Impact of Water-Based Binder on the Electrochemical Performance of P2-Na0.67Mn0.6Fe0.25Co0.15O2 Electrodes in Na-Ion Batteries


Abstract
:1. Introduction
2. Results
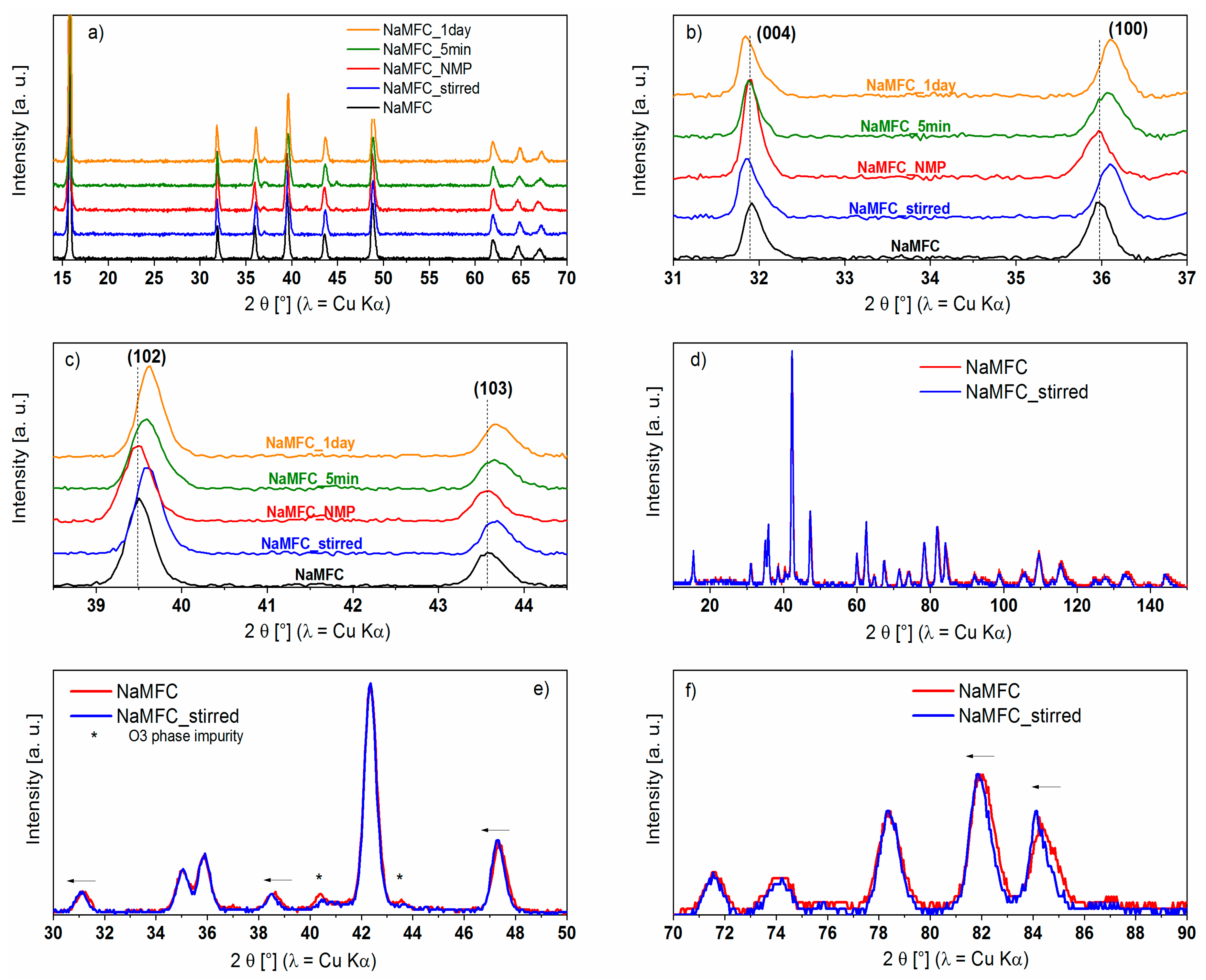
2.1. Influence of Water on NaMFC Powder
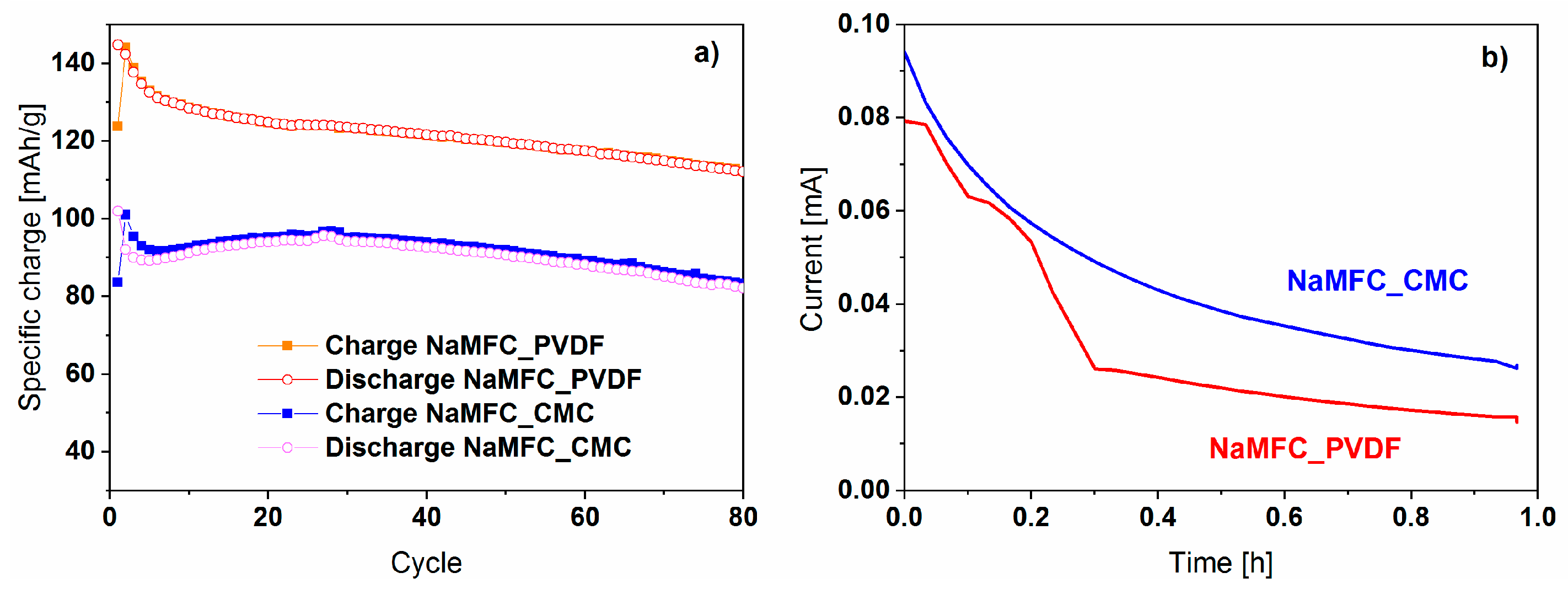
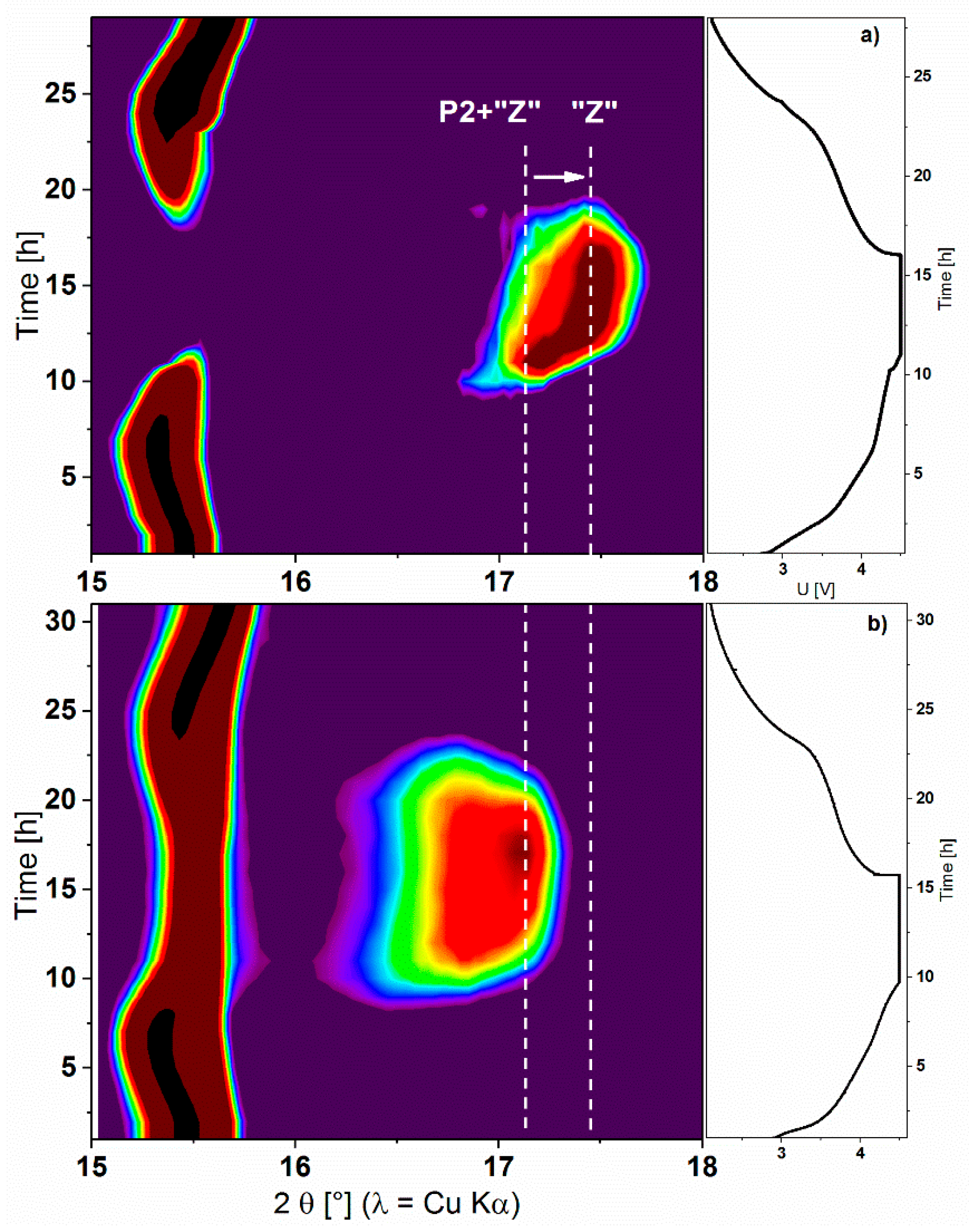
2.2. Effects of the Aqueous Slurry Preparation on the Electrochemical Performance
3. Discussion
4. Materials and Methods
4.1. Synthesis of P2-NaMFC
4.2. Electrode Preparation
4.3. Sample Preparation for Investigating the Effect of Water on NaMFC Powder
4.4. Titration
4.5. X-ray Diffraction (XRD)
4.6. Scanning Electron Microscopy (SEM) Coupled with Energy-Dispersive X-ray Spectroscopy (EDX)
4.7. Neutron Diffraction
4.8. Electrochemistry
4.9. Inductively Coupled Plasma–Optical Emission Spectrometry (ICP–OES)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chong, J.; Xun, S.; Zheng, H.; Song, X.; Liu, G.; Ridgway, P.; Wang, J.Q.; Battaglia, V.S. A comparative study of polyacrylic acid and poly(vinylidene difluoride) binders for spherical natural graphite/LiFePO4 electrodes and cells. J. Power Sources 2011, 196, 7707–7714. [Google Scholar] [CrossRef]
- Jeschull, F.; Brandell, D.; Wohlfahrt-Mehrens, M.; Memm, M. Water-Soluble Binders for Lithium-Ion Battery Graphite Electrodes: Slurry Rheology, Coating Adhesion, and Electrochemical Performance. Energy Technol. 2017, 5, 2108–2118. [Google Scholar] [CrossRef]
- Lestriez, B. Functions of polymers in composite electrodes of lithium ion batteries. C. R. Chim. 2010, 13, 1341–1350. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research development on sodium-ion batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Delmas, C.; Braconnier, J.-J.; Hagenmuller, P.-A. A new variety of LiCoO2 with an unusual oxygen packing obtained by exchange reaction. Mater. Res. Bull. 1982, 17, 117–123. [Google Scholar] [CrossRef]
- Liu, L.; Li, X.; Bo, S.-H.; Wang, Y.; Chen, H.; Twu, N.; Wu, D.; Ceder, G. High-Performance P2-Type Na2/3(Mn1/2Fe1/4)O2 Cathode Material with Superior Rate Capability for Na-Ion Batteries. Adv. Energy Mater. 2015, 5, 1500944. [Google Scholar] [CrossRef]
- Li, X.; Wu, D.; Zhou, Y.-N.; Liu, L.; Yang, X.-Q.; Ceder, G. O3-type Na(Mn0.25Fe0.25Co0.25Ni0.25)O2: A quaternary layered cathode compound for rechargeable Na ion batteries. Electrochem. Commun. 2014, 49, 51–54. [Google Scholar] [CrossRef]
- Buchholz, D.; Chagas, L.G.; Vaalma, C.; Wu, L.; Passerini, S. Water sensitivity of layered P2/P3-NaxNi0.22Co0.11Mn0.66O2 cathode material. J. Mater. Chem. A 2014, 2, 13415–13421. [Google Scholar] [CrossRef]
- Duffort, V.; Talaie, E.; Black, R.; Nazar, L.F. Uptake of CO2 in Layered P2-Na0.67Mn0.5Fe0.5O2: Insertion of Carbonate Anions. Chem. Mater. 2015, 27, 2515–2524. [Google Scholar] [CrossRef]
- Paulsen, J.M.; Dahn, J.R. Study of the layered manganese bronzes. Solid State Ion. 1999, 126, 3–24. [Google Scholar] [CrossRef]
- Sathiya, M.; Hemalatha, K.; Ramesha, K.; Tarascon, J.M.; Prakash, A.S. Synthesis, Structure, and Electrochemical Properties of the Layered Sodium Insertion Cathode Material: NaNi1/3Mn1/3Co1/3O2. Chem. Mater. 2012, 24, 1846–1853. [Google Scholar] [CrossRef]
- Shkrob, I.A.; Gilbert, J.A.; Phillips, P.J.; Klie, R.; Haasch, R.T.; Bareno, J.; Abraham, D.P. Chemical Weathering of Layered Ni-Rich Oxide Electrode Materials: Evidence for Cation Exchange. J. Electrochem. Soc. 2017, 164, A1489–A1498. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Dahn, J.R. Intercalation of Water in P2, T2 and O2 Structure Az[CoxNi1/3−xMn2/3]O2. Chem. Mater. 2001, 13, 1252–1257. [Google Scholar] [CrossRef]
- Dall’Asta, V.; Buchholz, D.; Chagas, L.G.; Dou, X.; Ferrara, C.; Quartarone, E.; Tealdi, C.; Passerini, S. Aqueous Processing of Na0.44MnO2 Cathode Material for the Development of Greener Na-Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 34891–34899. [Google Scholar] [CrossRef] [PubMed]
- Marino, C.; Marelli, E.; Villevieille, C. Impact of cobalt content in Na0.67MnxFeyCozO2 (x + y + z = 1), a cathode material for sodium ion batteries. RSC Adv. 2017, 7, 13851–13857. [Google Scholar] [CrossRef]
- Kadarmandalgi, S.G. Spot Test for Detection of Manganese. J. Chem. Educ. 1964, 41, 437–438. [Google Scholar] [CrossRef]
- Blesa, M.C.; Moran, E.; Menendez, N.; Tornero, J.D.; Torron, C. Hydrolysis of Sodium Orthoferrite. Mater. Res. Bull. 1993, 28, 837–847. [Google Scholar] [CrossRef]
- Polfus, J.M.; Yildiz, B.; Tuller, H.L.; Bredesen, R. Adsorption of CO2 and Facile Carbonate Formation on BaZrO3 Surfaces. J. Phys. Chem. C 2018, 122, 307–314. [Google Scholar] [CrossRef]
- Nam, K.W.; Kim, S.; Yang, E.; Jung, Y.; Levi, E.; Aurbach, D.; Choi, J.W. Critical Role of Crystal Water for a Layered Cathode Material in Sodium Ion Batteries. Chem. Mater. 2015, 27, 3721–3725. [Google Scholar] [CrossRef]
- Mortemard de Boisse, B.; Carlier, D.; Guignard, M.; Bourgeois, L.; Delmas, C. P2-NaxMn1/2Fe1/2O2 phase used as positive electrode in Na batteries: Structural changes induced by the electrochemical (de)intercalation process. Inorg. Chem. 2014, 53, 11197–11205. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, N.; Kajiyama, M.; Iwatate, J.; Nishikawa, H.; Hitomi, S.; Okuyama, R.; Usui, R.; Yamada, Y.; Komaba, S. P2-type Nax[Mn1/2Fe1/2]O2 made from earth-abundant elements for rechargeable Na batteries. Nat. Mater. 2012, 11, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Talaie, E.; Duffort, V.; Smith, H.L.; Fultz, B.; Nazar, L.F. Structure of the high voltage phase of layered P2-Na1/3−z[Mn1/2Fe1/2]O2 and the positive effect of Ni substitution on its stability. Energy Environ. Sci. 2015, 8, 2512–2523. [Google Scholar] [CrossRef]
- Kjeldgaard, S.; Birgisson, S.; Kielland, A.G.; Iversen, B.B. Operando powder X-ray diffraction study of P2-NaxNi0.3Mn0.7O2 cathode material during electrochemical cycling. J. Appl. Crystallogr. 2018, 51, 1304–1310. [Google Scholar] [CrossRef]
- Sharma, N.; Tapia-Ruiz, N.; Singh, G.; Armstrong, A.R.; Pramudita, J.C.; Brand, H.E.A.; Billaud, J.; Bruce, P.G.; Rojo, T. Rate Dependent Performance Related to Crystal Structure Evolution of Na0.67Mn0.8Mg0.2O2 in a Sodium-Ion Battery. Chem. Mater. 2015, 27, 6976–6986. [Google Scholar] [CrossRef]
- Bleith, P.; Kaiser, H.; Novák, P.; Villevieille, C. In situ X-ray diffraction characterisation of Fe0.5TiOPO4 and Cu0.5TiOPO4 as electrode material for sodium-ion batteries. Electrochim. Acta 2015, 176, 18–21. [Google Scholar] [CrossRef]
- Rodriguez-Carjaval, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B 1993, 192, 55–69. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Na/Co | Mn/Co | Fe/Co |
|---|---|---|---|
| NaMFC | 4.9 | 4.5 | 2.4 |
| NaMFC_stirred | 3.5 | 4.8 | 2.2 |
| Sample | NaMFC_5 min | NaMFC_1 Day | NaMFC_Stirred |
|---|---|---|---|
| Desodiation level [%] | 11 | 16 | 14 |
| Na content in NaMFC | 0.5963 | 0.5628 | 0.5762 |
| Sample | a (Å) | c (Å) | V (Å3) |
|---|---|---|---|
| NaMFC | 2.88200 (6) | 11.2095 (5) | 80.631 (4) |
| NaMFC_stirred | 2.88155 (6) | 11.2309 (4) | 80.760 (4) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marino, C.; Marelli, E.; Park, S.; Villevieille, C. Impact of Water-Based Binder on the Electrochemical Performance of P2-Na0.67Mn0.6Fe0.25Co0.15O2 Electrodes in Na-Ion Batteries. Batteries 2018, 4, 66. https://doi.org/10.3390/batteries4040066
Marino C, Marelli E, Park S, Villevieille C. Impact of Water-Based Binder on the Electrochemical Performance of P2-Na0.67Mn0.6Fe0.25Co0.15O2 Electrodes in Na-Ion Batteries. Batteries. 2018; 4(4):66. https://doi.org/10.3390/batteries4040066
Chicago/Turabian StyleMarino, Cyril, Elena Marelli, Sunkyu Park, and Claire Villevieille. 2018. "Impact of Water-Based Binder on the Electrochemical Performance of P2-Na0.67Mn0.6Fe0.25Co0.15O2 Electrodes in Na-Ion Batteries" Batteries 4, no. 4: 66. https://doi.org/10.3390/batteries4040066