of 1(MeOH)2 with thermal ellipsoids at 25% probability. Hydrogen atoms and MeOH solvent are omitted for clarity. Color code: green = Rh, red = O, gray = C, purple = Fe.
Figure 1.
Crystal structure of 1(MeOH)2 with thermal ellipsoids at 25% probability. Hydrogen atoms and MeOH solvent are omitted for clarity. Color code: green = Rh, red = O, gray = C, purple = Fe.

Figure 2.
Crystal structure of 2(MeOH)2 with thermal ellipsoids at 25% probability. Hydrogen atoms are omitted for clarity. Color code: green = Rh, red = O, gray = C, purple = Fe.
Figure 2.
Crystal structure of 2(MeOH)2 with thermal ellipsoids at 25% probability. Hydrogen atoms are omitted for clarity. Color code: green = Rh, red = O, gray = C, purple = Fe.

Figure 3.
Electronic structures and selected MOs of 1(MeOH)2 and 2(MeOH)2. Here, H and L represent the HOMO and LUMO, respectively.
Figure 3.
Electronic structures and selected MOs of 1(MeOH)2 and 2(MeOH)2. Here, H and L represent the HOMO and LUMO, respectively.

Figure 4.
Absorption spectra of 1 (black), 2 (green), fcaH (red), and [Rh2(piv)4] (blue) in 9:1 CHCl3-MeOH solution.
Figure 4.
Absorption spectra of 1 (black), 2 (green), fcaH (red), and [Rh2(piv)4] (blue) in 9:1 CHCl3-MeOH solution.

Figure 5.
CV diagrams of 1 (0.10 mM, black), 2 (0.50 mM, green), [Rh2(piv)4] (0.50 mM, blue), and fcaH (0.50 mM, red) in a 9:1 CHCl3-MeOH solution containing 0.10 M TBAPF6.
Figure 5.
CV diagrams of 1 (0.10 mM, black), 2 (0.50 mM, green), [Rh2(piv)4] (0.50 mM, blue), and fcaH (0.50 mM, red) in a 9:1 CHCl3-MeOH solution containing 0.10 M TBAPF6.

Figure 6.
Spin density distributions of 1(MeOH)2+ (left) and 2(MeOH)2+ (right).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic data of 1(MeOH)2 and 2(MeOH)2.
| 1(MeOH)2 | 2(MeOH)2 | |
|---|---|---|
| Chemical formula | C24H26Fe2O6Rh | C56H88Fe2O20Rh4 |
| Formula weight | 625.06 | 1604.60 |
| Crystal system | Monoclinic | Triclinic |
| Space group | P 21/n | P-1 |
| a (Å) | 13.8360(3) | 13.8890(3) |
| b (Å) | 9.5473(2) | 14.2283(3) |
| c (Å) | 17.6017(4) | 18.1715(4) |
| α (deg) | 90 | 105.982(2) |
| β (deg) | 104.691(2) | 91.795(2) |
| γ (deg) | 90 | 103.012(2) |
| V (Å3) | 2249.11(9) | 3346.82(13) |
| Z | 4 | 2 |
| Dcalc (g cm−3) | 1.846 | 1.592 |
| μ (mm−1) | 2.038 | 1.451 |
| F (000) | 1260.0 | 1632.0 |
| R1 (I > 2σ(I)) | 0.0248 | 0.0403 |
| wR2 (I > 2σ(I)) | 0.0559 | 0.1076 |
| R1 (all data) | 0.0305 | 0.0512 |
| wR2 (all data) | 0.0582 | 0.1138 |
| GOF on F2 | 1.064 | 1.071 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kataoka, Y.; Sato, K.; Yano, N.; Handa, M. Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes. Inorganics 2024, 12, 41. https://doi.org/10.3390/inorganics12020041
AMA Style
Kataoka Y, Sato K, Yano N, Handa M. Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes. Inorganics. 2024; 12(2):41. https://doi.org/10.3390/inorganics12020041
Chicago/Turabian StyleKataoka, Yusuke, Kozo Sato, Natsumi Yano, and Makoto Handa. 2024. "Ferrocene-Bearing Homoleptic and Heteroleptic Paddlewheel-Type Dirhodium Complexes" Inorganics 12, no. 2: 41. https://doi.org/10.3390/inorganics12020041
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.