
Rationale for the Cytogenomics of Cardiovascular Malformations Consortium: A Phenotype Intensive Registry Based Approach

Abstract
:1. Introduction
2. Experimental Section
2.1. Registry Organization
2.2. Study Population and Study Design
2.3. Cytogenetic Data Elements

{kind=link}
| Variable | Example |
|---|---|
| Platform | SNP |
| Chromosome | 22 |
| Cytolocation | 22q11.21 |
| Position Beginning | 17,074,487 |
| Position End | 19,806,645 |
| Version | HG18 |
| CNV Value | 1 |
| Gain/Loss | Loss |
| Gene | USP18, DGCR6, PRODH, DGCR2, DGCR14, TSSK2, GSC2, SLC25A1, CLTCL1, HIRA, MRPL40, UFD1L, CDC45L, CLDN5, SEPT5, GP1BB, TBX1, GNB1L, TXNRD2, COMT, ARVCF, DGCR8, TRMT2A, RANBP1, ZDHHC8, RTN4R, DGCR6L, GGTLC3, RIMBP3, ZNF74, SCARF2, MED15, PI4KA, SERPIND1, SNAP29, CRKL, LZTR1, THAP7, P2RX6, SLC7A4, BCRL2 |
| Inheritance | De novo |
| Size | 2732158 bp |
| Probes | N/A |
| Number of Probes | 1,140,419 |
| Analysis | arr 22q11.21(17,074,487 − 19,806,645) × 1 |
| Result | Consistent with a single copy loss of genomic segment or heterozygous deletion of approximately 2.73 Mb on chromosome 22q11.21. This finding confirms the concurrent FISH analysis, which showed a 22q11.2 deletion associated with DiGeorge syndrome. The deleted 22q11.21 region overlaps with the region associated with the 22q11.2 deletion syndrome (DiGeorge syndrome) and contains several disease genes including the TBX1 gene known to cause clinical phenotypes seen in patients with the syndrome. The deleted region is proximal to the SMARCB1 locus and no deletion of the SMARCB1 gene was detected in this assay. |
2.4. Detailed Cardiac and Extra-Cardiac Phenotyping
| The NBDPS describes each patient’s cardiovascular malformation (CVM) as a specific lesion and groups the lesion in fine, intermediate and coarse levels of detail to increase precision and allow flexibility in analyses [28] |
| The NBDPS describes complexity of CVM and patterns of associated anomalies |
| The CCVM Registry includes latent types of pediatric heart disease that have an established genetic etiology, including cardiomyopathy and aortopathy |
| The CCVM Registry includes vasculopathies and coronary artery abnormalities |
| The CCVM Registry includes subclinical CVMs, such as bicuspid aortic valve |
| The CCVM Registry includes persistent fetal connections when present beyond one year of life, such as patent ductus arteriosus |
| The CCVM Registry includes functional deficits that may identify emerging defects, such as left ventricular systolic dysfunction |
2.5. Data and Information Management
3. Results and Discussion
3.1. Study Population Enrollment
| Site | Current Cases | Yr1 | Yr2 | Yr3 | |
|---|---|---|---|---|---|
| BCM | 305 | 75 | 85 | 85 | |
| CCHMC | 174 | 55 | 60 | 60 | |
| Nationwide | 76 | 55 | 60 | 60 | |
| Emory/CHOA | 67 | 30 | 35 | 40 | |
| Utah | N/A | 15 | 20 | 20 | |
| Totals | 622 | 230 | 260 | 265 | 1377 |
3.2. Genetic Descriptors
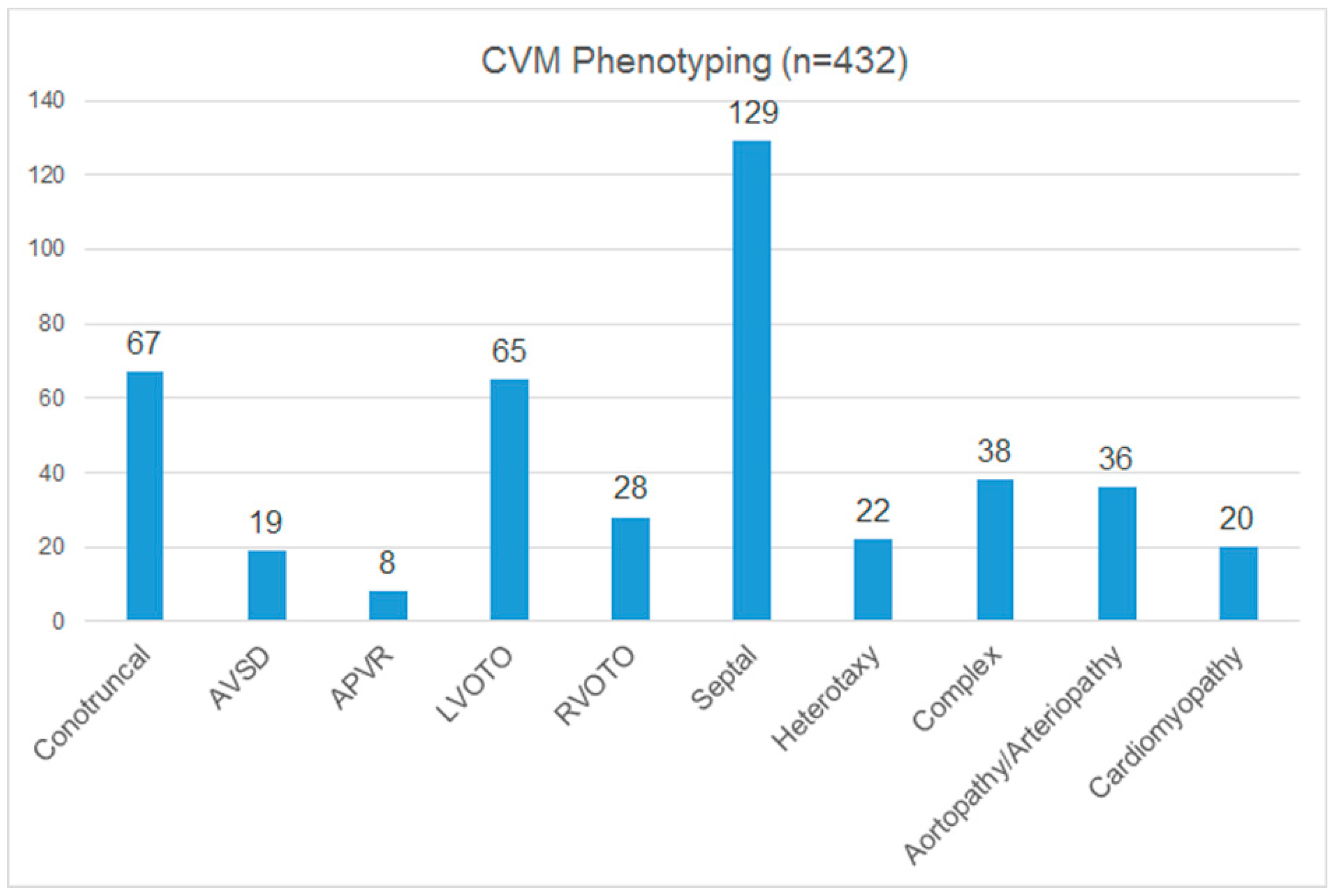
3.3. Cardiac Phenotyping
| Genetic Syndrome | N in Registry 1 | % of Registry Cases |
|---|---|---|
| 22q11.2 deletion syndrome (DiGeorge, Velocardiofacial syndrome) | 19 | 6.5% |
| 22q11.2 duplication syndrome | 3 | 1.0% |
| Williams-Beuren syndrome (7q11.23 deletion) | 12 | 4.1% |
| 7q11.23 duplication syndrome | 3 | 1.0% |
| Trisomy 21 | 1 | 0.3% |
| Total | 38 | 13.0% |

3.4. Proof of Principle: Anticipated Outcomes
3.4.1. Delineation of Cardiac Features for Well Characterized Genomic Disorders
3.4.2. Identification of Novel Loci that Confer CVM Susceptibility
3.4.3. Identification of Novel Genes Causing CVMs
3.5. Approaches for Genetic Identification of CVMs
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gillum, R.F. Epidemiology of congenital heart disease in the united states. Am. Heart J. 1994, 127, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Chaoui, R.; Korner, H.; Bommer, C.; Goldner, B.; Bierlich, A.; Bollmann, R. Prenatal diagnosis of heart defects and associated chromosomal aberrations. Ultraschall Med. 1999, 20, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Tennstedt, C.; Chaoui, R.; Korner, H.; Dietel, M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: Results of a seven year necropsy study. Heart (Br. Card. Soc.) 1999, 82, 34–39. [Google Scholar]
- Ferencz, C.; Boughman, J.A.; Neill, C.A.; Brenner, J.I.; Perry, L.W. Congenital cardiovascular malformations: Questions on inheritance. Baltimore-Washington Infant Study Group. J. Am. Coll. Cardiol. 1989, 14, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, F.; Larsen, L.A.; Zhang, L.; Tumer, Z.; Tommerup, N.; Chen, W.; Jacobsen, J.R.; Schubert, M.; Jurkatis, J.; Tzschach, A.; et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J. Med. Genet. 2008, 45, 704–709. [Google Scholar] [CrossRef]
- Lu, X.Y.; Phung, M.T.; Shaw, C.A.; Pham, K.; Neil, S.E.; Patel, A.; Sahoo, T.; Bacino, C.A.; Stankiewicz, P.; Kang, S.H.; et al. Genomic imbalances in neonates with birth defects: High detection rates by using chromosomal microarray analysis. Pediatrics 2008, 122, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W., Jr.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic basis for congenital heart defects: Current knowledge. A scientific statement from the American Heart Association congenital cardiac defects committee, Council on Cardiovascular Disease in the Young. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [PubMed]
- Øyen, N.; Poulsen, G.; Wohlfahrt, J.; Boyd, H.A.; Jensen, P.K.; Melbye, M. Recurrence of discordant congenital heart defects in families. Circ. Cardiovasc. Genet. 2010, 3, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Øyen, N.; Poulsen, G.; Boyd, H.A.; Wohlfahrt, J.; Jensen, P.K.; Melbye, M. Recurrence of congenital heart defects in families. Circulation 2009, 120, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Martin, L.J.; Rame-Gowda, S.; Tabangin, M.E.; Cripe, L.H.; Benson, D.W. Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 2007, 50, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Martin, L.J.; Rame-Gowda, S.; Tabangin, M.E.; Cripe, L.H.; Benson, D.W. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 2009, 53, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Pignatelli, R.; Lewin, M.; Ho, T.; Fernbach, S.; Menesses, A.; Lam, W.; Leal, S.M.; Kaplan, N.; Schliekelman, P.; et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet A 2005, 134A, 180–186. [Google Scholar] [CrossRef]
- McBride, K.L.; Zender, G.A.; Fitzgerald-Butt, S.M.; Koehler, D.; Menesses-Diaz, A.; Fernbach, S.; Lee, K.; Towbin, J.A.; Leal, S.; Belmont, J.W. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur. J. Hum. Genet. 2009, 17, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Pires, R.; Pires, L.M.; Vaz, S.O.; Maciel, P.; Anjos, R.; Moniz, R.; Branco, C.C.; Cabral, R.; Carreira, I.M.; Mota-Vieira, L. Screening of copy number variants in the 22q11.2 region of congenital heart disease patients from the São Miguel Island, Azores, revealed the second patient with a triplication. BMC Genet 2014, 15, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, T.; Wang, J.C.; Elnaggar, M.M.; Sanchez-Lara, P.; Ross, L.P.; Mahon, L.W.; Hafezi, K.; Deming, A.; Hinman, L.; Bruno, Y.; et al. Concurrent triplication and uniparental isodisomy: Evidence for microhomology-mediated break-induced replication model for genomic rearrangements. Eur. J. Hum. Genet 2015, 23, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, V.M.; Lupski, J.R. Duplications, deletions, and single-nucleotide variations: The complexity of genetic arithmetic. Genet Med. 2013, 15, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Carvalho, C.M.B.; Lupski, J.R. Complex human chromosomal and genomic rearrangements. Trends Genet 2009, 25, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet 2009, 10, 451–481. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Shaw, C.; Wang, X.; Patel, A.; Patterson, L.W.; Kolodziejska, K.; Szafranski, P.; Ou, Z.; Tian, Q.; Kang, S.H.; et al. Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Hum. Mol. Genet 2013, 22, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, L.G.; Bejjani, B.A.; Torchia, B.; Kirkpatrick, S.; Coppinger, J.; Ballif, B.C. The identification of microdeletion syndromes and other chromosome abnormalities: Cytogenetic methods of the past, new technologies for the future. Am. J. Med. Genet C Semin. Med. Genet 2007, 145C, 335–345. [Google Scholar] [CrossRef]
- Lander, J.; Ware, S.M. Copy Number Variation in Congenital Heart Defects. Curr. Genet Med. Rep. 2014, 2, 168–178. [Google Scholar] [CrossRef]
- Cowan, J.; Ware, S.M. Genetics and Genetic Testing in Congenital Heart Disease. Clin. Perinatol. 2015, in press. [Google Scholar]
- Abbott, M.E. Statistics of Congenital Cardiac Disease: 400 Cases Analyzed. J. Med. Res. 1908, 19, 77–81. [Google Scholar] [PubMed]
- Fyler, D.C.; Rudolph, A.M.; Wittenborg, M.H.; Nadas, A.S. Ventricular septal defect in infants and children; a correlation of clinical, physiologic, and autopsy data. Circulation 1958, 18, 833–851. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Young. Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.B. Pathogenetic mechanisms of congenital cardiovascular malformations revisited. Semin. Perinatol. 1996, 20, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Botto, L.D.; Lin, A.E.; Riehle-Colarusso, T.; Malik, S.; Correa, A. National Birth Defects Prevention Study. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gnzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Ferencz, C.; Rubin, J.; Loffredo, C.; Wilson, P. Genetic and environmental risk factors of major cardiovascular malformations: The Baltimore-Washington Infant Study 1981–1989. In Perspectives in Pediatric Cardiology; Futura Publishing: Armonk, NY, USA, 1997; Volume 5, pp. 1–189. [Google Scholar]
- Connor, J.A.; Hinton, R.B.; Miller, E.M.; Sund, K.L.; Ruschman, J.G.; Ware, S.M. Genetic testing practices in infants with congenital heart disease. Congenit. Heart Dis. 2014, 9, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Geddes, G.C.; Butterly, M.; Sajan, I. FISH for 22q11.2 Deletion Not Cost-Effective for Infants with Congenital Heart Disease with Microarray. Pediatr. Cardiol. 2015, 36, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.S.; Liang, L.; Edwards, J.; Brandt, T.; Mei, H.; Sharp, A.J.; Hsu, D.T.; Newburger, J.W.; Ohye, R.G.; Chung, W.K.; et al. Effect of copy number variants on outcomes for infants with single ventricle heart defects. Circ. Cardiovasc. Genet. 2013, 6, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.C.; Pereira, A.C.; Lin, J.C.; DePalma, S.R.; Israel, S.J.; Mesquita, S.M.; Ergul, E.; Conta, J.H.; Korn, J.M.; McCarroll, S.A.; et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009, 41, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Parrott, A.; James, J.; Goldenberg, P.; Hinton, R.B.; Miller, E.; Shikany, A.; Aylsworth, A.S.; Kaiser-Rogers, K.; Ferns, S.J.; Lalani, S.R.; et al. Aortopathy in the 7q11.23 microduplication syndrome. Am. J. Med. Genet A 2015, 167A, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Lalani, S.R.; Ware, S.M.; Wang, X.; Zapata, G.; Tian, Q.; Franco, L.M.; Jiang, Z.; Bucasas, K.; Scott, D.A.; Campeau, P.M.; et al. MCTP2 is a dosage-sensitive gene required for cardiac outflow tract development. Hum. Mol. Genet 2013, 22, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Mislankar, M.; Misra, C.; Huang, N.; Dajusta, D.G.; Harrison, S.M.; McBride, K.L.; Baker, L.A.; Garg, V. Genetic abnormalities in FOXP1 are associated with congenital heart defects. Hum. Mutat 2013, 34, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Ware, S.M.; Shikany, A.; Landis, B.J.; James, J.F.; Hinton, R.B. Twins with progressive thoracic aortic aneurysm, recurrent dissection and ACTA2 mutation. Pediatrics 2014, 134, e1218–e1223. [Google Scholar] [CrossRef] [PubMed]
- Parrott, A.; Ware, S.M. The Role of the Geneticist and Genetic Counselor in an ACHD Clinic. Prog. Pediatr. Cardiol. 2012, 34, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Somers, A.E.; Ware, S.M.; Collins, K.; Jefferies, J.L.; He, H.; Miller, E.M. Provision of cardiovascular genetic counseling services: Current practice and future directions. J. Genet Couns. 2014, 23, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Mavroudis, C.; Jacobs, M.L.; Lacour-Gayet, F.G.; Tchervenkov, C.I.; William Gaynor, J.; Clarke, D.R.; Spray, T.L.; Maruszewski, B.; Stellin, G.; et al. Lessons learned from the data analysis of the second harvest (1998–2001) of the Society of Thoracic Surgeons (STS) Congenital Heart Surgery Database. Eur. J. Cardiothorac. Surg. 2004, 26, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Pediatric Cardiac Genomics Consortium; Gelb, B.; Brueckner, M.; Chung, W.; Goldmuntz, E.; Kaltman, J.; Kaski, J.P.; Kim, R.; Kline, J.; Mercer-Rosa, L.; Porter, G.; et al. The Congenital Heart Disease Genetic Network Study: Rationale, design, and early results. Circ. Res. 2013, 112, 698–706. [Google Scholar]
- McBride, K.L.; Ware, S.M. Modifying Mendel: Approaches for identification of susceptibility alleles for human cardiovascular malformations. Circ. Cardiovasc. Genet 2012, 5, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Arrington, C.B.; Bleyl, S.B.; Matsunami, N.; Bonnell, G.D.; Otterud, B.E.; Nielsen, D.C.; Stevens, J.; Levy, S.; Leppert, M.F.; Bowles, N.E. Exome analysis of a family with pleiotropic congenital heart disease. Circ. Cardiovasc. Genet 2012, 5, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Hershberger, R.E.; Caleshu, C.; Ellinor, P.T.; Garcia, J.G.; Herrington, D.M.; Ho, C.Y.; Johnson, J.A.; Kittner, S.J.; Macrae, C.A.; et al. American Heart Association Advocacy Coordinating Committee. Genetics and cardiovascular disease: A policy statement from the American Heart Association. Circulation 2012, 126, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.E.; Salbert, B.A.; Belmont, J.; Smoot, L. Total is more than the sum of the parts: Phenotyping the heart in cardiovascular genetics clinics. Am. J. Med. Genet A 2004, 131, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Teot, L.A.; Sposto, R.; Khayat, A.; Qualman, S.; Reaman, G.; Parham, D.; Children’s Oncology Group. The problems and promise of central pathology review: Development of a standardized procedure for the Children’s Oncology Group. Pediatr. Dev. Pathol. 2007, 10, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinton, R.B.; McBride, K.L.; Bleyl, S.B.; Bowles, N.E.; Border, W.L.; Garg, V.; Smolarek, T.A.; Lalani, S.R.; Ware, S.M. Rationale for the Cytogenomics of Cardiovascular Malformations Consortium: A Phenotype Intensive Registry Based Approach. J. Cardiovasc. Dev. Dis. 2015, 2, 76-92. https://doi.org/10.3390/jcdd2020076
Hinton RB, McBride KL, Bleyl SB, Bowles NE, Border WL, Garg V, Smolarek TA, Lalani SR, Ware SM. Rationale for the Cytogenomics of Cardiovascular Malformations Consortium: A Phenotype Intensive Registry Based Approach. Journal of Cardiovascular Development and Disease. 2015; 2(2):76-92. https://doi.org/10.3390/jcdd2020076
Chicago/Turabian StyleHinton, Robert B., Kim L. McBride, Steven B. Bleyl, Neil E. Bowles, William L. Border, Vidu Garg, Teresa A. Smolarek, Seema R. Lalani, and Stephanie M. Ware. 2015. "Rationale for the Cytogenomics of Cardiovascular Malformations Consortium: A Phenotype Intensive Registry Based Approach" Journal of Cardiovascular Development and Disease 2, no. 2: 76-92. https://doi.org/10.3390/jcdd2020076