

Mixer Design and Flow Rate as Critical Variables in Flow Chemistry Affecting the Outcome of a Chemical Reaction: A Review
N.D. Zelinsky Institute of Organic Chemistry of the Russian Academy of Sciences, Leninsky Prosp., 47, Moscow 119991, Russia
*
Author to whom correspondence should be addressed.
Inventions 2023, 8(5), 128; https://doi.org/10.3390/inventions8050128
Submission received: 31 August 2023
/
Revised: 6 October 2023
/
Accepted: 10 October 2023
/
Published: 16 October 2023
(This article belongs to the Special Issue Innovative Research and Applications in Hydrodynamics and Flow Control)
Abstract
:Flow chemistry offers several advantages for performing chemical reactions and has become an important area of research. It may seem that sufficient knowledge has already been acquired on this topic to understand how to choose the design of microreactor/micromixer and flow rate in order to achieve the desired outcome of a reaction. However, some experimental data are difficult to explain based on commonly accepted concepts of chemical reactivity and performance of microfluidic systems. In this mini review, we attempt to identify such data and offer a rational explanation of unusual results based on the supramer approach. We demonstrate that variation in flow regime (determined by mixer design and flow rate) can either improve or worsen the reactivity and lead to completely different products, including stereoisomers. It is not necessary to mix the reagents with maximum efficiency. The real challenge is to mix reagents the right way since at a too high or too low flow rate (in the particular mixer), the molecules of reagents are incorrectly presented on the surface of supramers, leading to altered stereoselectivity, or form tight supramers, in which most of the molecules are located inside the supramer core and are inaccessible for attack, leading to low yields.
1. Introduction
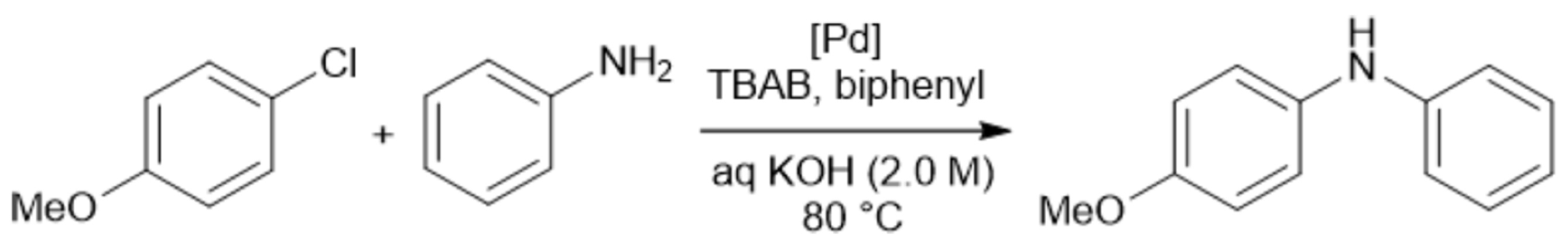
Over the past decades, chemistry in flow became a powerful tool of modern organic and inorganic chemistry, and many publications devoted to the study of chemical reactions in flow appeared. Dozens of reviews have been published on flow chemistry. Here, we mentioned only some of them [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20], in which a reader can find further references on specific topics. The most comprehensive coverage and critical analysis of the recent flow chemistry accounts can be found in an outstanding review [11], in which an in-depth analysis of almost every aspect of performing chemical experiments in flow accompanied by invaluable advice on how one can control the reaction parameters to achieve the desired result is presented.
Flow chemistry (aka microfluidics [2]) offers several advantages for performing chemical reactions such as minimizing of accident risks and cheaper production of chemicals to name a few [7,11,13,17,18,19]:
- Flow chemistry facilitates the optimization of reactions at a much faster rate, resulting in a reduction in costs and time. Reaction time, temperature, ratio of reagents, concentration, and reagents themselves can all be rapidly modified. One reaction can be followed by another, separated by solvent, increasing throughput. Automated flow chemistry allows for rapid variation of reaction conditions on a very small scale (≤100 μL).
- Performing reactions in flow can be safer than in batch reactors, since only a small portion of reactive or hazardous material is transformed to product at any particular moment.
- The use of flow chemistry has the advantage of enhanced heat and mass transfer, which can result in faster reaction rates and higher yields.
- Flow reactors provide excellent reaction selectivity and allow for preparation of products that are not accessible under batch conditions.
Successful attempts to generalize the basic principles and fundamentals of flow chemistry [11] and to standardize the description of reactions in flow [14] have been advanced. Ten key issues in modern flow chemistry have been identified and discussed in detail [6]. Various designs of microreactors [3] and their classification [13] have been considered (for a more detailed discussion, vide infra). A state-of-the-art, future perspectives and challenges in creating reconfigurable microfluidic platforms, which would allow a user to easily tailor a microfluidic system for a specific application and “to tune a microscale experiment with the capacity to make real-time decisions”, has been reviewed [16].
There are many examples of the successful use of flow reactors for conducting reactions of various classes [1,11,13,15,18], including those useful for production of active pharmaceutical ingredients [17]. Functional group interconversion reactions have been successfully performed in continuous flow reactors [15]. Flow technology was found to be unexpectedly efficient in supramolecular chemistry [12,20]. A new paradigm for synthesis of natural products including carbohydrates has been proposed [4,5,9,10], which is based on performing traditional organic reactions under microfluidic conditions.
Among many parameters [11] the design of the microreactor/micromixer [11,21,22,23,24,25] and flow rate used are considered as the key factors that determine the outcome of a reaction performed in flow. One may think that sufficient knowledge has already been acquired [11] on this topic to understand how to select appropriate experimental conditions to achieve the desired reaction result.
However, only few researchers publish the “negative” results that could demonstrate their “inability” to forecast the result of a particular experiment; many researchers tend to modify the initial objectives of research after obtaining the positive results (“much of good science is opportunistic and revisionist”, as G. Whitesides put it [26]). For this understandable reason, reports of detailed studies of the influence of the design of the microreactor/micromixer and/or flow rate on the outcome of the real-life reactions (as opposed to the studies of “test reactions” in flow, vide infra), performed by practicing organic chemists (rather than by experts in chemical engineering), are rare. Conditions for successful practically important chemical transformations in flow are mostly reported.
This situation creates a “research gap” between reports on successes or failures of reactions, performed under specific conditions in flow, and understanding the reasons lying behind. Moreover, some experimental data are difficult to explain based on commonly accepted concepts of chemical reactivity (in general) and performance of microfluidic systems (in particular). In this mini review, we attempt to identify such data and offer a rational explanation of unusual results based on the supramer approach (vide infra) [27].
2. Mixing Performance
Typical microfluidic systems are featured by low Reynolds numbers (Re, [28]) (Re ≤ 1000 [1,8,29]) that would generally suggest a laminar flow, even in reactors in which biphasic reactions proceed [30], and guarantee that there is no turbulence and therefore no back-mixing in the reactor [8]. This notion may create the wrong impression that mass transfer of reactants in microfluidic systems is controlled by diffusion only; in fact, convection/advection can also contribute significantly to the mass transfer in these systems (vide infra).
It is clear, even from general considerations, that the reagents need to be mixed well for the effective reaction to occur [29,31,32,33,34,35,36,37]. This review is limited to discussion of passive [29,35,36] mixing that does not rely on external energy input. This mixing is realized within mixing devices (micromixers), which often serve also as microreactors (for this reason, hereinafter we denote them as “microreactors/micromixers”).
Many studies [21,29,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92] were dedicated to modeling various types of micromixers/microreactors and determining their mixing efficiency (mixing quality) theoretically and/or experimentally using a set of test reactions [39,57,63]. This widely used “chemical” approach for accessing the mixing efficiency, unlike purely physical approaches like flow imaging, is based on the implicit supposition that the dependence of outcome of a chemical reaction, performed in flow, on mixing conditions is determined by mixing efficiency of the micromixer/microreactor used. We emphasize that most experimental studies of mixing efficiency of different micromixers/microreactors are in fact providing information on chemical reactivity in the reactions performed under flow conditions using these micromixers/microreactors. This means that the observed dependence of conversion of the starting compound and product yield on mixing conditions may reflect not only the mixing issues (as usually believed) but also the influence of other (unknown) factors that are affected by changing the mixing mode. This opens the way for alternative views on chemical reactivity as exemplified by application of the supramer approach [27] to the flow chemistry (vide infra).
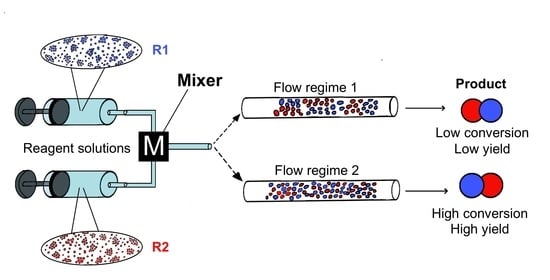
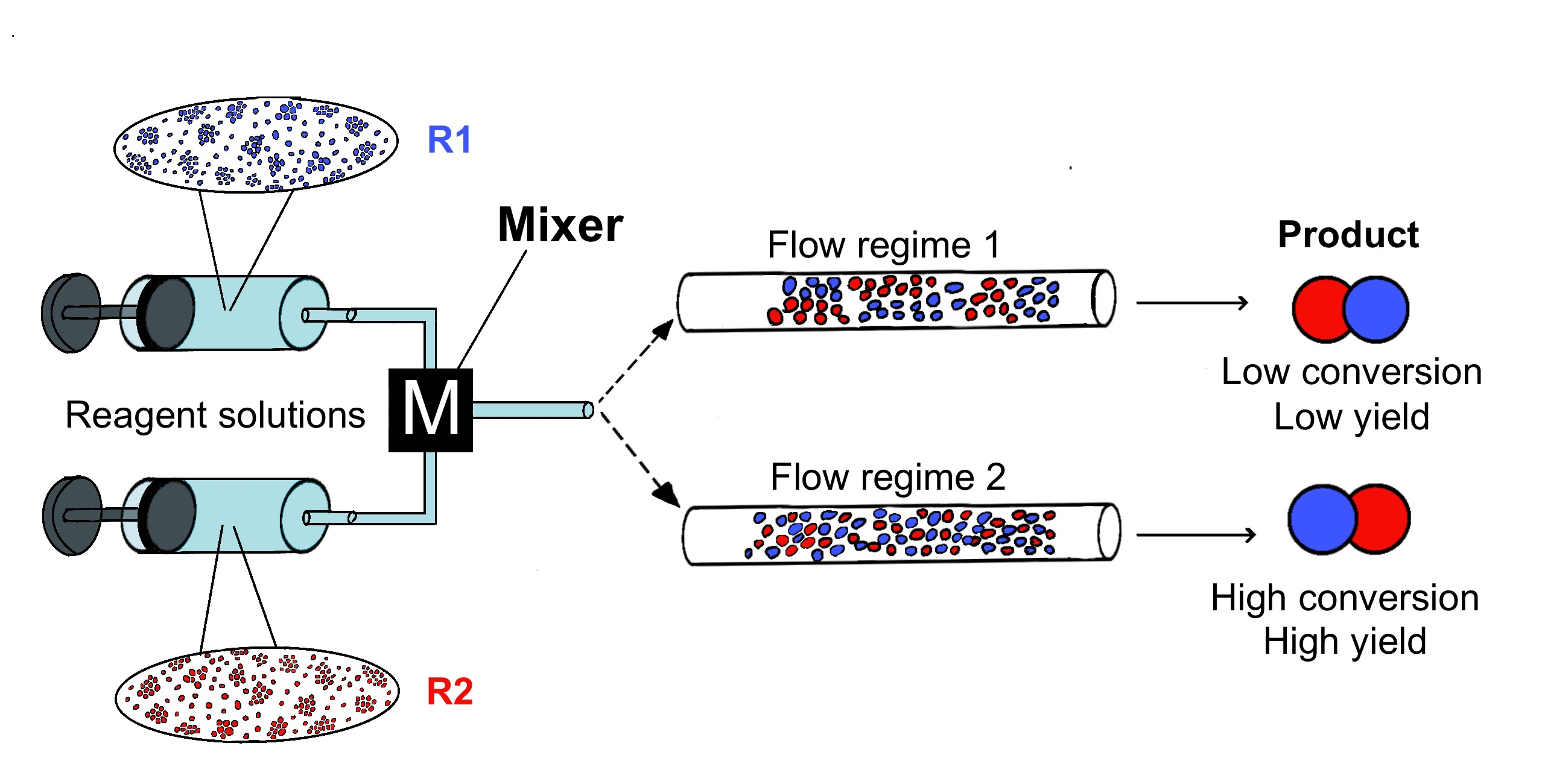
The mixing performance of different types of mixers has been previously compared [21,35,36,43,53,57,69,70,72,79,93]. The main message for the practicing synthetic chemist from these studies is that the mixing quality is controlled by the flow regime in the mixing device. Importantly, the flow regimes may differ considerably for different mixers; for the same mixer flow regimes may also vary depending on the flow rate (an example is shown in Figure 1).
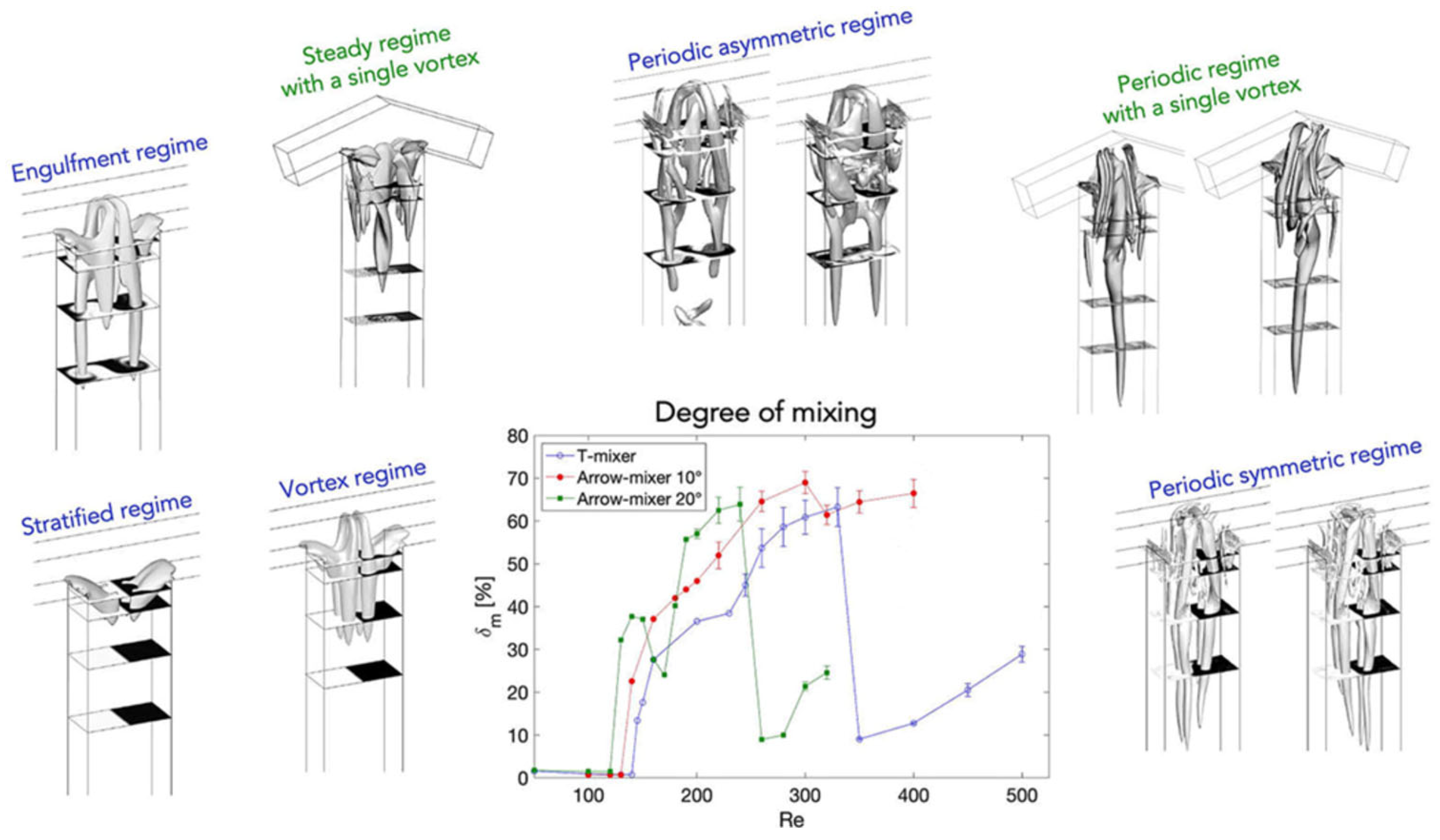
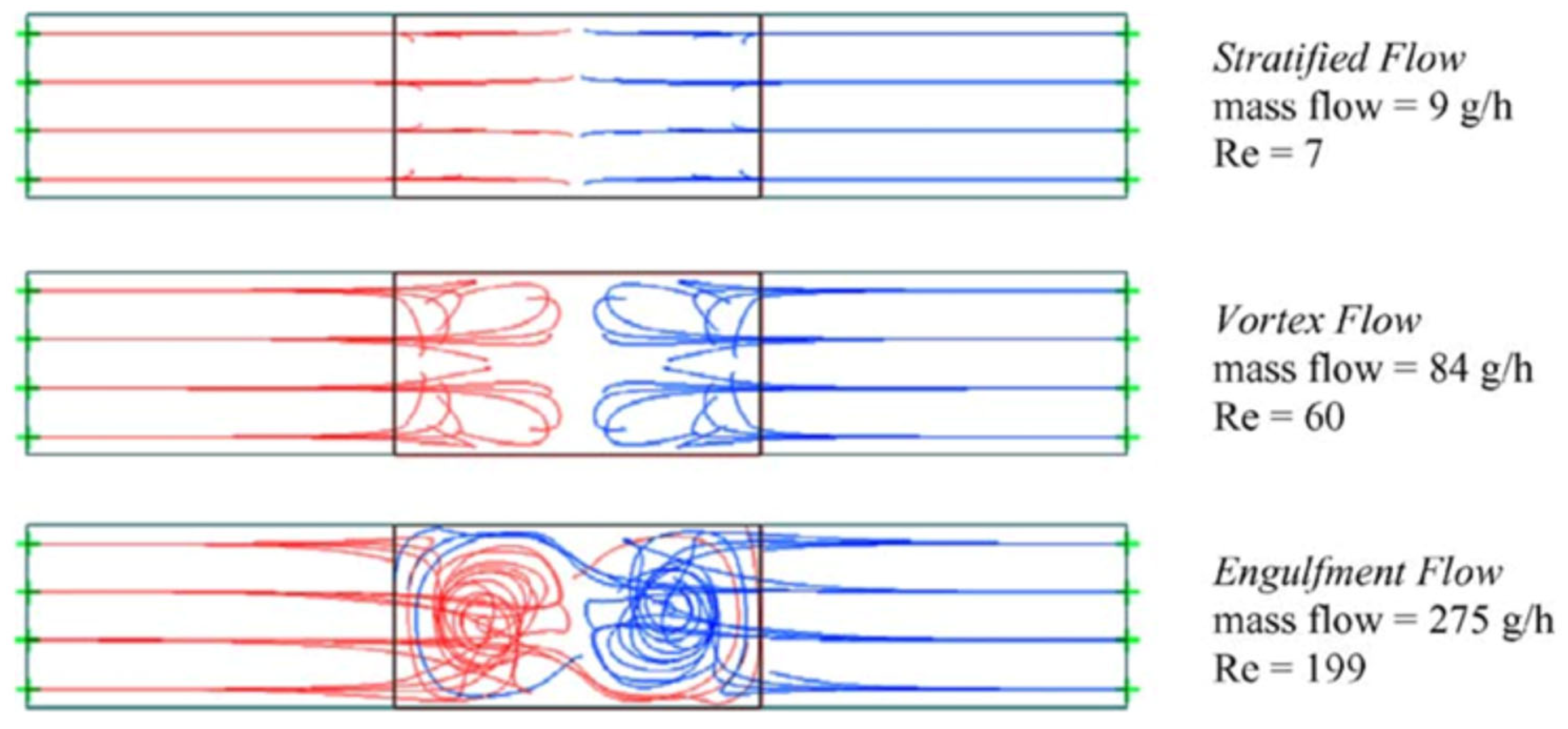
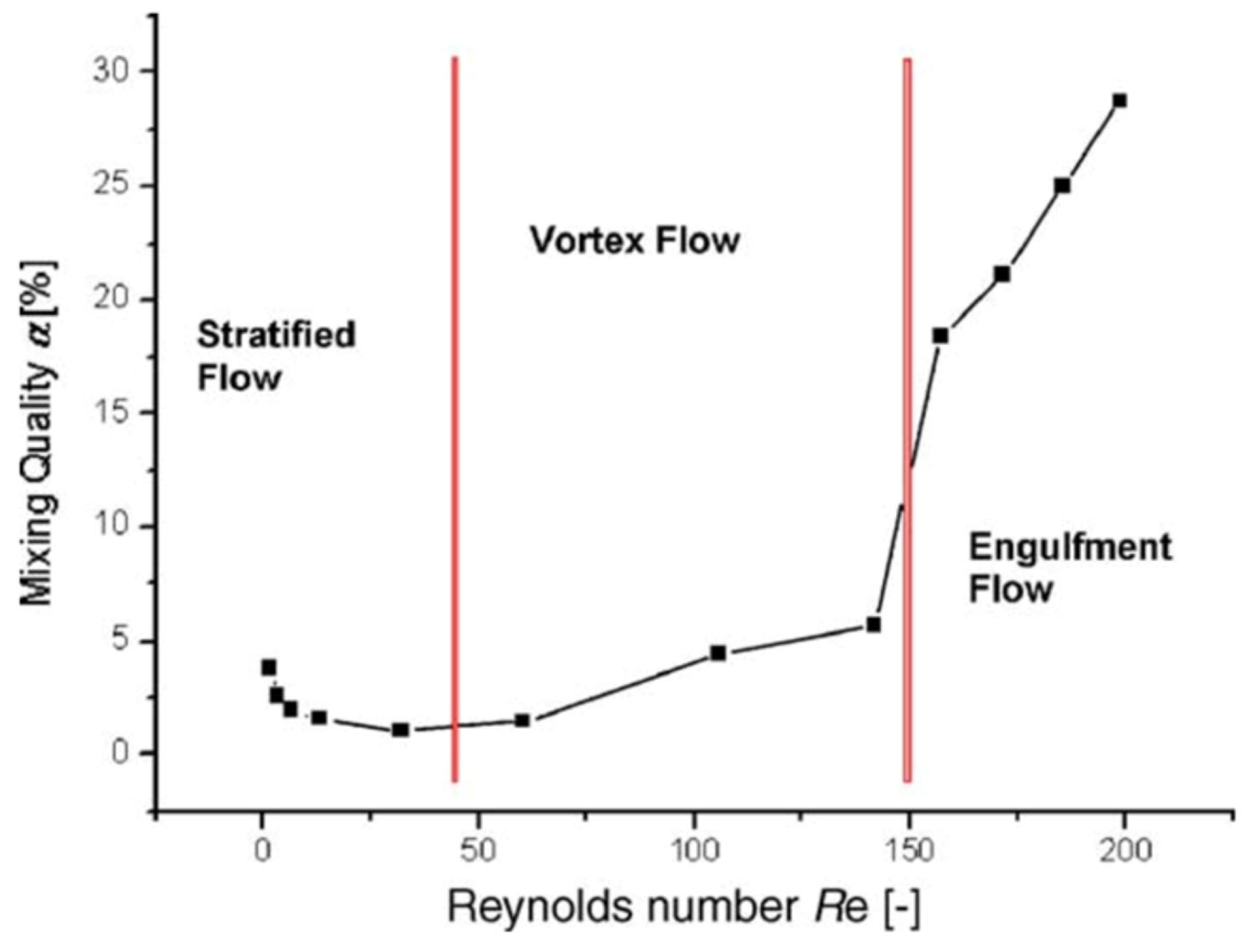
In most studies mainly the Reynolds number was used to describe the flow regime (but occasionally other numbers were used [94]). However, the Reynolds number alone is not necessarily always trustworthy for estimating mixing quality in micromixers/microreactors. It was demonstrated that mixing quality of a simple and widely used T-shaped micromixer sharply increases in the “engulfment flow regime”, featured by the increased vorticity (Figure 2) hence mixing quality, which occurs at the high flow rates even in the low Reynolds number range (Re ~ 200) (Figure 3) [43]. It was concluded that the Reynolds number is not suitable for identification of the flow regime, since it varies a lot for different mixer geometries under similar flow regimes [43].
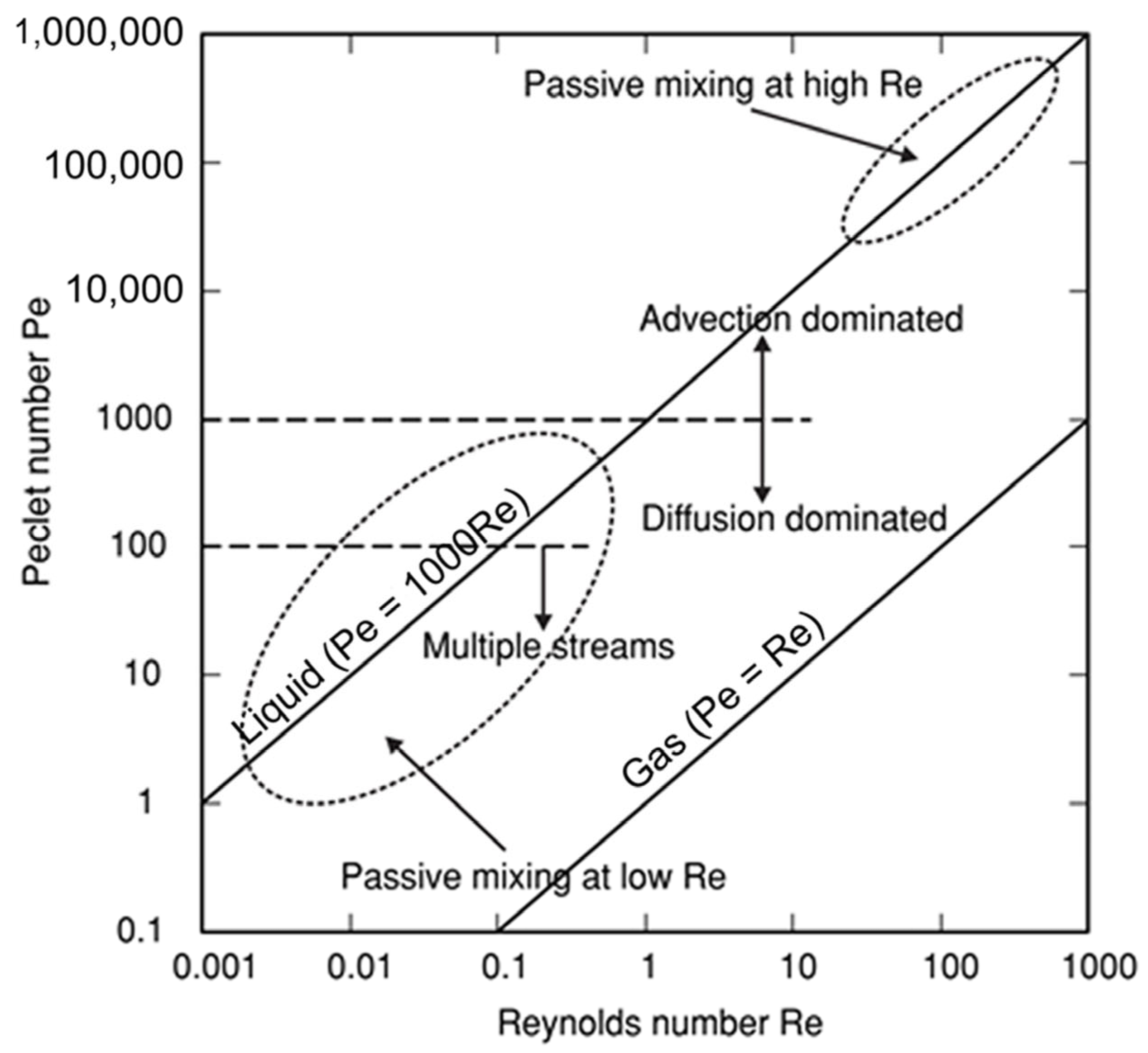
Using examples of micromixers/microreactors of varying complexity (from the most studied simple T-shaped micromixers [35,43,44,46,48,49,53,58,60,69,70,72,74,76,79,80,81,82,83,84,85,88,91] to more sophisticated “split-and-recombine” microreactors [35,36,37,47,51,70,91,95,96]), it was shown that regardless of the value of the Reynolds number, mixing can be effective at least in some flow regimes. Especially effective is the so called “chaotic mixing/advection” [3,29,31,42,96]. Operation conditions of passive mixers based on chaotic advection can be distributed around the characteristic lines in the Pe–Re diagram for a wide range of Reynolds numbers (see Figure 4). Importantly, the passive micromixers work well either at low Reynolds numbers and low Peclet numbers (bottom left corner in Figure 4) or at high Reynolds numbers in the transition regime to turbulence (top right corner in Figure 4). Noteworthy, chaotic micromixers can be designed for use in a wide range of Reynolds numbers. Importantly, mixing with chaotic advection does not depend on the Péclet number [29].
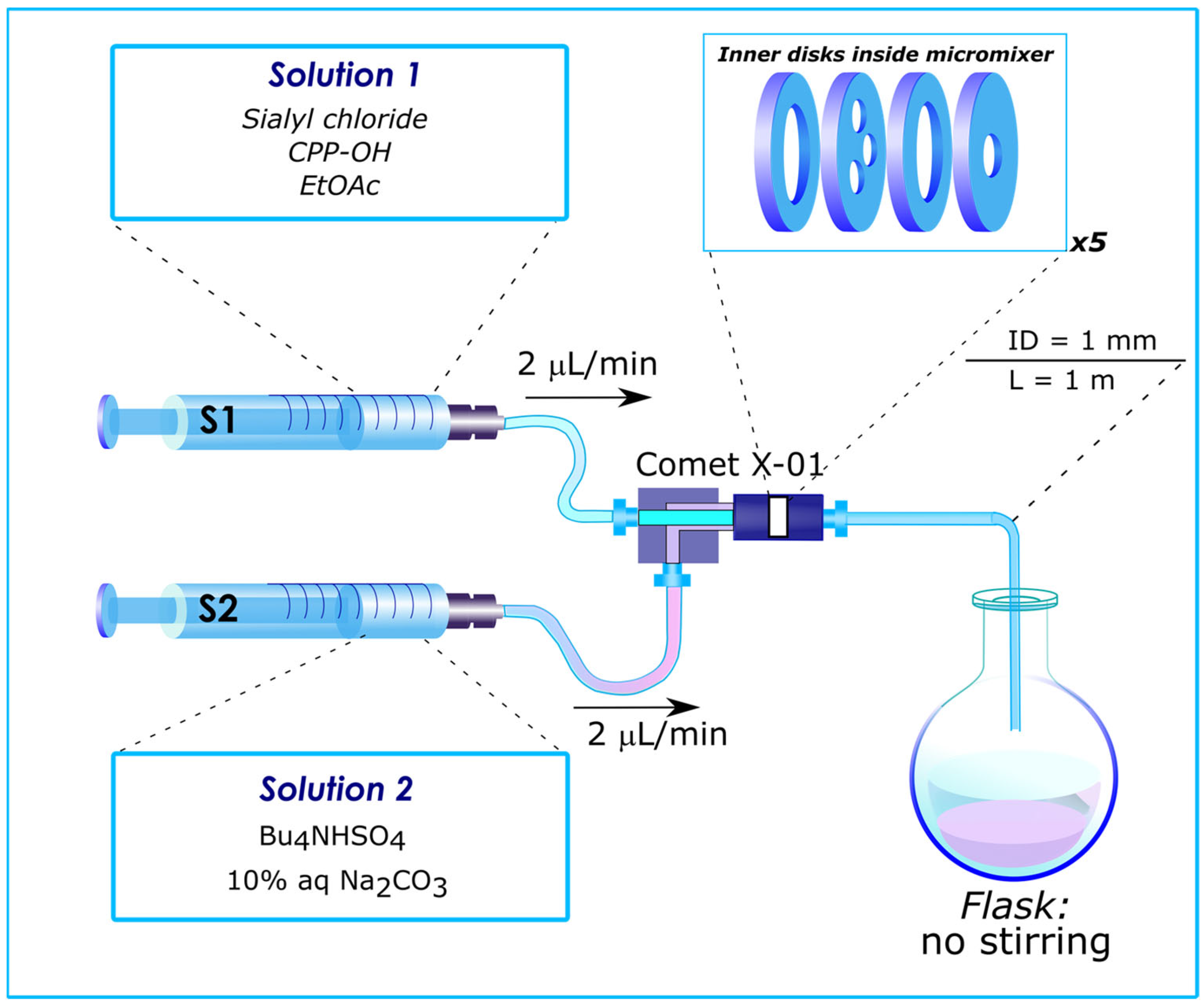
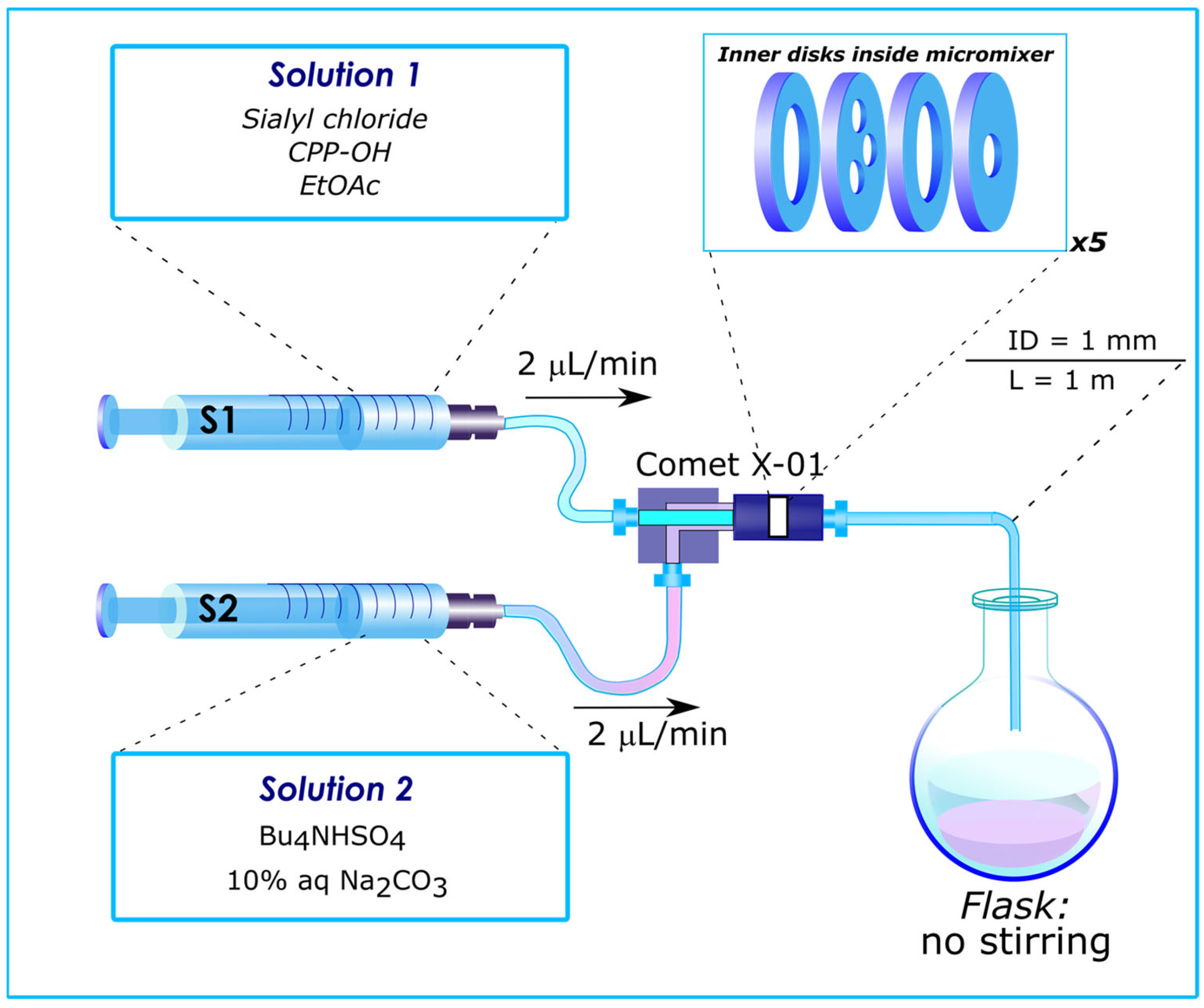
Among many different micromixers designed for microflow systems, a Comet X-01 micromixer should be highlighted. This mixer has previously been used to perform and optimize various reactions [4,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114]. Inside this mixer, the flow is split into three and merged back into one (repeated five times) (see Figure 5 and supporting information file for Ref. [113]) remotely resembling that in the previously mentioned “split-and-recombine” microreactor [35,36,37,47,51,70,91,95,96]) featured by “chaotic mixing” [3,29,31,42,96], which ensures efficient mixing regardless of the Reynolds number [3]. The unique design of the Comet X-01 micromixer made possible the discovery of unprecedented results described later in this review (vide infra).
Concluding this section, we must stress that current knowledge suggests the importance of mixing issues in chemical reactions to be related to the relative values of the mixing times as compared to the reaction times. For a detailed discussion and recommendations for choosing the design of the commercially available micromixers suitable for specific applications see [57].
The effects of mixing on the outcome of the sufficiently slow chemical reaction performed in flow can usually be neglected. If the reaction time is significantly longer than the mixing time, it can be considered that the reaction takes place under homogeneous environment (“homogeneous concentration field”). In such a situation, enhancing the mixing performance cannot increase the conversion rate [57].
At the same time, fast and ultrafast reactions (as compared to mixing of reagents) should be extremely sensitive to changes in mixing efficiency which is determined by the flow regime realized in the mixer of specific design at the indicated flow rate. In this situation “the reaction takes place in a heterogeneous concentration field with multilamellae structures which are determined by the mixing process. The length scales of these structures as well as the time scale of their generation can have a strong influence on the conversion rate. In the case of a reaction system with competitive parallel or consecutive reactions, mixing time can also affect the selectivity of this system, especially if parallel reactions proceed at a different time scale” [57].
For this reason, many studies report the influence of both parameters (mixer design and the flow rate) on the reaction outcome, and they will be discussed together in the next section. Note that until recently, most of the reports on flow chemistry dealt with conversion of the starting material and product(s) yield as measures of the reaction progress. Issues of stereoselective formation of stereoisomers were mainly neglected; the rare examples will be mentioned below.
3. Influence of Mixer Design and Flow Rate on Reaction Outcome
The flow rate plays an important role in flow reactions. There is a lot of evidence that flow rate can influence the reaction yield. There are cases when the yield increases both with a decrease [110,115] and with an increase [116,117,118,119] in the flow rate.
A change of flow rate with constant path length after mixing expectedly results in the change of reagents residence time (vide infra). A decrease in flow rate usually leads to an increase in product yield due to increase in residence time. However, in some cases due to increased residence time, the number of side processes leading to formation of undesirable products may also increase leading to a decrease in the yield of the target product. There are situations in which an increase in the flow rate (hence, the Reynolds number) leads to an increase in product yield [113]. However, increasing the flow rate is not a panacea: if the flow rate is too high, then the reactants simply will not have enough time to react inside the reactor. It is generally accepted that for the optimal course of the reaction, it is necessary to find a golden mean between these extremes.
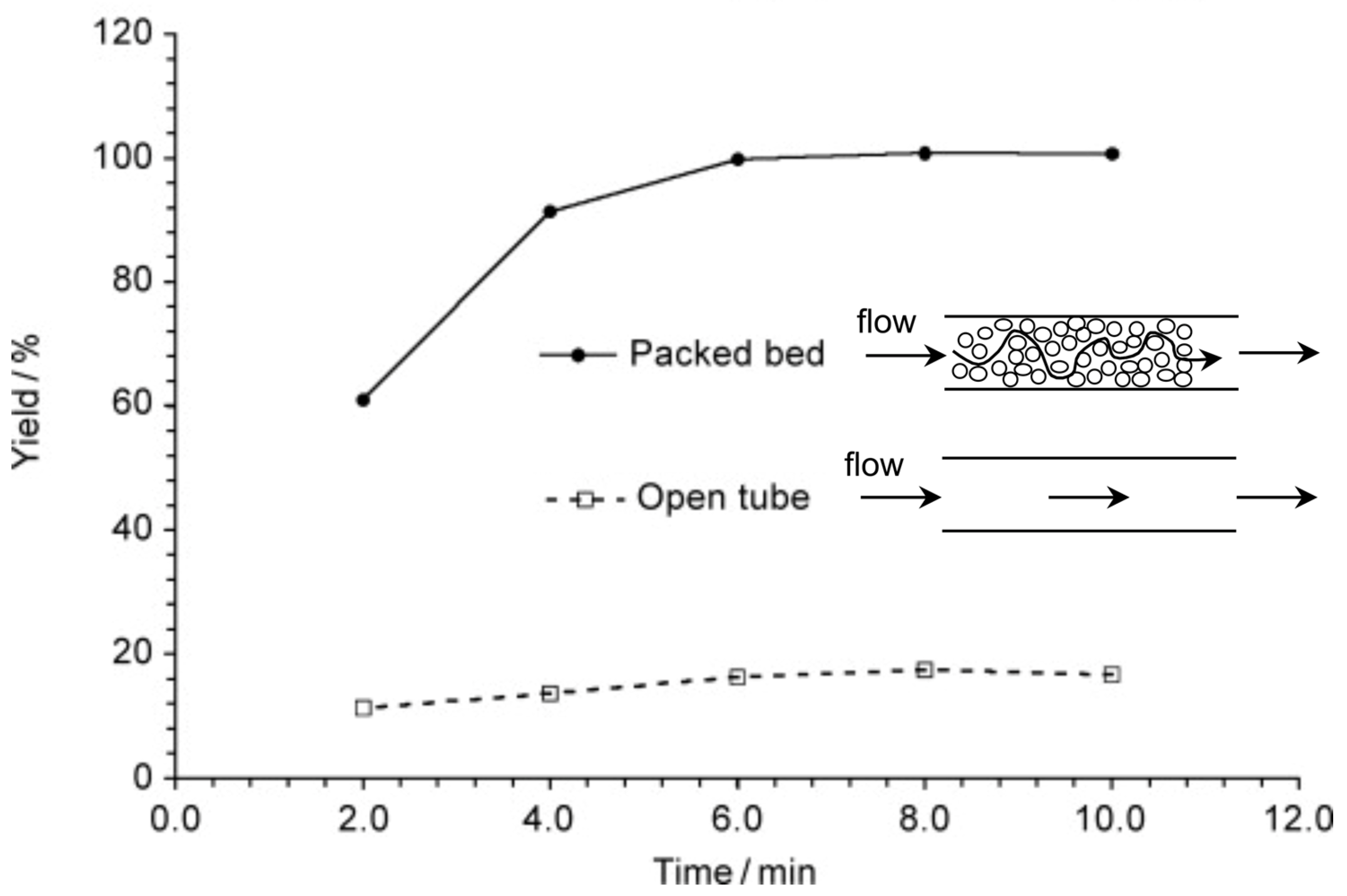
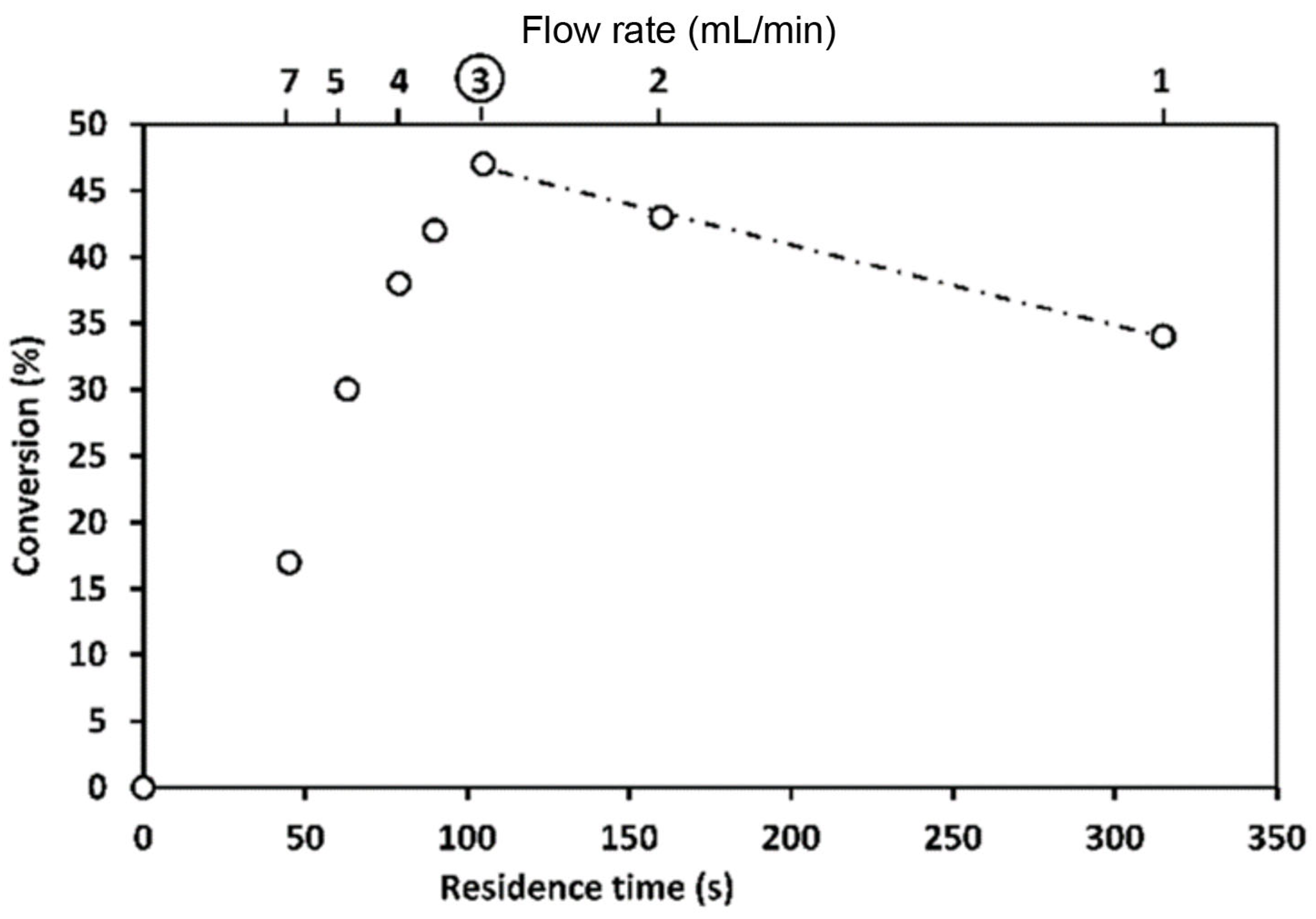
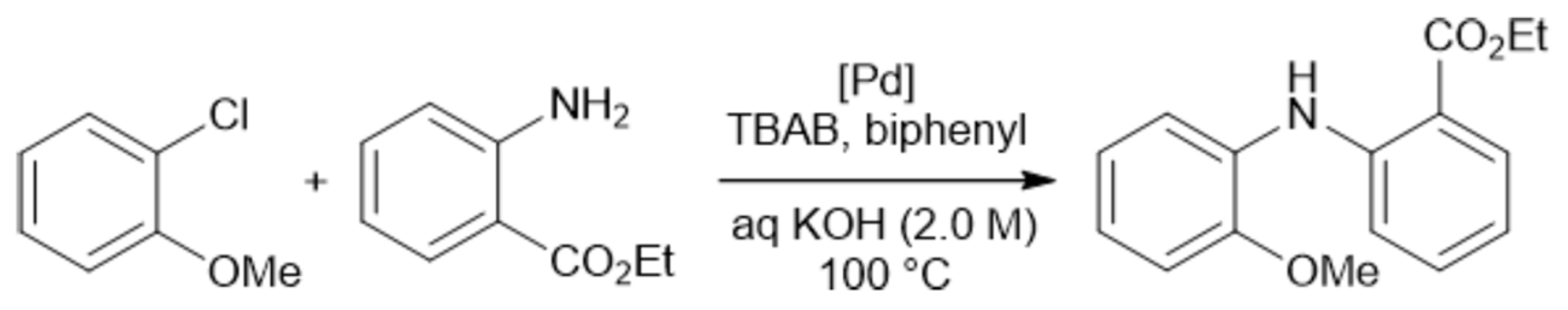
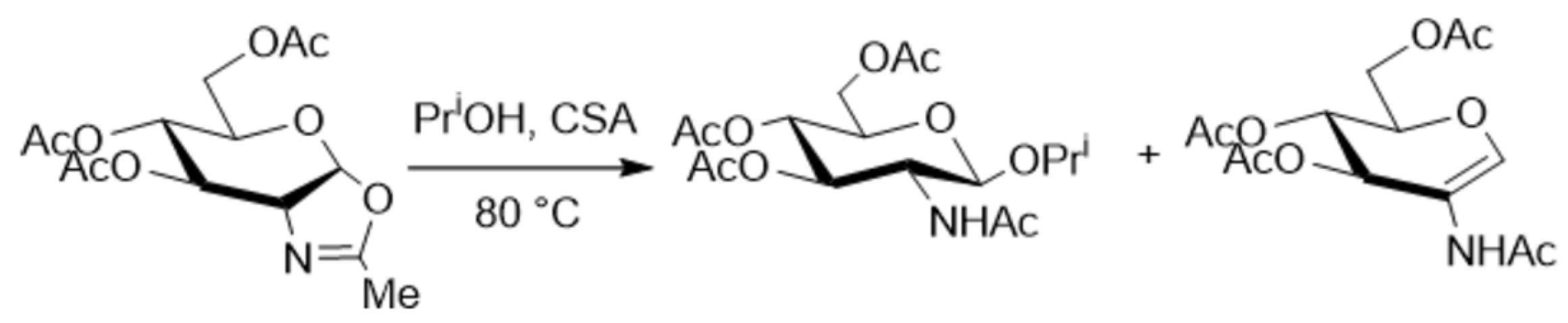
The crucial role of mixing efficiency, determined by the mixer design (vide supra), for chemical reactions performed under flow conditions was demonstrated for C–N cross-coupling reactions (Scheme 1 and Scheme 2) [120]. In one case, the capillary reactor contained grains around which the reaction solutions flow. In this case, in the thin films formed between the grains, mixing occurs efficiently, and the reaction is complete within 6 min. In another case, there was a free capillary in which conditions for effective mixing were not created. As a result, the yield after the same time was relatively small (Figure 6).
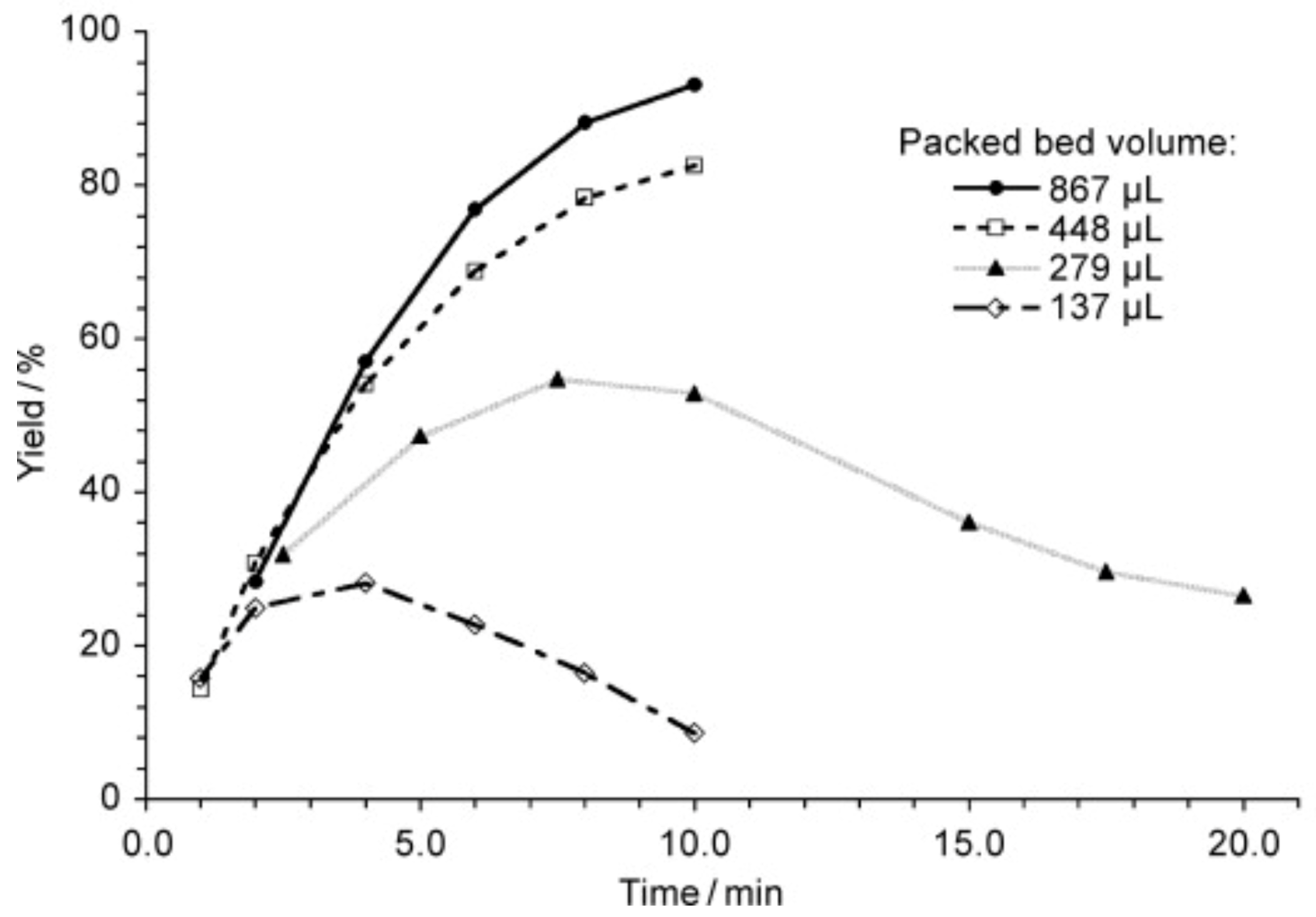
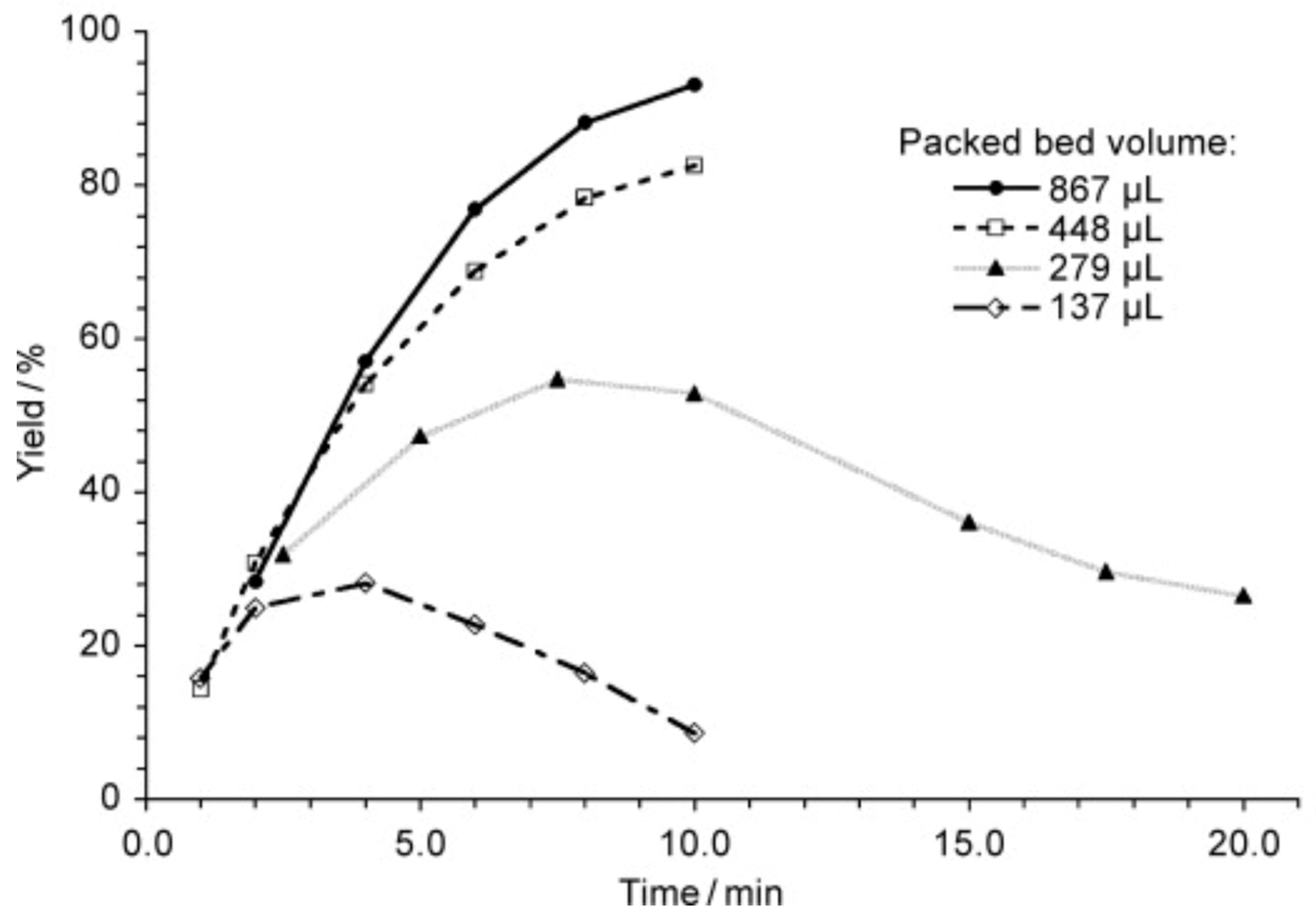
More than that, a study of a similar reaction [120] showed that the yield depends on packed bed volume (Scheme 2, Figure 7). When the size of the packed bed was decreased, the yield of the reaction was decreased too. Note the maxima on the plots of the yield against the residence time. Since it was demonstrated that the product was not degrading over time the authors attributed the decrease in yield to changes in mixing performance at low flow rates (larger residence time).
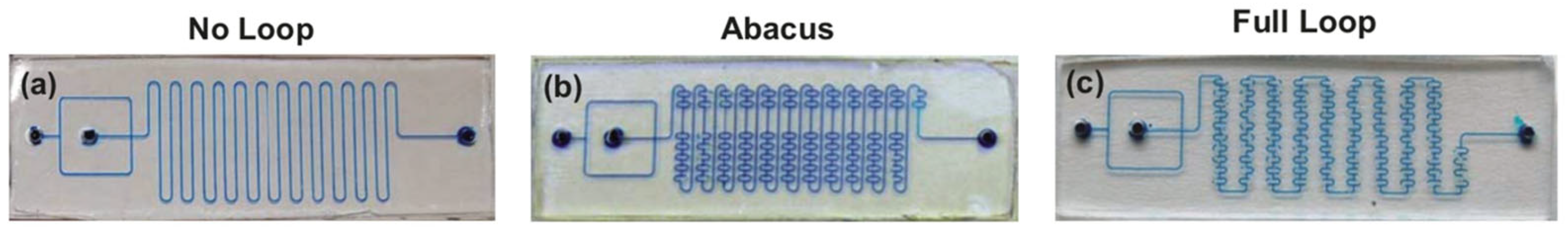
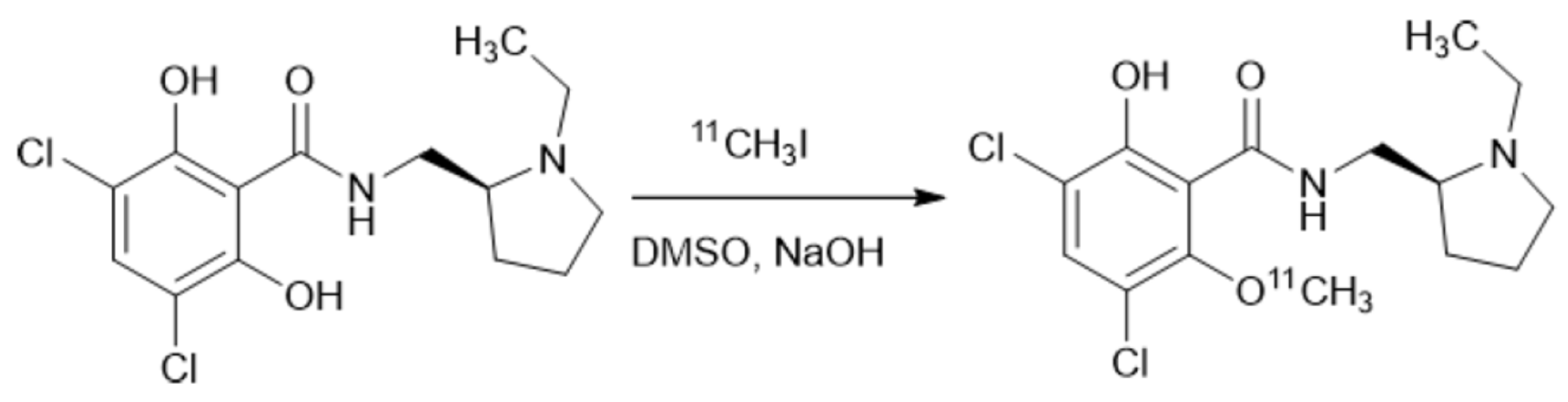
An increase in the mixing efficiency often leads to an improvement of the reaction outcome. A remarkable example is the preparation of [11C]raclopride under flow conditions by the methylation reaction (Scheme 3) [121]. During the study of the impact of the micromixer/microreactor design on this process, mixers with a consistently increasing number of additional loops (Figure 8a–c) were used, which were expected to increase the mixing efficiency and improve the reaction outcome. It was shown that reaction with non-radioactive CH3I resulted in higher yields in the “abacus” micromixer than in the “no loop” micromixer (5.8% and 1.3% yields, respectively). At the stage of using radioactive 11CH3I as the methylation reagent it was also shown that the “full loop” microreactor is better than the “abacus” micromixer (relative radioactivity 25.6 and 16.0, respectively).
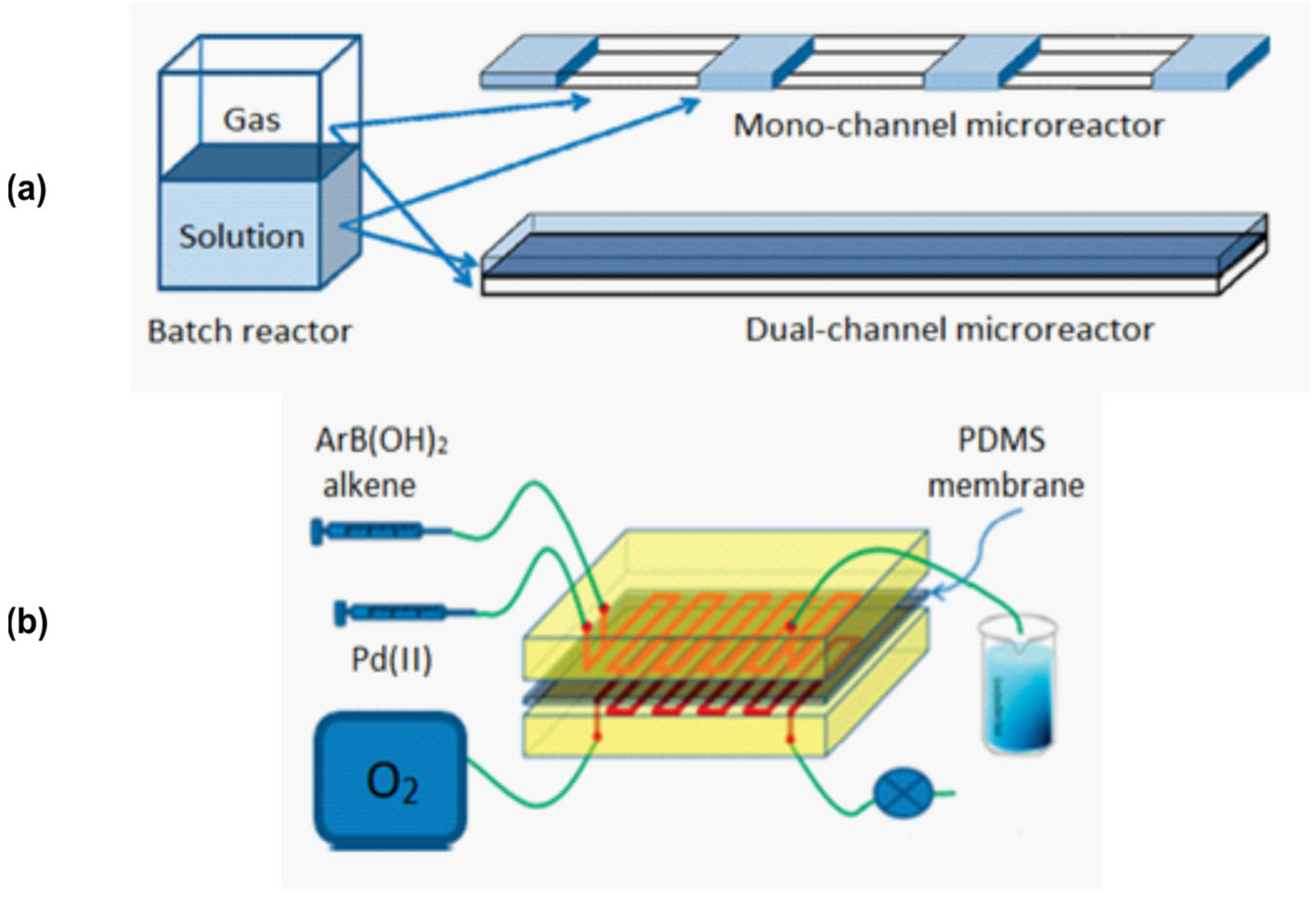
The outcome of two-phase reactions carried out in flow may also depend strongly on the mixing method. This was shown using a Heck reaction where two modes of oxygen delivery to the reaction solution were compared (Figure 9) [122]. The main idea of this design is that the flows of liquid and gas occur not in one capillary, but in two parallel ones (Figure 9a). This can be achieved using the setup shown in Figure 9b: two parallel capillaries are separated by the PDMS membrane. This setup allows one to adjust the flow rate for each phase independently. Dual-channel design compared to slug flow showed higher convenience and higher yields: ≥75% in dual-channel and ≤65% in mono-channel modes (1H NMR data).
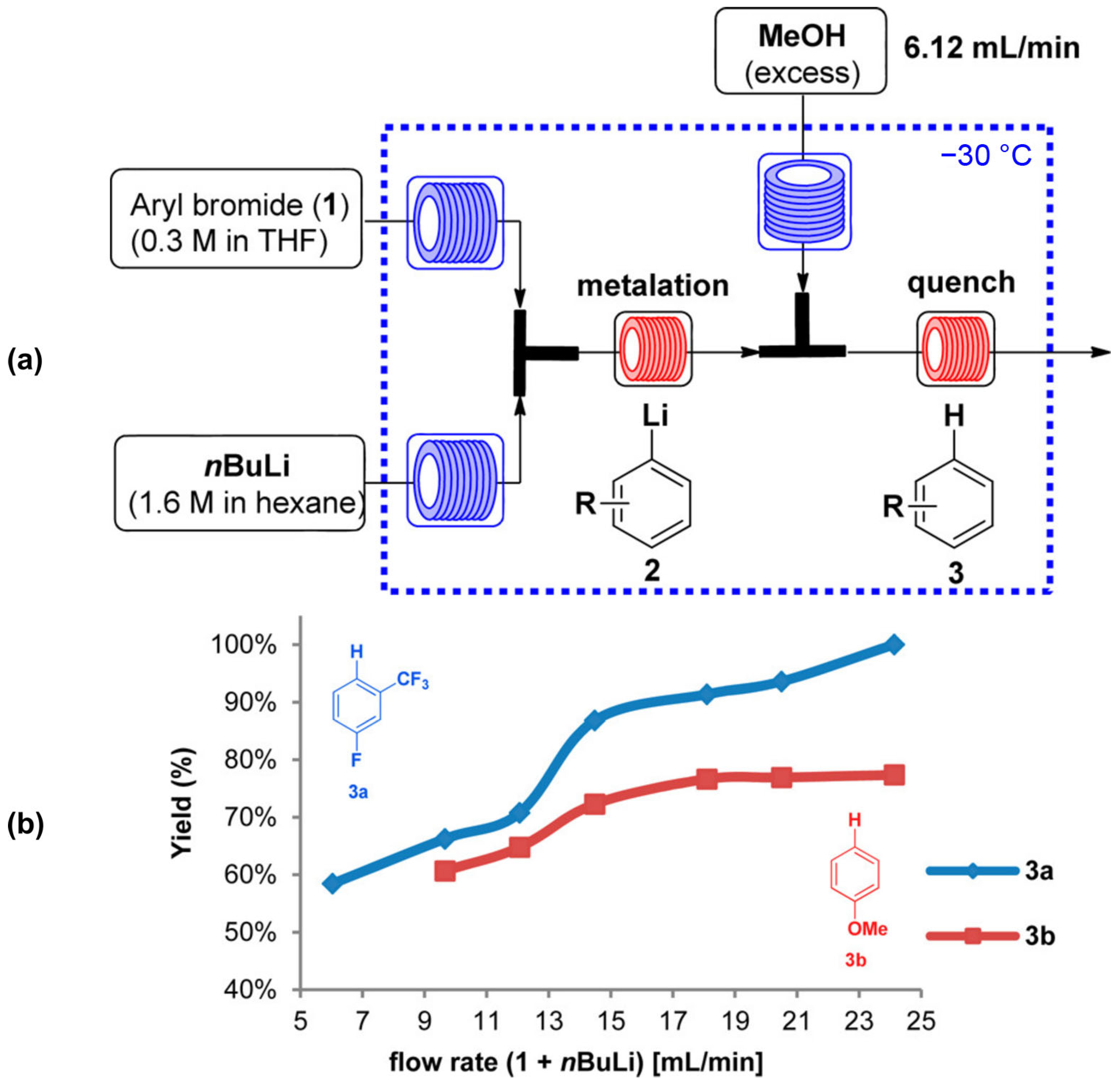
The mixing efficiency of a T-shaped micromixer is known to strongly depend on the flow rate [57]. When such mixer was used for a study of efficiency of metalation of substituted bromobenzenes with butyl lithium followed by quenching with methanol (Figure 10a) the inflection points on the plots of the yields of both electron-rich and electron-poor compounds were observed at 14 mL/min flow rate suggesting that the increased mixing efficiency within the T-shaped micromixer at a high flow rate, hence higher product yield, is due to a change in the flow regime [118] (Figure 10b; compare with Figure 3).
The design of the mixer plays an important role in the processes, during which further transformation of the product as a result of side reactions is possible. This is well illustrated by the Friedel–Crafts reaction (Scheme 4) [123]. In this example, sequential alkylation of the aromatic system occurs. The monosubstituted (main) and disubstituted (side) products were obtained in different ratios when different micromixers were used for mixing reagents. In a simple T-shaped micromixer the yields of the main and the side products were 36% and 31%, respectively. When the YM-1 mixer [45] was used the yields of the main and the side products became 50% and 14%, respectively. The best result was achieved in the IMM mixer [21,35]: the yields of the main and the side products were 92% and 4%, respectively.
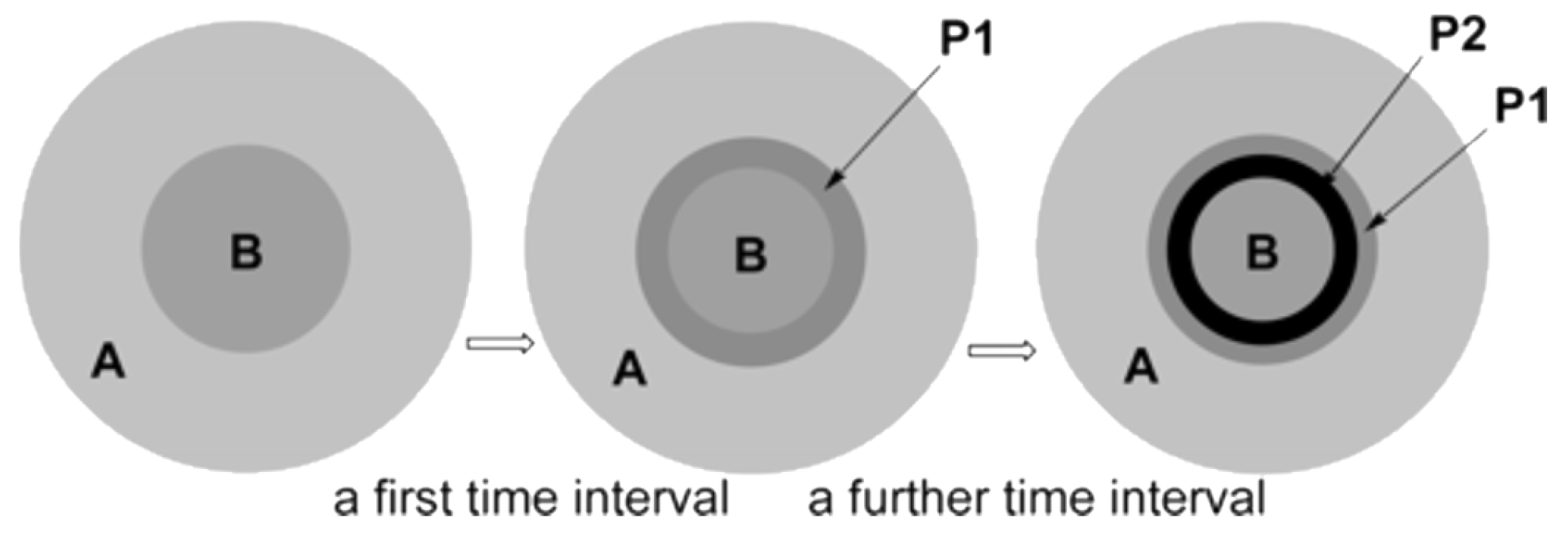
Such a strong influence of the mixer design (hence the mixing efficiency) was explained [123] from the point of view of the model proposed by Rys [124] (see also a discussion in [57] on competitive consecutive reactions). This model can be used to describe multistep rapid reactions whose rate is limited by the diffusion of reagents (Figure 11). At the first time interval after mixing, regions with an increased concentration of reagent molecules (B) surrounded by a solution of another reagent (A) are formed. At the points of contact, the first product (P1) is formed, which, without effective mixing, does not have time to diffuse into the outer sphere and continues to react to give P2 at the further time interval.
During a study of Lewis acid (TMSOTf) promoted microfluidic glycosylation of a 4,6-galactose diol with a 5-azido-sialyl imidate in EtCN at −78 °C (bath temperature) a noticeable influence of the mixer design on the outcome of glycosylation was detected for the first time [100]. Both employed micromixers (IMM and Comet X-01) allowed highly efficient synthesis of the target disaccharide (yields were virtually quantitative in both cases). However, stereoselectivity was higher when the IMM micromixer was used (α/β = 95:5) as compared to stereoselectivity achieved in glycosylation with the Comet X-01 micromixer (α/β = 88:12). The authors explained this difference in selectivity by higher temperature in the second case, leading to lower stereoselectivity, “probably due to poorer heat removal of Comet X-01” (the reaction is exotermic) [100].
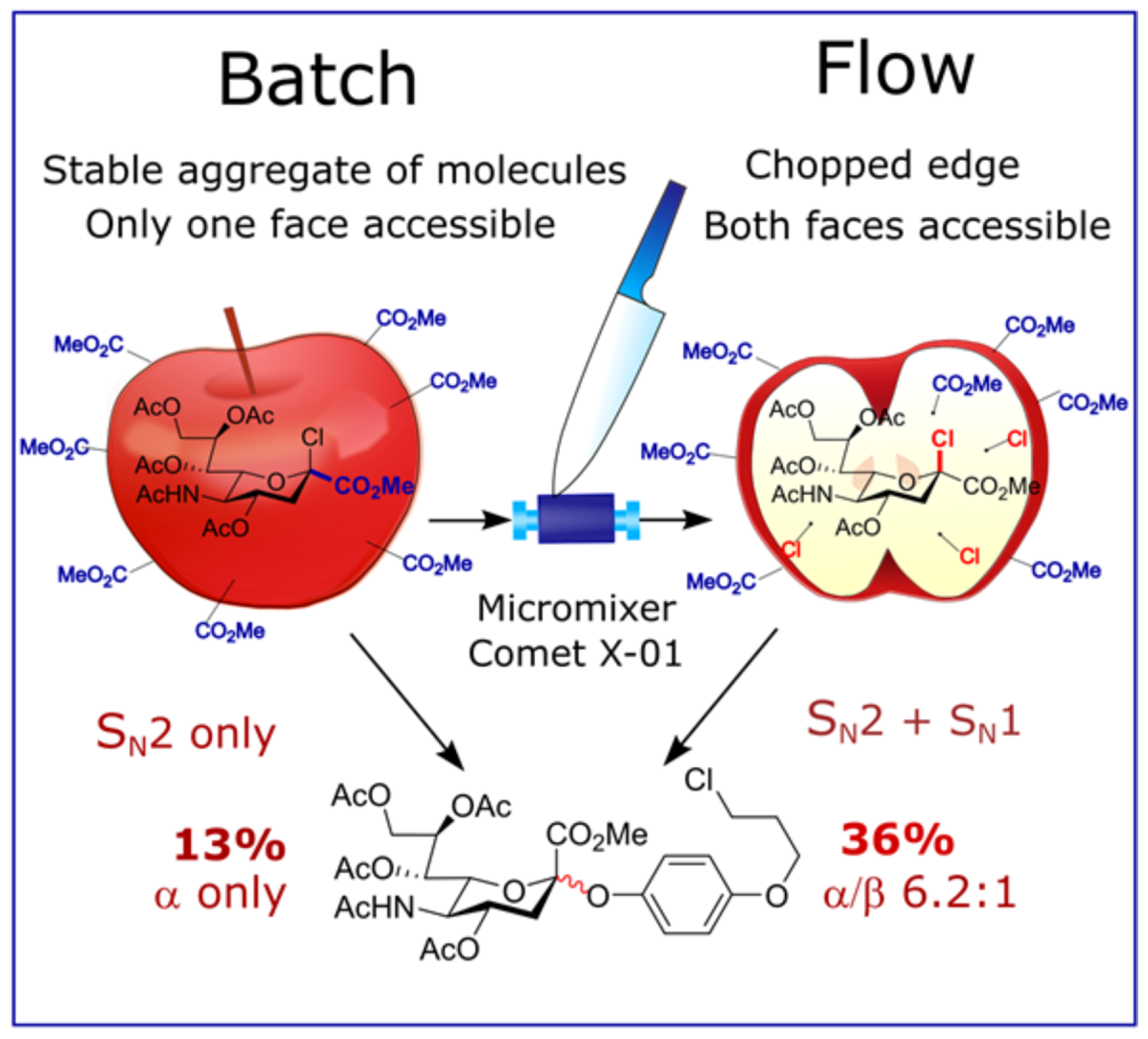
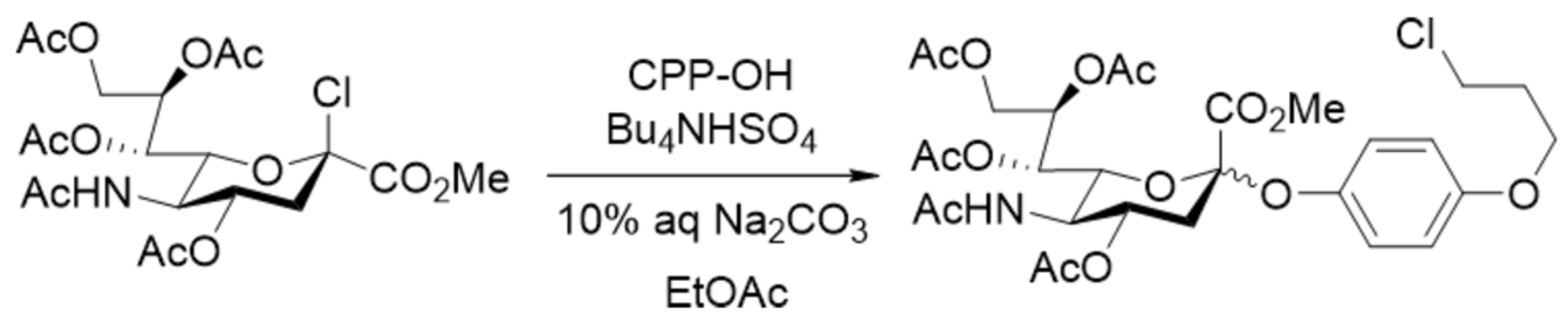
The influence of the mixer design on the result of glycosylation was also observed in the sialylation reaction of a substituted phenol (Scheme 5) performed at room temperature under conditions of phase transfer catalysis [113,114]. Here, two different micromixers were used: a Comet X-01 micromixer (vide supra), where flow splits into three and joins together several times (see Figure 5), and a simple T-shaped micromixer. At 2 μL/min flow rate the product yields in both cases turned out to be similar: 36% and 33%, respectively. However, the difference in stereoselectivity (α/β) was more significant: 6.2:1 and 13.3:1, respectively. Note that the same reaction performed in batch is stereospecific providing a kinetically controlled α-isomer as the only product of glycosylation [113].
The effect of the flow rate on the yield and on the stereoselectivity of glycosylation was observed in this sialylation reaction (Scheme 5) [113]. In this case, with the increase of the flow rate from 2 to 250 μL/min, the yield and stereoselectivity decreased from 36% to 11% and from α/β = 6.2:1 to α/β = 1.9:1, respectively. However, after further increase of the flow rate to 1000 μL/min the yield and stereoselectivity increased to 37% and α/β = 3.8:1, respectively. Therefore, both high (1000 μL/min) and low (2 μL/min) flow rates result in similar yields and stereoselectivities, but intermediate flow rates give worse results. The influence of flow rate and mixer design on the product yield could be easily rationalized within the framework of the mixing-centered concept, discussed above, which emphasizes the importance of the adequate flow regime for efficient mixing of reagents.
However, the unexpected loss of stereoselectivity under microfluidic conditions and its dramatic dependence on mixing conditions (mixer design and flow rate), hence flow regime, does not fit the current knowledge [125,126,127,128] on the origin of stereoselectivity of glycosylation. It is unclear how flow rate or mixer design could affect the stereoselectivity of reactions that involve “isolated” molecules.
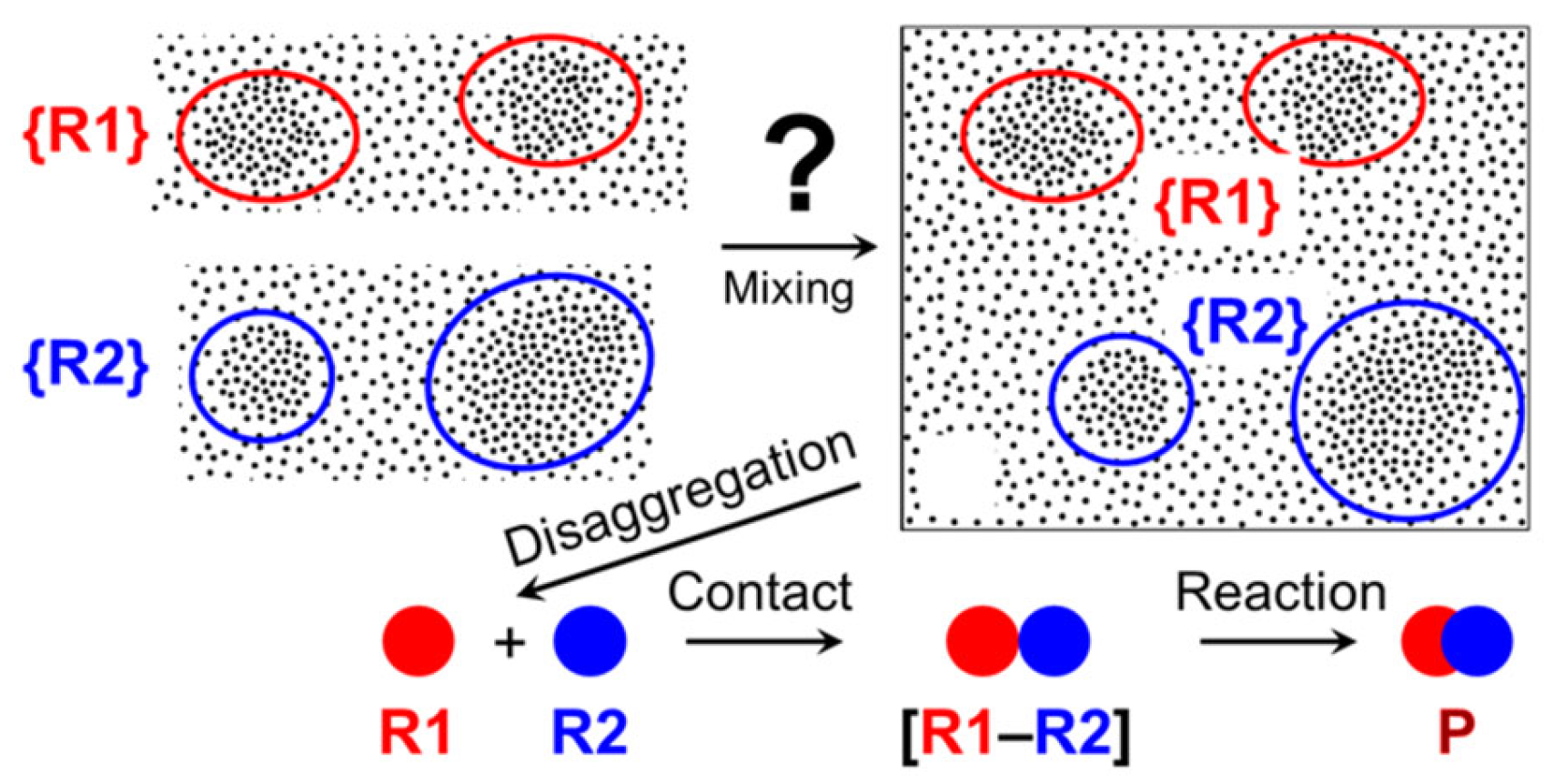
However, the found phenomenon can be rationally discussed within the framework of supramer hypothesis (supramer approach) [27], according to which the real reacting species in many cases are supramolecular aggregates (supramers) rather than single molecules of reacting substances (the “molecular species”). Recent studies revealed [129,130,131,132,133,134,135,136,137] that even macroscopically homogeneous aqueous and non-aqueous solutions of low-molecular-mass non-amphiphilic compounds can contain nano- and mesoscale heterogeneities (size from ca. 1 nm to 102–103 nm) that are kinetically stable, although very small interaction energy, which does not exceed kBT [138], is involved. The most probable reason for their existence and omnipresence is the “solvophobicity-driven mesoscale” structuring [136] promoted by even minute amounts of “solvophobic admixtures” [27,136] that are present in most commercially available compounds [133] since “no truly pure chemicals exist” [136].
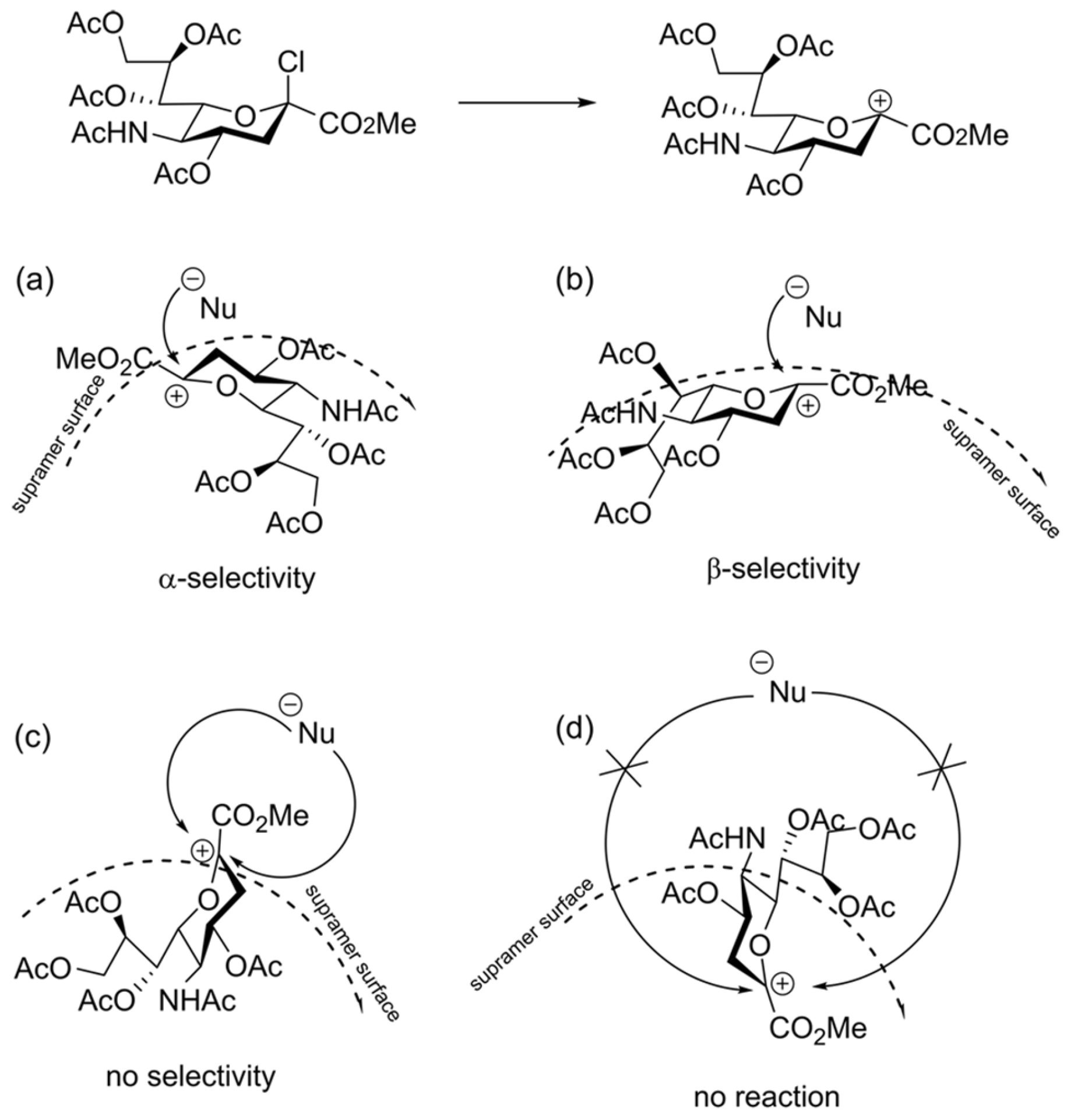
Considering the presence of such heterogeneities, supramers in our terminology [27], which belong to the realm of soft matter, we have been developing the supramer approach (vide supra) for the description of chemical reactions (see Refs. [27,139,140,141] and references cited therein). According to the supramer approach, the “presentation” (spatial orientation) of molecules on the surface of supramers of glycosyl donor (or glycosyl cation formed from it) may play an important role in determining reaction stereoselectivity (see Figure 12).
The first stage of the reaction is formation of glycosyl cation which next reacts with nucleophile (Figure 12). Apparently, this stage is the most important one, since the presentation of the glycosyl cation on the surface of the supramer would determine the face of the glycosyl cation on which the nucleophile attack will occur leading to different stereoisomers of the product (Figure 12a–c). It is also possible that the cationic center is hidden inside the supramer (Figure 12d), which would prevent the attack of the nucleophile and lead to a decrease in reactivity of the starting compound (lowering conversion) and the product yield. Similar processes of orientation of molecules on a surface apparently occur during the formation of J-aggregates [142,143,144,145,146,147] for which the dependence of the resulting types of aggregates on the mixing method was established [148,149].
According to the explanation suggested [113], the efficiency of disaggregation/rearrangement (see Figure 13) of sialyl chloride supramers [27] varies under different mixing modes (in particular, at different flow rates). This makes the SN1-like pathway within SN1–SN2-continuum of glycosylation mechanisms [150,151] possible under microfluidic conditions, leading to formation of both anomers of CPP sialoside (Scheme 5).
According to the authors [113,114], the β-anomer is formed only if sialyl chloride supramers undergo disaggregation (Figure 14), which seems to be more effective in Comet X-01 micromixer (apparently featured by “chaotic mixing” [3,29,31,42,96]) than in T-shaped micromixer. This result suggests that high efficiency of mixing is not a panacea for improving the reaction outcome.
We must emphasize that this is the first documented example [113,114] of the influence of flow regime on stereoselectivity of a chemical reaction (that does not involve epimerization of the product), which has additionally been rationally explained.
An increase in the syn/anti ratio (from 8:1 to 18:1) of the product upon increase in the flow rate has been reported for organocatalyzed 1,4-addition of aldehydes to nitroolefins. The authors explained this by epimerization of the product by the catalyst, which becomes more profound at increased residence times that inevitably accompany lower flow rates [152].
Continuous telescoped two-carbon homologation of esters to α,β-unsaturated esters was performed under microfluidic conditions, which incorporated an ester reduction, phosphonate deprotonation, and Horner–Wadsworth–Emmons olefination into a single, uninterrupted system. The authors are puzzled by the fact that the E/Z selectivity of the olefination in flow was noticeably higher than that under batch conditions using similar reagents [153].
All other previous studies of reactive mixing (see, e.g., [39,57,63]) were concerned with conversion of the starting compound and product yield as the measure of mixing quality. It is difficult to surmise from basic chemical principles [154] that flow regime could alter stereoselectivity of glycosylation, which is considered to be governed (for kinetically controlled reactions, to which glycosylation belongs) by relative rates of reagent attack on Re- and Si-faces of glycosyl cation, a real glycosylating agent in these systems [114]. One can speculate that the breakdown of symmetry (leading to inequality of these rates) could occur in the presence of an external chiral stimulus. Note that vortex-controlled generation of supramolecular chirality has been documented for batch systems [148,149,155,156,157].
The recently discovered [20] phenomenon of enhanced formation of non-covalent bonds under flow conditions, thus easing the self-assembly of new supramolecular structures, might also be related to rearrangements of supramers of reactants induced by shear stress in flow as discussed above (see Figure 14). Specifically, we hypothesize that supramers of reactants disaggregate (partially or completely) (see Figure 13 and Figure 14) under flow conditions in a dynamical fashion creating the fragmented supramers (and, ultimately, the “molecular species”) that are (1) more reactive compared to the parent supramers, present in the starting solutions, and (2) capable of forming supramolecular architectures different from those formed “passively” [20] in the flask from the parent supramers. In line with the discussion above, we propose that the ease of supramolecular assembly in flow would dramatically depend on the flow regime, hence on the mixer design and the flow rate used.
Similar reasoning might explain the well-known phenomenon of decreasing the reaction time in flow as compared to that of the same reaction performed in batch. From the supramer point of view, shear stress in flow might induce fragmentation of the parent supramers, which exist in solution before the reaction, leading to formation of more reactive species and making the reaction faster. Traditional explanation of this considerable decrease in reaction time (e.g., from 24 h in batch to 7 min in flow [152]) would involve listing beneficial features of the microfluidic technique such as improved mass transfer due to efficient mixing, fast heat transfer, and residence time control to name a few (see, e.g., [5,18,19]).
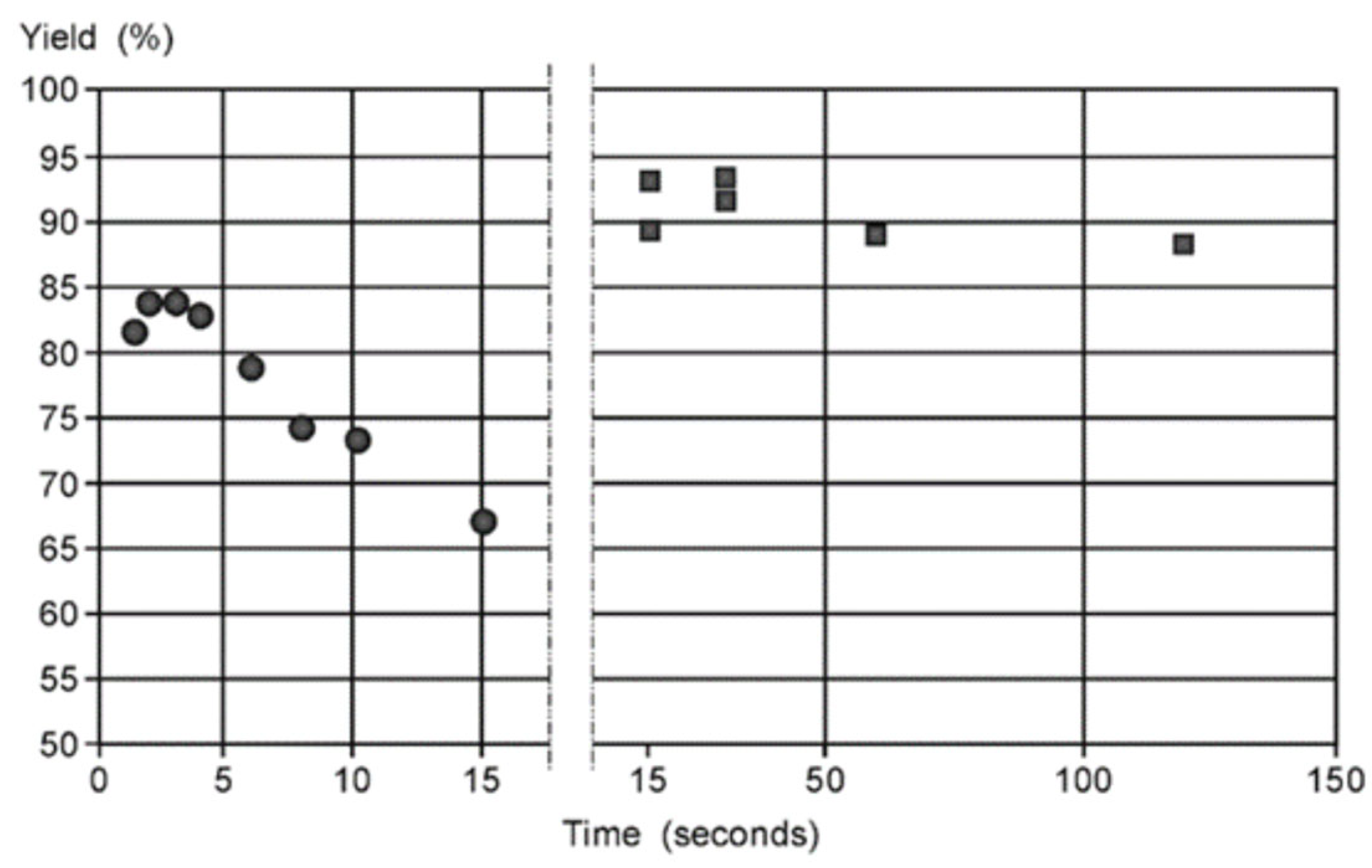
The design of the mixer and flow rate can strongly determine the reactivity of a substance. This phenomenon was demonstrated using glycosylation of isopropyl alcohol with oxazoline as the glycosyl donor (Scheme 6) carried out in a Comet X-01 micromixer (Figure 15) [110]. As expected, at high flow rates, the yields of both products were low—the reaction did not have enough time to occur. With a decrease in the flow rate, the yields expectedly increased. But at some point, at very low flow rates (flow rate ≤43 μL/h), the acid-labile oxazoline stopped reacting and the starting reagents were recovered. This apparent loss of reactivity was observed only when Comet X-01 micromixer was used. However, when a Comet X-01 micromixer was replaced with a T-shaped micromixer (at 30 μL/h flow rate), the expected reaction products, glycoside and glycal, appeared in the reaction mixture. Thus, the change in mixer design (from Comet X-01 micromixer to T-shaped micromixer) apparently altered the flow regime resulting in better mixing of reagents, if we follow the mixing-centered concept, discussed above.
However, within the framework of the supramer approach [27] (vide supra), this result apparently suggests a higher disaggregation of the supramers of reagents under these conditions (using a T-shaped micromixer), which allows the chemical reaction between them to occur. Apparently, at very low flow rates (30 μL/h) in the Comet X-01 micromixer, the molecules of this glycosyl donor are located inside the core of tight supramers (similar to those discovered recently [113,114,139,158]) or presented on the surface of supramers in such a way that the anomeric position becomes inaccessible for attack (which is manifested as an enhanced stability of oxazoline under the action of acid), similarly to the situation shown in Figure 12 for glycosyl cation derived from sialyl chloride. Based on these facts, we conclude that the flow rate in combination with change in the mixer design in this case directly affects the reactivity of molecules probably by changing the flow regime which makes disaggregation (see Figure 13 and Figure 14) of the supramers of reagents (im)possible thus altering the observed reactivity pattern.
Similar observations were made earlier for another reaction [159], where it has been proposed that the observed decrease in conversion at low flow rates is related to the change in the flow regime from “dispersed flow regime” (good mixing) to “stratified flow regime” (bad mixing) (Figure 16) supported by mixing experiments with non-reacting substances. Indeed, this phenomenon of “the dependence of mass transfer on the two-phase flow patterns” was studied in single- and two-phase flow systems [61,64]. However, as in the previous example, one can speculate that a change in the flow rate leads to a change in the flow regime, which modulates the efficiency of disaggregation of the supramers of reagents. As a result, at low flow rates, some of the molecules are located inside the core of tight supramers [113,114,139,158] and cannot react. As the flow rate increases, the efficiency of disaggregation increases, and the conversion rate increases accordingly. With a further increase in the flow rate, the conversion decreases, which may be due either to insufficient contact time of the reagents or to generation of supramers of different type formed during disaggregation of the parent supramers.
An interesting example of dramatic reactivity enhancing upon increase in flow rate (from 0.3 mL/min to 5 mL/min) was reported for in-flow metalation of an acrylate ester containing thiophene moiety with 2,2,6,6-tetramethylpiperidinylmagnesium chloride lithium chloride complex at −25 °C. While at low flow rates only low conversions could be achieved with long residence times (5 or 15 min), at high flow rates almost full conversion was achieved within 1 min leading to isolation of the desired product in 72% yield [117].
4. Influence of Residence Time on the Reaction Outcome
The flow rate changes can affect the reaction outcome also by simply changing the residence time (time of contact = time of flowing reagents together without changing the flow rate) without apparent influence of the mixing efficiency. In fact, it may be difficult to separate these effects experimentally.
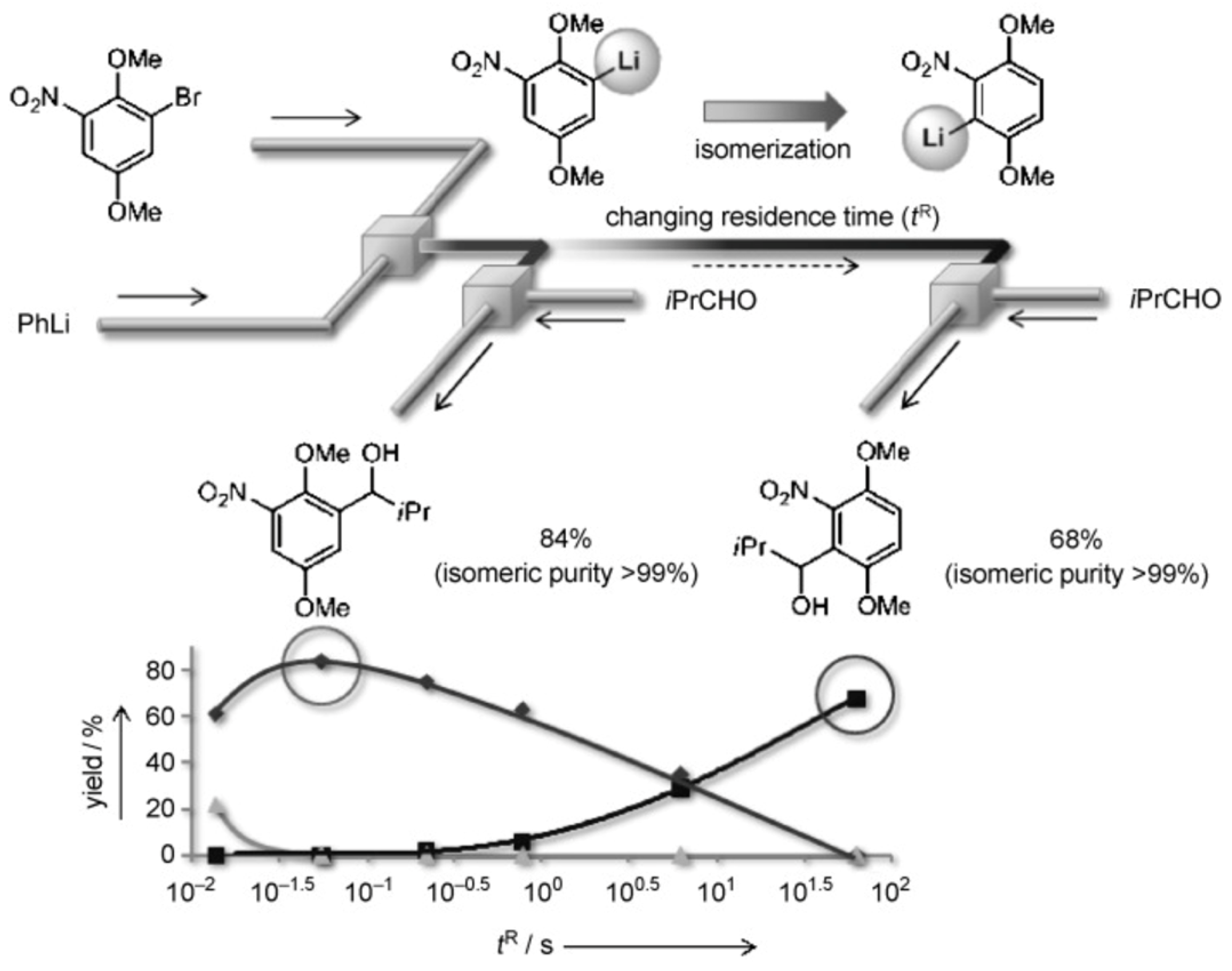
There are a number of examples when an increase in residence time of reagents leads to an increase in the reaction yield as expected for batch reactions [160]. However, with too long times, selectivity decreases [161] or by-products appear [119]. Special attention should be paid to examples when it is possible to obtain various products in high yields by varying the residence time. A striking example of such an approach is the Br-Li exchange reaction followed by the reaction of the lithiated species with electrophiles performed in flow (Figure 17) [162]. There, kinetically formed 5-substitued product is selectively formed at residence time 0.06 s (yield 84%). Increasing residence time to 63 s gives thermodynamically preferred 3-substitued product in yield 68%. In both cases only traces of another isomer were observed.
5. Conclusions
In conclusion, we demonstrated that variation in flow regime (determined by a combination of mixer design and flow rate) can either improve or worsen the reactivity and lead to completely different products, including stereoisomers. We would like to stress that it is not necessary to homogenize the reaction mixture as much as possible or to mix the reagents with maximum efficiency. The real challenge is to mix reagents the right way. To achieve this in a flow reactor one should combine the appropriate values of flow rate and the designs of microreactor.
In terms of the supramer approach [27], at too high or too low a flow rate (in the particular mixer), the molecules of reagents are incorrectly presented on the surface of supramers, leading to altered stereoselectivity, or form tight supramers (similar to those discovered recently [113,114,139,158]), in which most of the molecules are located inside the supramer core and are inaccessible for attack, leading to low conversions and yields. The flow rate at which these problems do not exist can be considered optimal.
We can discuss the efficiency of mixing in a similar way. On the one hand, efficiently mixed reagents can react faster and with greater conversion, giving higher yields. On the other hand, the most efficiently mixed system loses the effect of the asymmetry of the supramer surface (since all supramers are disaggregated), which eliminates a possibility of controlling stereoselectivity of the reaction and allows all side processes to proceed.
Chemistry is definitely a statistical science. There are myriads of molecules in the reaction mixture, each of which can react (in one or many ways) or not. For each specific molecule, the reaction proceeds conditionally instantaneously, provided that the reaction partners (who have the fundamental ability to react) have approached the right faces. If it were possible to find a tool or a method of influencing molecules, in which only the approaches we need would be encountered, then product yields and stereoselectivities of any reactions would be high. Experimental data suggest that, for reactions conducted in flow, the flow rate and the design of the microreactor can serve as such tools.
Author Contributions
Conceptualization, I.V.M. and L.O.K.; methodology, I.V.M. and L.O.K.; investigation, I.V.M.; writing—original draft preparation, I.V.M.; writing—review and editing, L.O.K.; visualization, I.V.M. and L.O.K.; supervision, L.O.K.; project administration, L.O.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jahnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Chemistry in microstructured reactors. Angew. Chem. Int. Ed. 2004, 43, 406. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- deMello, A.J. Control and detection of chemical reactions in microfluidic systems. Nature 2006, 442, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Fukase, K. Acid-mediated reactions under microfluidic conditions: A new strategy for practical synthesis of biofunctional natural products. Beilstein J. Org. Chem. 2009, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Fukase, K. Renaissance of Traditional Organic Reactions under Microfluidic Conditions: A New Paradigm for Natural Products Synthesis. Org. Process Res. Dev. 2009, 13, 983–990. [Google Scholar] [CrossRef]
- Wegner, J.; Ceylan, S.; Kirschning, A. Ten key issues in modern flow chemistry. Chem. Commun. 2011, 47, 4583–4592. [Google Scholar] [CrossRef] [PubMed]
- Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. Novel Process Windows for Enabling, Accelerating, and Uplifting Flow Chemistry. ChemSusChem 2013, 6, 746–789. [Google Scholar] [CrossRef] [PubMed]
- Elvira, K.S.; Casadevall i Solvas, X.; Wootton, R.C.R.; demello, A.J. The past, present and potential for microfluidic reactor technology in chemical synthesis. Nat. Chem. 2013, 5, 905–915. [Google Scholar] [CrossRef]
- Fukase, K.; Shimoyama, A.; Manabe, Y. Effective Synthesis of Oligosaccharide under Microfiuidic Conditions. J. Synth. Org. Chem. Jpn. 2015, 73, 452–459. [Google Scholar] [CrossRef]
- Fukase, K.; Tanaka, K.; Fujimoto, Y.; Shimoyama, A.; Manabe, Y. Sugar synthesis by microfluidic techniques. In Glycochemical Synthesis; Hung, S.-C., Zulueta, M.M.L., Eds.; Wiley Online Books; Wiley: Hoboken, NJ, USA, 2016; pp. 205–219. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Sevim, S.; Sorrenti, A.; Franco, C.; Furukawa, S.; Pané, S.; deMello, A.J.; Puigmartí-Luis, J. Self-assembled materials and supramolecular chemistry within microfluidic environments: From common thermodynamic states to non-equilibrium structures. Chem. Soc. Rev. 2018, 47, 3788–3803. [Google Scholar] [CrossRef] [PubMed]
- Guidi, M.; Seeberger, P.H.; Gilmore, K. How to approach flow chemistry. Chem. Soc. Rev. 2020, 49, 8910–8932. [Google Scholar] [CrossRef]
- Hone, C.A.; Kappe, C.O. Towards the Standardization of Flow Chemistry Protocols for Organic Reactions. Chem. Methods 2021, 1, 454–467. [Google Scholar] [CrossRef]
- Leslie, A.; Joseph, A.M.; Baumann, M. Functional Group Interconversion Reactions in Continuous Flow Reactors. Curr. Org. Chem. 2021, 25, 2217–2231. [Google Scholar] [CrossRef]
- Paratore, F.; Bacheva, V.; Bercovici, M.; Kaigala, G.V. Reconfigurable microfluidics. Nat. Rev. Chem. 2021, 6, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Burange, A.S.; Osman, S.M.; Luque, R. Understanding flow chemistry for the production of active pharmaceutical ingredients. iScience 2022, 25, 103892. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, L.; Wen, Z.; Noel, T. A field guide to flow chemistry for synthetic organic chemists. Chem. Sci. 2023, 14, 4230–4247. [Google Scholar] [CrossRef] [PubMed]
- Monbaliu, J.-C.M.; Legros, J. Will the next generation of chemical plants be in miniaturized flow reactors? Lab Chip 2023, 23, 1349–1357. [Google Scholar] [CrossRef]
- Numata, M.; Kanzaki, C. Supramolecular Chemistry of a Moving Solution: Flow Drives New Non-covalent Bond Formation. Chem. Lett. 2023, 52, 602–610. [Google Scholar] [CrossRef]
- Ehrfeld, W.; Hessel, V.; Löwe, H. (Eds.) Micromixers. In Microreactors; Wiley: Hoboken, NJ, USA, 2000; pp. 41–85. [Google Scholar] [CrossRef]
- Suryawanshi, P.L.; Gumfekar, S.P.; Bhanvase, B.A.; Sonawane, S.H.; Pimplapure, M.S. A review on microreactors: Reactor fabrication, design, and cutting-edge applications. Chem. Eng. Sci. 2018, 189, 431–448. [Google Scholar] [CrossRef]
- Bojang, A.A.; Wu, H.-S. Design, fundamental principles of fabrication and applications of microreactors. Processes 2020, 8, 891. [Google Scholar] [CrossRef]
- Dong, Z.; Wen, Z.; Zhao, F.; Kuhn, S.; Noel, T. Scale-up of micro- and milli-reactors: An overview of strategies, design principles and applications. Chem. Eng. Sci. X 2021, 10, 100097. [Google Scholar] [CrossRef]
- Kucherov, F.A.; Romashov, L.V.; Ananikov, V.P. Development of 3D + G printing for the design of customizable flow reactors. Chem. Eng. J. 2022, 430, 132670. [Google Scholar] [CrossRef]
- Whitesides, G.M. Whitesides’ Group: Writing a Paper. Adv. Mater. 2004, 16, 1375–1377. [Google Scholar] [CrossRef]
- Kononov, L.O. Chemical reactivity and solution structure: On the way to a paradigm shift? RSC Adv. 2015, 5, 46718–46734. [Google Scholar] [CrossRef]
- Reynolds, O. XXIX. An experimental investigation of the circumstances which determine whether the motion of water shall be direct or sinuous, and of the law of resistance in parallel channels. Philos. Trans. R. Soc. Lond. 1883, 174, 935–982. [Google Scholar] [CrossRef]
- Nguyen, N.-T.; Wu, Z. Micromixers—A review. J. Micromech. Microeng. 2005, 15, R1–R16. [Google Scholar] [CrossRef]
- Hisamoto, H.; Saito, T.; Tokeshi, M.; Hibara, A.; Kitamori, T. Fast and high conversion phase-transfer synthesis exploiting the liquid–liquid interface formed in a microchannel chip. Chem. Commun. 2001, 2662–2663. [Google Scholar] [CrossRef]
- Ottino, J.M. Mixing, Chaotic Advection, and Turbulence. Annu. Rev. Fluid Mech. 1990, 22, 207–254. [Google Scholar] [CrossRef]
- Ottino, J.M. Mixing and chemical reactions a tutorial. Chem. Eng. Sci. 1994, 49, 4005–4027. [Google Scholar] [CrossRef]
- Bałdyga, J.; Bourne, J.R. Turbulent Mixing and Chemical Reactions; Wiley: Chichester, UK, 1999; p. 896. [Google Scholar]
- Bourne, J.R. Mixing and the Selectivity of Chemical Reactions. Org. Process Res. Dev. 2003, 7, 471–508. [Google Scholar] [CrossRef]
- Hessel, V.; Löwe, H.; Schönfeld, F. Micromixers—A review on passive and active mixing principles. Chem. Eng. Sci. 2005, 60, 2479–2501. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Chang, C.-L.; Wang, Y.-N.; Fu, L.-M. Microfluidic Mixing: A Review. Int. J. Mol. Sci. 2011, 12, 3263–3287. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Lemenand, T.; Della Valle, D.; Peerhossaini, H. Static mixers: Mechanisms, applications, and characterization methods—A review. Chem. Eng. Res. Des. 2014, 92, 205–228. [Google Scholar] [CrossRef]
- Arimond, J.; Erwin, L. A simulation of a motionless mixer. Chem. Eng. Commun. 1985, 37, 105–126. [Google Scholar] [CrossRef]
- Ehrfeld, W.; Golbig, K.; Hessel, V.; Löwe, H.; Richter, T. Characterization of Mixing in Micromixers by a Test Reaction: Single Mixing Units and Mixer Arrays. Ind. Eng. Chem. Res. 1999, 38, 1075–1082. [Google Scholar] [CrossRef]
- Wörz, O.; Jäckel, K.P.; Richter, T.; Wolf, A. Microreactors—A New Efficient Tool for Reactor Development. Chem. Eng. Technol. 2001, 24, 138–142. [Google Scholar] [CrossRef]
- Johnson, T.J.; Ross, D.; Locascio, L.E. Rapid Microfluidic Mixing. Anal. Chem. 2002, 74, 45–51. [Google Scholar] [CrossRef]
- Stroock, A.D.; Dertinger, S.K.W.; Ajdari, A.; Mezić, I.; Stone, H.A.; Whitesides, G.M. Chaotic Mixer for Microchannels. Science 2002, 295, 647–651. [Google Scholar] [CrossRef]
- Engler, M.; Kockmann, N.; Kiefer, T.; Woias, P. Numerical and experimental investigations on liquid mixing in static micromixers. Chem. Eng. J. 2004, 101, 315–322. [Google Scholar] [CrossRef]
- Kockmann, N.; Engler, M.; Woias, P. Theoretische und experimentelle Untersuchungen der Mischvorgänge in T-förmigen Mikroreaktoren—Teil 3: Konvektives Mischen und chemische Reaktionen. Chem. Ing. Tech. 2004, 76, 1777–1783. [Google Scholar] [CrossRef]
- Mae, K.; Maki, T.; Hasegawa, I.; Eto, U.; Mizutani, Y.; Honda, N. Development of a new micromixer based on split/recombination for mass production and its application to soap free emulsifier. Chem. Eng. J. 2004, 101, 31–38. [Google Scholar] [CrossRef]
- Bothe, D.; Stemich, C.; Warnecke, H.-J. Fluid mixing in a T-shaped micro-mixer. Chem. Eng. Sci. 2006, 61, 2950–2958. [Google Scholar] [CrossRef]
- Hardt, S.; Pennemann, H.; Schönfeld, F. Theoretical and experimental characterization of a low-Reynolds number split-and-recombine mixer. Microfluid. Nanofluid. 2006, 2, 237–248. [Google Scholar] [CrossRef]
- Kockmann, N.; Kiefer, T.; Engler, M.; Woias, P. Convective mixing and chemical reactions in microchannels with high flow rates. Sens. Actuators B 2006, 117, 495–508. [Google Scholar] [CrossRef]
- Kockmann, N.; Dreher, S.; Woias, P. Unsteady Laminar Flow Regimes and Mixing in T-Shaped Micromixers. In Proceedings of the ASME 2007 5th International Conference on Nanochannels, Microchannels, and Minichannels, Puebla, Mexico, 18–20 June 2007; ASME: Puebla, Mexico, 2007; pp. 671–678. [Google Scholar] [CrossRef]
- Kockmann, N. Transport Phenomena in Micro Process Engineering, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2008; p. 365. [Google Scholar] [CrossRef]
- Fang, W.-F.; Yang, J.-T. A novel microreactor with 3D rotating flow to boost fluid reaction and mixing of viscous fluids. Sens. Actuators B 2009, 140, 629–642. [Google Scholar] [CrossRef]
- Singh, M.K.; Anderson, P.D.; Meijer, H.E.H. Understanding and Optimizing the SMX Static Mixer. Macromol. Rapid Commun. 2009, 30, 362–376. [Google Scholar] [CrossRef]
- Dreher, S.; Engler, M.; Kockmann, N.; Woias, P. Theoretical and Experimental Investigations of Convective Micromixers and Microreactors for Chemical Reactions. In Micro and Macro Mixing: Analysis, Simulation and Numerical Calculation; Bockhorn, H., Mewes, D., Peukert, W., Warnecke, H.-J., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 325–346. [Google Scholar] [CrossRef]
- Suh, Y.K.; Kang, S. A Review on Mixing in Microfluidics. Micromachines 2010, 1, 82–111. [Google Scholar] [CrossRef]
- Kockmann, N.; Roberge, D.M. Scale-up concept for modular microstructured reactors based on mixing, heat transfer, and reactor safety. Chem. Eng. Process. 2011, 50, 1017–1026. [Google Scholar] [CrossRef]
- Zhdanov, V.; Chorny, A. Development of macro- and micromixing in confined flows of reactive fluids. Int. J. Heat Mass Transf. 2011, 54, 3245–3255. [Google Scholar] [CrossRef]
- Schwolow, S.; Hollmann, J.; Schenkel, B.; Roeder, T. Application-Oriented Analysis of Mixing Performance in Microreactors. Org. Process Res. Dev. 2012, 16, 1513–1522. [Google Scholar] [CrossRef]
- Orsi, G.; Roudgar, M.; Brunazzi, E.; Galletti, C.; Mauri, R. Water-ethanol mixing in T-shaped microdevices. Chem. Eng. Sci. 2013, 95, 174–183. [Google Scholar] [CrossRef]
- Wunderlich, B.; Nettels, D.; Benke, S.; Clark, J.; Weidner, S.; Hofmann, H.; Pfeil, S.H.; Schuler, B. Microfluidic mixer designed for performing single-molecule kinetics with confocal detection on timescales from milliseconds to minutes. Nat. Protoc. 2013, 8, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Guo, L.; Huang, T.; Wen, L.; Chen, J. Experimental and CFD studies on the intensified micromixing performance of micro-impinging stream reactors built from commercial T-junctions. Chem. Eng. Sci. 2014, 119, 124–133. [Google Scholar] [CrossRef]
- Woitalka, A.; Kuhn, S.; Jensen, K.F. Scalability of mass transfer in liquid–liquid flow. Chem. Eng. Sci. 2014, 116, 1–8. [Google Scholar] [CrossRef]
- Ghotli, R.A.; Abdul Aziz, A.R.; Ibrahim, S. Liquid-liquid mass transfer studies in various stirred vessel designs. Rev. Chem. Eng. 2015, 31, 329–343. [Google Scholar] [CrossRef]
- Jasińska, M. Test Reactions to Study Efficiency of Mixing. Chem. Eng. Process. 2015, 36, 171–208. [Google Scholar] [CrossRef]
- Wu, K.-J.; Nappo, V.; Kuhn, S. Hydrodynamic Study of Single- and Two-Phase Flow in an Advanced-Flow Reactor. Ind. Eng. Chem. Res. 2015, 54, 7554–7564. [Google Scholar] [CrossRef]
- Luo, J.-Z.; Luo, Y.; Chu, G.-W.; Arowo, M.; Xiang, Y.; Sun, B.-C.; Chen, J.-F. Micromixing efficiency of a novel helical tube reactor: CFD prediction and experimental characterization. Chem. Eng. Sci. 2016, 155, 386–396. [Google Scholar] [CrossRef]
- Plouffe, P.; Bittel, M.; Sieber, J.; Roberge, D.M.; Macchi, A. On the scale-up of micro-reactors for liquid–liquid reactions. Chem. Eng. Sci. 2016, 143, 216–225. [Google Scholar] [CrossRef]
- Gobert, S.R.L.; Kuhn, S.; Braeken, L.; Thomassen, L.C.J. Characterization of Milli- and Microflow Reactors: Mixing Efficiency and Residence Time Distribution. Org. Process Res. Dev. 2017, 21, 531–542. [Google Scholar] [CrossRef]
- Kockmann, N. Transport Phenomena and Chemical Reactions in Modular Microstructured Devices. Heat Transf. Eng. 2017, 38, 1316–1330. [Google Scholar] [CrossRef]
- Rahimi, M.; Azimi, N.; Parsamogadam, M.A.; Rahimi, A.; Masahy, M.M. Mixing performance of T, Y, and oriented Y-micromixers with spatially arranged outlet channel: Evaluation with Villermaux/Dushman test reaction. Microsyst. Technol. 2017, 23, 3117–3130. [Google Scholar] [CrossRef]
- Reckamp, J.M.; Bindels, A.; Duffield, S.; Liu, Y.C.; Bradford, E.; Ricci, E.; Susanne, F.; Rutter, A. Mixing Performance Evaluation for Commercially Available Micromixers Using Villermaux-Dushman Reaction Scheme with the Interaction by Exchange with the Mean Model. Org. Process Res. Dev. 2017, 21, 816–820. [Google Scholar] [CrossRef]
- Zhou, M.; Bai, D.; Zong, Y.; Zhao, L.; Thornock, J.N. Numerical investigation of turbulent reactive mixing in a novel coaxial jet static mixer. Chem. Eng. Process. 2017, 122, 190–203. [Google Scholar] [CrossRef]
- Ansari, M.A.; Kim, K.-Y.; Kim, S.M. Numerical and Experimental Study on Mixing Performances of Simple and Vortex Micro T-Mixers. Micromachines 2018, 9, 204. [Google Scholar] [CrossRef]
- Levesque, F.; Rogus, N.J.; Spencer, G.; Grigorov, P.; McMullen, J.P.; Thaisrivongs, D.A.; Davies, I.W.; Naber, J.R. Advancing Flow Chemistry Portability: A Simplified Approach to Scaling Up Flow Chemistry. Org. Process Res. Dev. 2018, 22, 1015–1021. [Google Scholar] [CrossRef]
- Mariotti, A.; Galletti, C.; Mauri, R.; Salvetti, M.V.; Brunazzi, E. Steady and unsteady regimes in a T-shaped micro-mixer: Synergic experimental and numerical investigation. Chem. Eng. J. 2018, 341, 414–431. [Google Scholar] [CrossRef]
- Rossetti, I. Continuous flow (micro-)reactors for heterogeneously catalyzed reactions: Main design and modelling issues. Catal. Today 2018, 308, 20–31. [Google Scholar] [CrossRef]
- Su, Y.; Song, Y.; Xiang, L. Continuous-Flow Microreactors for Polymer Synthesis: Engineering Principles and Applications. Top. Curr. Chem. 2018, 376, 44. [Google Scholar] [CrossRef]
- Li, W.; Xia, F.; Qin, H.; Zhang, M.; Li, W.; Zhang, J. Numerical and experimental investigations of micromixing performance and efficiency in a pore-array intensified tube-in-tube microchannel reactor. Chem. Eng. J. 2019, 370, 1350–1365. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, N.; Chen, J.; Rodgers, V.G.J.; Brisk, P.; Grover, W.H. Finding the optimal design of a passive microfluidic mixer. Lab Chip 2019, 19, 3618–3627. [Google Scholar] [CrossRef] [PubMed]
- Camarri, S.; Mariotti, A.; Galletti, C.; Brunazzi, E.; Mauri, R.; Salvetti, M.V. An Overview of Flow Features and Mixing in Micro T and Arrow Mixers. Ind. Eng. Chem. Res. 2020, 59, 3669–3686. [Google Scholar] [CrossRef]
- Cheng, D.; Chen, F.-E. Experimental and Numerical Studies of the Phase-Transfer-Catalyzed Wittig Reaction in Liquid-Liquid Slug-Flow Microchannels. Ind. Eng. Chem. Res. 2020, 59, 4397–4410. [Google Scholar] [CrossRef]
- Desir, P.; Chen, T.-Y.; Bracconi, M.; Saha, B.; Maestri, M.; Vlachos, D.G. Experiments and computations of microfluidic liquid-liquid flow patterns. React. Chem. Eng. 2020, 5, 39–50. [Google Scholar] [CrossRef]
- Hosseini, F.; Rahimi, M. Computational fluid dynamics and experimental investigations on liquid-liquid mass transfer in T-type microchannels with different mixing channel barrier shapes. Sep. Sci. Technol. 2020, 55, 3502–3516. [Google Scholar] [CrossRef]
- Mariotti, A.; Antognoli, M.; Galletti, C.; Mauri, R.; Salvetti, M.V.; Brunazzi, E. The role of flow features and chemical kinetics on the reaction yield in a T-shaped micro-reactor. Chem. Eng. J. 2020, 396, 125223. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Li, F.; Li, L.; Ge, X.; Zhang, S.; Qiu, T. Scale-up of microreactor: Effects of hydrodynamic diameter on liquid-liquid flow and mass transfer. Chem. Eng. Sci. 2020, 226, 115838. [Google Scholar] [CrossRef]
- Zhang, H.; Kopfmüller, T.; Achermann, R.; Zhang, J.; Teixeira, A.; Shen, Y.; Jensen, K.F. Accessing multidimensional mixing via 3D printing and showerhead micromixer design. AIChE J. 2020, 66, e16873. [Google Scholar] [CrossRef]
- Gill, K.K.; Gibson, R.; Yiu, K.H.C.; Hester, P.; Reis, N.M. Microcapillary film reactor outperforms single-bore mesocapillary reactors in continuous flow chemical reactions. Chem. Eng. J. 2021, 408, 127860. [Google Scholar] [CrossRef]
- Manzano Martínez, A.N.; Jansen, R.; Walker, K.; Assirelli, M.; van der Schaaf, J. Experimental and modeling study on meso- and micromixing in the rotor–stator spinning disk reactor. Chem. Eng. Res. Des. 2021, 173, 279–288. [Google Scholar] [CrossRef]
- Kravtsova, A.Y.; Ianko, P.E.; Kashkarova, M.V.; Bilsky, A.V.; Kravtsov, Y.V.; Naumov, I.V. Conditions of the Effective Mixing in a T-Type Micromixer at Low Reynolds Numbers. In Processes in GeoMedia—Volume V; Chaplina, T., Ed.; Springer International Publishing: Cham, Switzerland, 2022; Volume 5, pp. 35–41. [Google Scholar] [CrossRef]
- Trabzon, L.; Karimian, G.; Khosroshahi, A.R.; Gül, B.; Bakhshayesh, A.G.; Kocak, A.F.; Akyıldız, D.; Aldi, Y.E. High-throughput nanoscale liposome formation via electrohydrodynamic-based micromixer. Phys. Fluids 2022, 34, 102011. [Google Scholar] [CrossRef]
- Jafarpour, A.M.; Khosroshahi, A.R.; Hanifi, M.; Moghanlou, F.S. Experimental study on the performance of a mini-scale Y-type mixer with two liquid metal-enabled pumps. Phys. Fluids 2022, 34, 112110. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, M.; Zheng, C.; Li, H. Numerical Simulation of Mixing Characteristics in a Split-and-Recombine Microchannel. Heat Transf. Eng. 2023, 44, 1563–1577. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, N.; Liu, H.; Tang, J.; Wang, C.; Wang, R.; Yang, X. Modification of Meso-Micromixing Interaction Reaction Model in Continuous Reactors. Processes 2023, 11, 1576. [Google Scholar] [CrossRef]
- Falk, L.; Commenge, J.M. Performance comparison of micromixers. Chem. Eng. Sci. 2010, 65, 405–411. [Google Scholar] [CrossRef]
- Rehage, H.; Kind, M. The first Damköhler number and its importance for characterizing the influence of mixing on competitive chemical reactions. Chem. Eng. Sci. 2021, 229, 116007. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, K.H.; Fang, W.F.; Tsai, S.H.; Fang, J.M.; Yang, J.T. Flash synthesis of carbohydrate derivatives in chaotic microreactors. Chem. Eng. J. 2011, 174, 421–424. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Fang, W.-F.; Liu, Y.-C.; Yang, J.-T. Analysis of chaos and FRET reaction in split-and-recombine microreactors. Microfluid. Nanofluid. 2011, 11, 339–352. [Google Scholar] [CrossRef]
- Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka, S.I.; Fukase, K. Large-scale synthesis of immunoactivating natural product, pristane, by continuous microfluidic dehydration as the key step. Org. Lett. 2007, 9, 299–302. [Google Scholar] [CrossRef]
- Tanaka, K.; Mori, Y.; Fukase, K. Practical Synthesis of a Manβ(1-4)GlcNTroc Fragment via Microfluidic β-Mannosylation. J. Carbohydr. Chem. 2009, 28, 1–11. [Google Scholar] [CrossRef]
- Uchinashi, Y.; Nagasaki, M.; Zhou, J.; Tanaka, K.; Fukase, K. Reinvestigation of the C5-acetamide sialic acid donor for α-selective sialylation: Practical procedure under microfluidic conditions. Org. Biomol. Chem. 2011, 9, 7243–7248. [Google Scholar] [CrossRef] [PubMed]
- Uchinashi, Y.; Tanaka, K.; Manabe, Y.; Fujimoto, Y.; Fukase, K. Practical and Efficient Method for α-Sialylation with an Azide Sialyl Donor Using a Microreactor. J. Carbohydr. Chem. 2014, 33, 55–67. [Google Scholar] [CrossRef]
- Audiger, L.; Watts, K.; Elmore, S.C.; Robinson, R.I.; Wirth, T. Ritter Reactions in Flow. ChemSusChem 2012, 5, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Graz, M.; Saunders, R.; Evans, G.J.S.; Kaul, S.; Wirth, T. Flow Synthesis of Symmetrical Di- and Trisulfides Using Phase-Transfer Catalysis. J. Flow Chem. 2013, 3, 118–121. [Google Scholar] [CrossRef]
- Pradipta, A.R.; Tsutsui, A.; Ogura, A.; Hanashima, S.; Yamaguchi, Y.; Kurbangalieva, A.; Tanaka, K. Microfluidic Mixing of Polyamine with Acrolein Enables the Detection of the [4 + 4] Polymerization of Intermediary Unsaturated Imines: The Properties of a Cytotoxic 1,5-Diazacyclooctane Hydrogel. Synlett 2014, 25, 2442–2446. [Google Scholar] [CrossRef]
- Umezu, S.; Yoshiiwa, T.; Tokeshi, M.; Shindo, M. Generation of ynolates via reductive lithiation using flow microreactors. Tetrahedron Lett. 2014, 55, 1822–1825. [Google Scholar] [CrossRef]
- Matthew, H.; Thomas, W. Safe use of nitromethane for aldol reactions in flow. J. Flow Chem. 2016, 6, 202–205. [Google Scholar] [CrossRef]
- Nagasaki, M.; Manabe, Y.; Minamoto, N.; Tanaka, K.; Silipo, A.; Molinaro, A.; Fukase, K. Chemical synthesis of a complex-type N-glycan containing a core fucose. J. Org. Chem. 2016, 81, 10600–10616. [Google Scholar] [CrossRef]
- Ogasawara, S.; Hayashi, Y. Multistep Continuous-Flow Synthesis of (–)-Oseltamivir. Synthesis 2017, 49, 424–428. [Google Scholar] [CrossRef]
- Ikawa, T.; Masuda, S.; Akai, S. Microflow Fluorinations of Benzynes: Efficient Synthesis of Fluoroaromatic Compounds. Chem. Pharm. Bull. 2018, 66, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Koge, T.; Katsuragi, S.; Akai, S.; Oishi, T. Flow Synthesis of (R)- and (S)-(E)-1-Iodohexa-1,5-dien-3-ol: Chiral Building Blocks for Natural Product Synthesis. Chem. Lett. 2018, 47, 1116–1118. [Google Scholar] [CrossRef]
- Myachin, I.V.; Orlova, A.V.; Kononov, L.O. Glycosylation in flow: Effect of the flow rate and type of the mixer. Russ. Chem. Bull. 2019, 68, 2126–2129. [Google Scholar] [CrossRef]
- Kondo, M.; Wathsala, H.D.P.; Sako, M.; Hanatani, Y.; Ishikawa, K.; Hara, S.; Takaai, T.; Washio, T.; Takizawa, S.; Sasai, H. Exploration of flow reaction conditions using machine-learning for enantioselective organocatalyzed Rauhut–Currier and [3 + 2] annulation sequence. Chem. Commun. 2020, 56, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Fukumoto, S.; Iwase, T.; Mizota, I.; Shimizu, M.; Hachiya, I. Umpolung Reactions of α-Tosyloximino Esters in a Flow System. Synlett 2020, 31, 1930–1936. [Google Scholar] [CrossRef]
- Myachin, I.V.; Mamirgova, Z.Z.; Stepanova, E.V.; Zinin, A.I.; Chizhov, A.O.; Kononov, L.O. Black swan in phase transfer catalysis: Influence of mixing mode on the stereoselectivity of glycosylation. Eur. J. Org. Chem. 2022, 2022, e202101377. [Google Scholar] [CrossRef]
- Myachin, I.V.; Kononov, L.O. Phase-transfer catalyzed microfluidic glycosylation: A small change in concentration results in a dramatic increase in stereoselectivity. Catalysts 2023, 13, 313. [Google Scholar] [CrossRef]
- Miller, P.W.; Long, N.J.; De Mello, A.J.; Vilar, R.; Passchier, J.; Gee, A. Rapid formation of amides via carbonylative coupling reactions using a microfluidic device. Chem. Commun. 2006, 546–548. [Google Scholar] [CrossRef]
- Webb, D.; Jamison, T.F. Diisobutylaluminum Hydride Reductions Revitalized: A Fast, Robust, and Selective Continuous Flow System for Aldehyde Synthesis. Org. Lett. 2012, 14, 568–571. [Google Scholar] [CrossRef]
- Ganiek, M.A.; Becker, M.R.; Ketels, M.; Knochel, P. Continuous Flow Magnesiation or Zincation of Acrylonitriles, Acrylates, and Nitroolefins. Application to the Synthesis of Butenolides. Org. Lett. 2016, 18, 828–831. [Google Scholar] [CrossRef]
- Hafner, A.; Meisenbach, M.; Sedelmeier, J. Flow Chemistry on Multigram Scale: Continuous Synthesis of Boronic Acids within 1 s. Org. Lett. 2016, 18, 3630–3633. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, A.; Tomita, R.; Kurimoto, K.; Yamamura, H. Selective deprotection of trityl group on carbohydrate by microflow reaction inhibiting migration of acetyl group. Synth. Commun. 2016, 46, 556–562. [Google Scholar] [CrossRef]
- Naber, J.R.; Buchwald, S.L. Packed-Bed Reactors for Continuous-Flow C–N Cross-Coupling. Angew. Chem. Int. Ed. 2010, 49, 9469–9474. [Google Scholar] [CrossRef] [PubMed]
- Haroun, S.; Sanei, Z.; Jivan, S.; Schaffer, P.; Ruth, T.J.; Li, P.C.H. Continuous-flow synthesis of [11C]raclopride, a positron emission tomography radiotracer, on a microfluidic chip. Can. J. Chem. 2013, 91, 326–332. [Google Scholar] [CrossRef]
- Park, C.P.; Kim, D.-P. Dual-Channel Microreactor for Gas–Liquid Syntheses. J. Am. Chem. Soc. 2010, 132, 10102–10106. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J.I. Control of extremely fast competitive consecutive reactions using micromixing. Selective Friedel-Crafts aminoalkylation. J. Am. Chem. Soc. 2005, 127, 11666–11675. [Google Scholar] [CrossRef] [PubMed]
- Rys, P. Disguised chemical selectivities. Acc. Chem. Res. 1976, 9, 345–351. [Google Scholar] [CrossRef]
- Roy, R.; Tropper, F.D.; Cao, S.; Kim, J.M. Anomeric Group Transformations Under Phase-Transfer Catalysis. In Phase-Transfer Catalysis; Halpern, M., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 659, pp. 163–180. [Google Scholar] [CrossRef]
- Boons, G.J.; Demchenko, A.V. Recent advances in O-sialylation. Chem. Rev. 2000, 100, 4539–4566. [Google Scholar] [CrossRef]
- De Meo, C.; Jones, B.T. Chapter two—Chemical synthesis of glycosides of N-acetylneuraminic acid. Adv. Carbohydr. Chem. Biochem. 2018, 75, 215–316. [Google Scholar] [CrossRef]
- De Meo, C.; Goeckner, N. 2.07—Synthesis of glycosides of sialic acid. In Comprehensive Glycoscience, 2nd ed.; Barchi, J.J., Vidal, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 2, pp. 228–266. [Google Scholar] [CrossRef]
- Sedlák, M. Large-scale supramolecular structure in solutions of low molar mass compounds and mixtures of liquids: I. Light scattering characterization. J. Phys. Chem. B 2006, 110, 4329–4338. [Google Scholar] [CrossRef]
- Sedlák, M. Large-scale supramolecular structure in solutions of low molar mass compounds and mixtures of liquids: II. Kinetics of the formation and long-time stability. J. Phys. Chem. B 2006, 110, 4339–4345. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, M. Large-scale supramolecular structure in solutions of low molar mass compounds and mixtures of liquids. III. Correlation with molecular properties and interactions. J. Phys. Chem. B 2006, 110, 13976–13984. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, M.; Rak, D. Large-Scale Inhomogeneities in Solutions of Low Molar Mass Compounds and Mixtures of Liquids: Supramolecular Structures or Nanobubbles? J. Phys. Chem. B 2013, 117, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, M.; Rak, D. On the origin of mesoscale structures in aqueous solutions of tertiary butyl alcohol: The mystery resolved. J. Phys. Chem. B 2014, 118, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- Rak, D.; Ovadová, M.; Sedlák, M. (Non)existence of bulk nanobubbles: The role of ultrasonic cavitation and organic solutes in water. J. Phys. Chem. Lett. 2019, 10, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Rak, D.; Sedlák, M. On the mesoscale solubility in liquid solutions and mixtures. J. Phys. Chem. B 2019, 123, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Rak, D.; Sedlák, M. Solvophobicity-Driven Mesoscale Structures: Stabilizer-Free Nanodispersions. Langmuir 2023, 39, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.; Anisimov, M.A. Phase behavior and mesoscale solubilization in aqueous solutions of hydrotropes. Fluid Phase Equilib. 2014, 362, 170–176. [Google Scholar] [CrossRef]
- Zemb, T.; Kunz, W. Weak aggregation: State of the art, expectations and open questions. Curr. Opin. Colloid Interface Sci. 2016, 22, 113–119. [Google Scholar] [CrossRef]
- Orlova, A.V.; Laptinskaya, T.V.; Malysheva, N.N.; Kononov, L.O. Light scattering in non-aqueous solutions of low-molecular-mass compounds: Application for supramer analysis of reaction solutions. J. Solution Chem. 2020, 49, 629–644. [Google Scholar] [CrossRef]
- Orlova, A.V.; Kononov, L.O. Polarimetry as a method for studying the structure of aqueous carbohydrate solutions: Correlation with other methods. RENSIT 2020, 12, 95–106. [Google Scholar] [CrossRef]
- Orlova, A.V.; Ahiadorme, D.A.; Laptinskaya, T.V.; Kononov, L.O. Supramer analysis of 2,3,5-tri-O-benzoyl-α-D-arabinofuranosyl bromide solutions in different solvents: Supramolecular aggregation of solute molecules in 1,2-dichloroethane mediated by halogen bonds. Russ. Chem. Bull. 2021, 70, 2214–2219. [Google Scholar] [CrossRef]
- Jelley, E.E. Spectral Absorption and Fluorescence of Dyes in the Molecular State. Nature 1936, 138, 1009–1010. [Google Scholar] [CrossRef]
- Jelley, E.E. Molecular, Nematic and Crystal States of I: I-Diethyl-Cyanine Chloride. Nature 1937, 139, 631. [Google Scholar] [CrossRef]
- Scheibe, G. Über die Veränderlichkeit der Absorptionsspektren in Lösungen und die Nebenvalenzen als ihre Ursache. Angew. Chem. 1937, 50, 212–219. [Google Scholar] [CrossRef]
- Mei, J.; Hong, Y.; Lam, J.W.Y.; Qin, A.; Tang, Y.; Tang, B.Z. Aggregation-Induced Emission: The Whole Is More Brilliant than the Parts. Adv. Mater. 2014, 26, 5429–5479. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.; Ankri, R. Theoretical Models, Preparation, Characterization and Applications of Cyanine J-Aggregates: A Minireview. ChemistryOpen 2022, 11, e202200103. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Shuai, Z. Molecular mechanism of aggregation-induced emission. Aggregate 2021, 2, e91. [Google Scholar] [CrossRef]
- Ribo, J.M.; El-Hachemi, Z.; Arteaga, O.; Canillas, A.; Crusats, J. Hydrodynamic Effects in Soft-matter Self-assembly: The Case of J-Aggregates of Amphiphilic Porphyrins. Chem. Rec. 2017, 17, 713–724. [Google Scholar] [CrossRef]
- Maeda, T.; Mori, T.; Ikeshita, M.; Ma, S.C.; Muller, G.; Ariga, K.; Naota, T. Vortex Flow-controlled Circularly Polarized Luminescence of Achiral Pt(II) Complex Aggregates Assembled at the Air-Water Interface. Small Methods 2022, 6, 2200936. [Google Scholar] [CrossRef]
- Adero, P.O.; Amarasekara, H.; Wen, P.; Bohé, L.; Crich, D. The experimental evidence in support of glycosylation mechanisms at the SN1–SN2 interface. Chem. Rev. 2018, 118, 8242–8284. [Google Scholar] [CrossRef] [PubMed]
- Crich, D. En route to the transformation of glycoscience: A chemist’s perspective on internal and external crossroads in glycochemistry. J. Am. Chem. Soc. 2021, 143, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Ötvös, S.B.; Mándity, I.M.; Fülöp, F. Highly efficient 1,4-addition of aldehydes to nitroolefins: Organocatalysis in continuous flow by solid-supported peptidic catalysts. ChemSusChem 2012, 5, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.; Jamison, T.F. A Continuous Homologation of Esters: An Efficient Telescoped Reduction-Olefination Sequence. Org. Lett. 2012, 14, 2465–2467. [Google Scholar] [CrossRef] [PubMed]
- Andreana, P.R.; Crich, D. Guidelines for O-glycoside formation from first principles. ACS Cent. Sci. 2021, 7, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Ribó, J.M.; Crusats, J.; Sagués, F.; Claret, J.; Rubires, R. Chiral Sign Induction by Vortices During the Formation of Mesophases in Stirred Solutions. Science 2001, 292, 2063–2066. [Google Scholar] [CrossRef] [PubMed]
- Crusats, J.; El-Hachemi, Z.; Ribó, J.M. Hydrodynamic effects on chiral induction. Chem. Soc. Rev. 2010, 39, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Yang, D.; Duan, P.; Liu, M. Towards homochiral supramolecular entities from achiral molecules by vortex mixing-accompanied self-assembly. Chem. Sci. 2019, 10, 2718–2724. [Google Scholar] [CrossRef]
- Nagornaya, M.O.; Orlova, A.V.; Stepanova, E.V.; Zinin, A.I.; Laptinskaya, T.V.; Kononov, L.O. The use of the novel glycosyl acceptor and supramer analysis in the synthesis of sialyl-α(2–3)-galactose building block. Carbohydr. Res. 2018, 470, 27–35. [Google Scholar] [CrossRef]
- Penverne, C.; Hazard, B.; Rolando, C.; Penhoat, M. Scale-up Study of Benzoic Acid Alkylation in Flow: From Microflow Capillary Reactor to a Milliflow Reactor. Org. Process Res. Dev. 2017, 21, 1864–1868. [Google Scholar] [CrossRef]
- Nagaki, A.; Kenmoku, A.; Moriwaki, Y.; Hayashi, A.; Yoshida, J.-i. Cross-Coupling in a Flow Microreactor: Space Integration of Lithiation and Murahashi Coupling. Angew. Chem. Int. Ed. 2010, 49, 7543–7547. [Google Scholar] [CrossRef]
- Tomida, Y.; Nagaki, A.; Yoshida, J.-i. Asymmetric Carbolithiation of Conjugated Enynes: A Flow Microreactor Enables the Use of Configurationally Unstable Intermediates before They Epimerize. J. Am. Chem. Soc. 2011, 133, 3744–3747. [Google Scholar] [CrossRef]
- Nagaki, A.; Kim, H.; Yoshida, J.-i. Nitro-Substituted Aryl Lithium Compounds in Microreactor Synthesis: Switch between Kinetic and Thermodynamic Control. Angew. Chem. Int. Ed. 2009, 48, 8063–8065. [Google Scholar] [CrossRef]
Figure 1.
Summary of the steady flow regimes and mixing quality (δ) as a function of the Reynolds number (Re) in T-shaped and arrow-shaped micromixers. Adapted with permission from [79].
Figure 1.
Summary of the steady flow regimes and mixing quality (δ) as a function of the Reynolds number (Re) in T-shaped and arrow-shaped micromixers. Adapted with permission from [79].
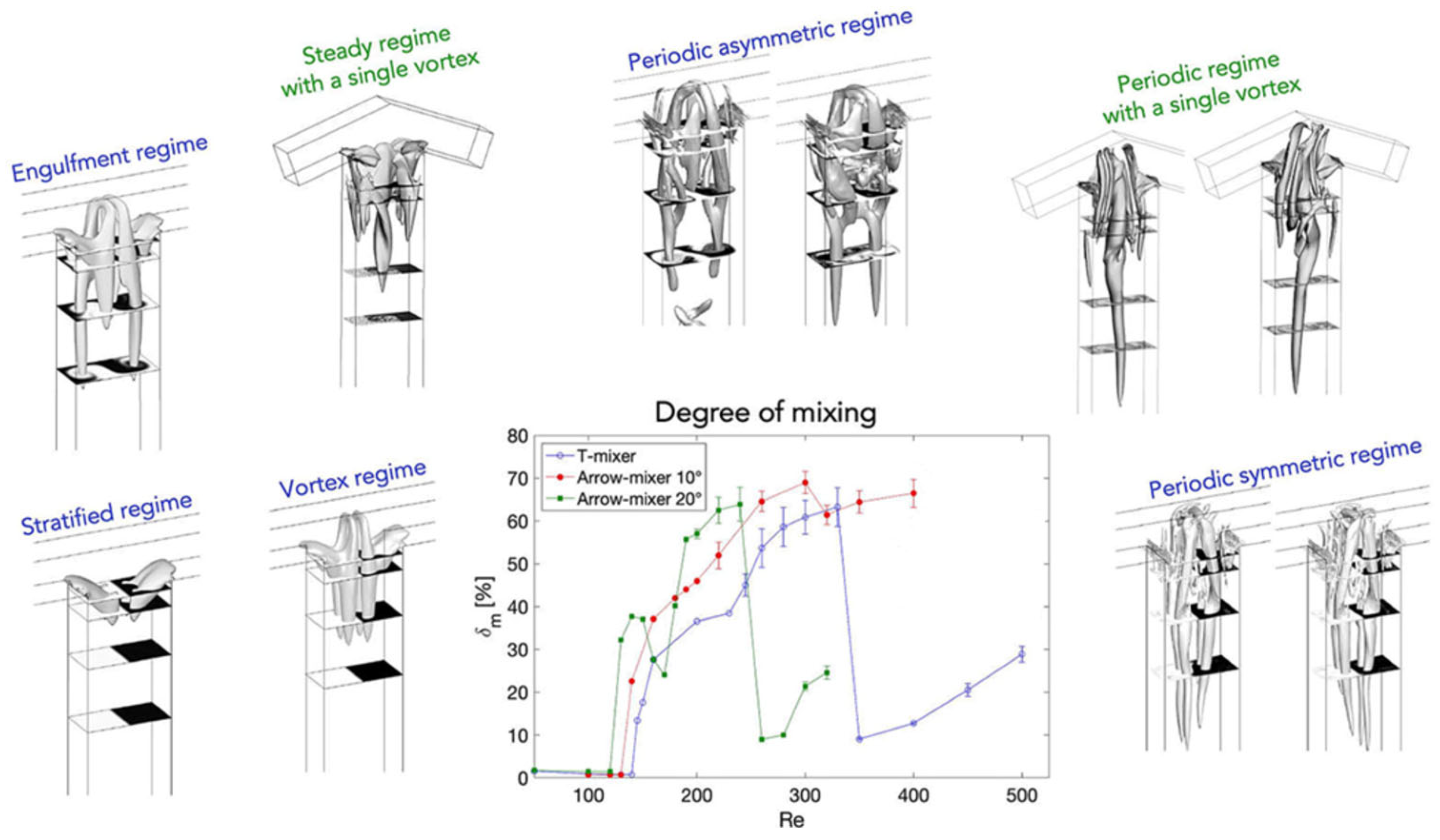
Figure 2.
Streamlines inside a T-shaped micromixer (channel widths 300 and 600 μm) at different mass flows. View is into the mixing channel (left (red) and right (blue) are the inlet channels). Reproduced from [43] with permission from Elsevier.
Figure 2.
Streamlines inside a T-shaped micromixer (channel widths 300 and 600 μm) at different mass flows. View is into the mixing channel (left (red) and right (blue) are the inlet channels). Reproduced from [43] with permission from Elsevier.
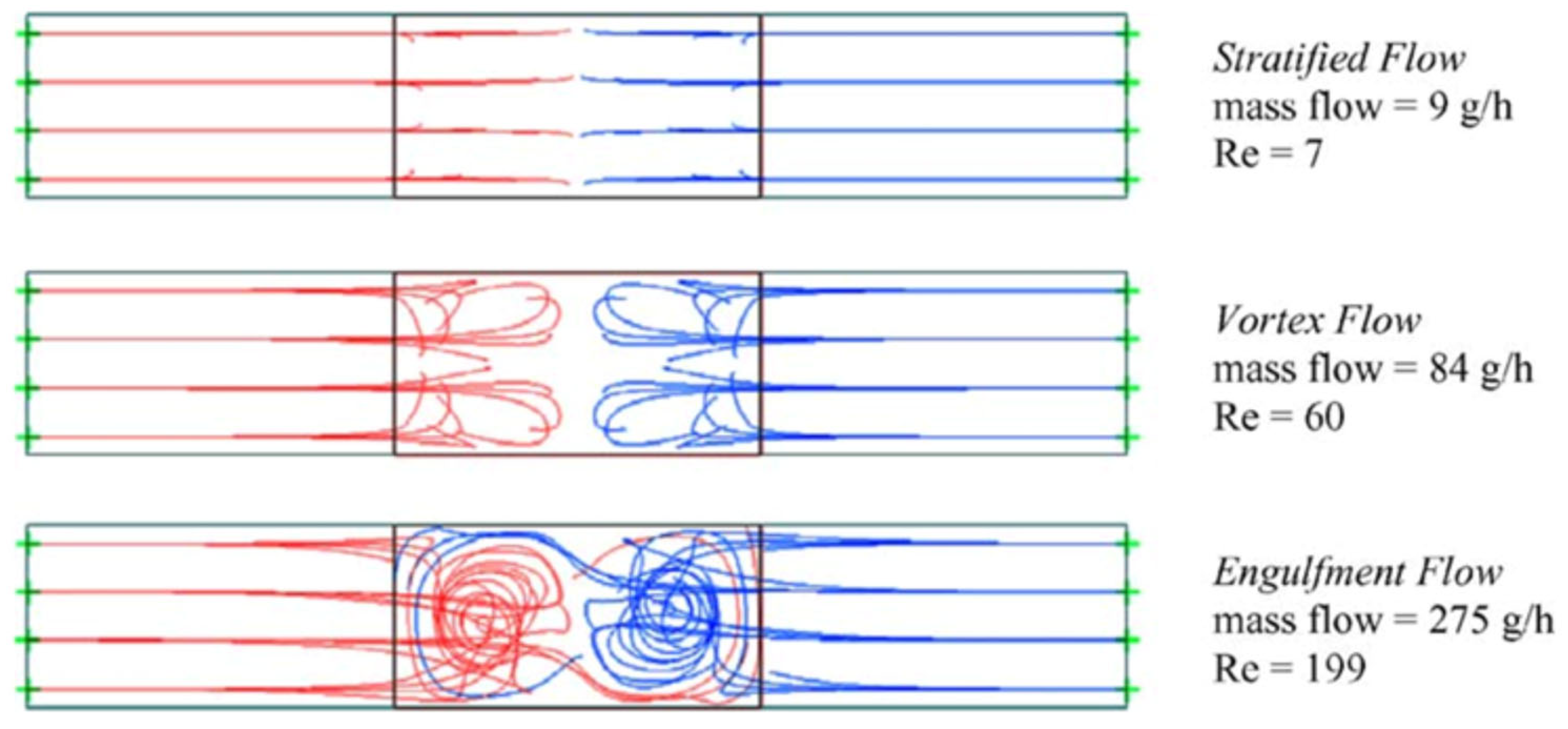
Figure 3.
Mixing quality (α) over the Reynolds number (Re) for a T-shaped micromixer. Reproduced from [43] with permission from Elsevier.
Figure 3.
Mixing quality (α) over the Reynolds number (Re) for a T-shaped micromixer. Reproduced from [43] with permission from Elsevier.
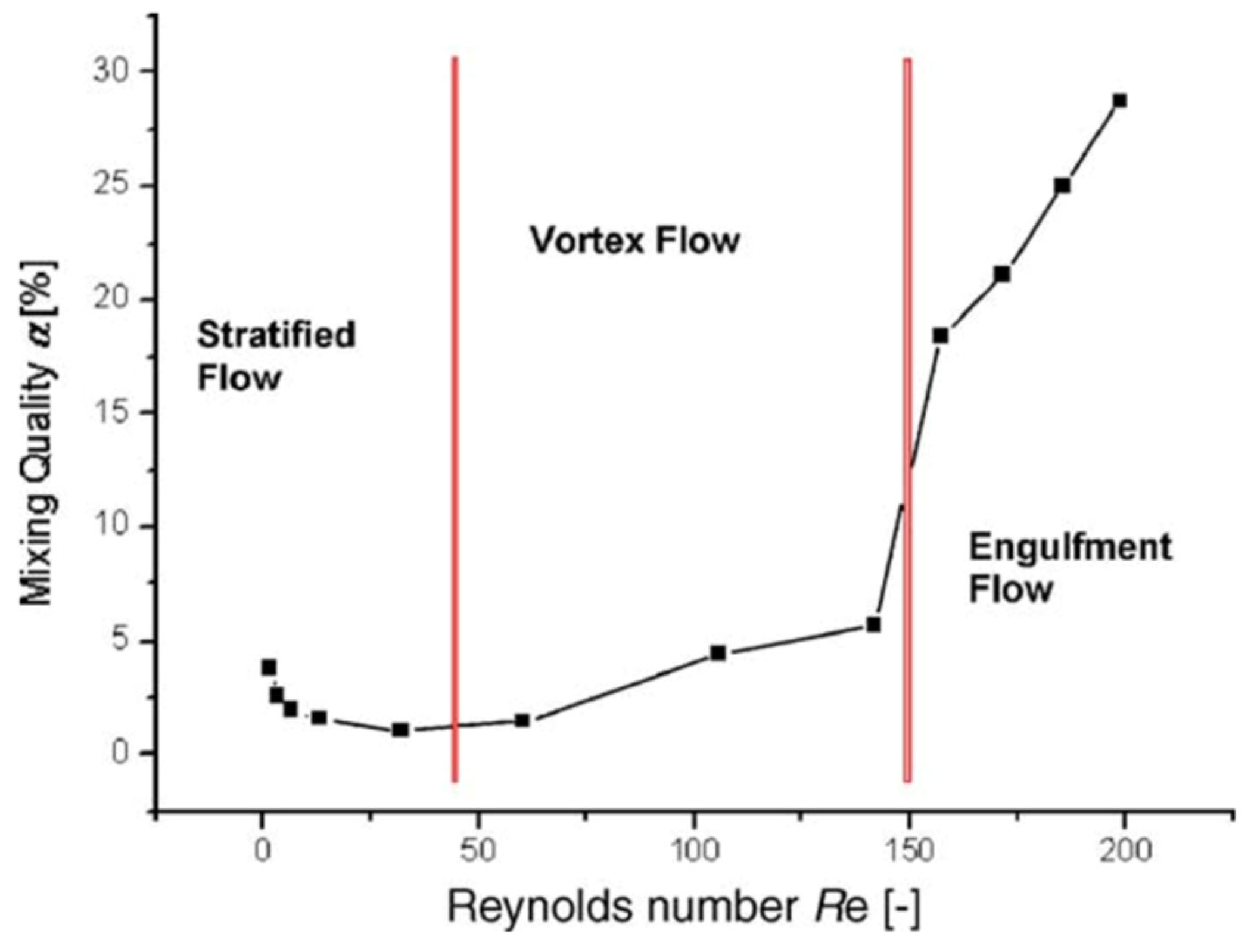
Figure 4.
The Pe–Re diagram. The Reynolds number (Re) describes the flow regime in the mixing channel (Re is a convenient parameter for predicting whether a flow regime will be laminar or turbulent), while the Péclet number (Pe) represents the ratio between convection/advection and diffusion (when Pe << 1, then diffusive effects dominate over convective/advective transport, whilst the opposite is generally true for Pe >> 1). Adapted with permission from [29]. © IOP Publishing. All rights reserved.
Figure 4.
The Pe–Re diagram. The Reynolds number (Re) describes the flow regime in the mixing channel (Re is a convenient parameter for predicting whether a flow regime will be laminar or turbulent), while the Péclet number (Pe) represents the ratio between convection/advection and diffusion (when Pe << 1, then diffusive effects dominate over convective/advective transport, whilst the opposite is generally true for Pe >> 1). Adapted with permission from [29]. © IOP Publishing. All rights reserved.
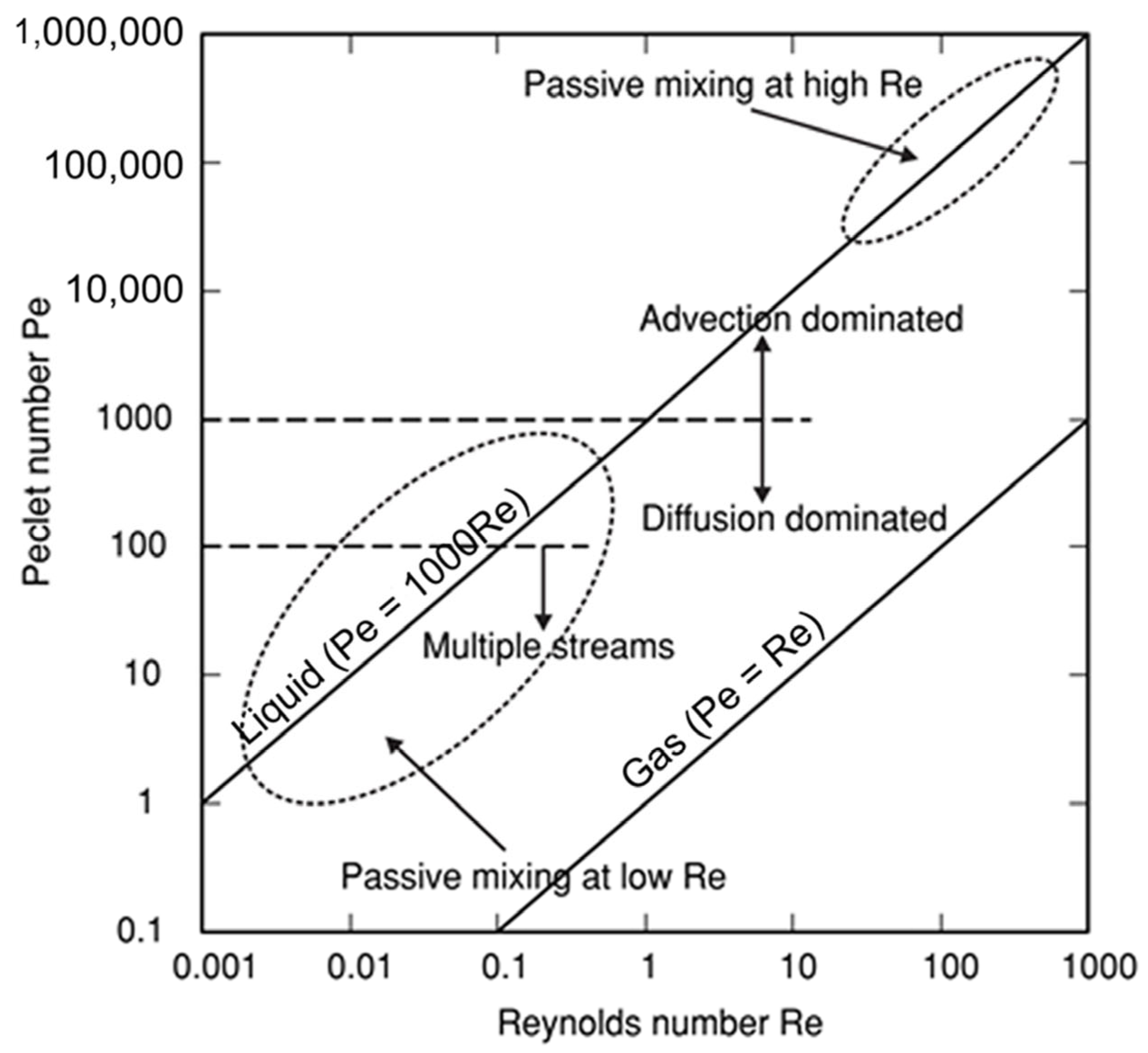
Figure 5.
Flow reactor, based on Comet X-01 micromixer, used for glycosylation experiments. S1, S2—syringes. Internal volume of Comet X-01 micromixer—0.1 mL, that of outlet capillary—0.785 mL. Reproduced with permission from [114].
Figure 5.
Flow reactor, based on Comet X-01 micromixer, used for glycosylation experiments. S1, S2—syringes. Internal volume of Comet X-01 micromixer—0.1 mL, that of outlet capillary—0.785 mL. Reproduced with permission from [114].
Scheme 1.
Palladium-catalyzed C–N cross-coupling reaction performed in flow with packed bed and open tube reactor (see Figure 6) [120].
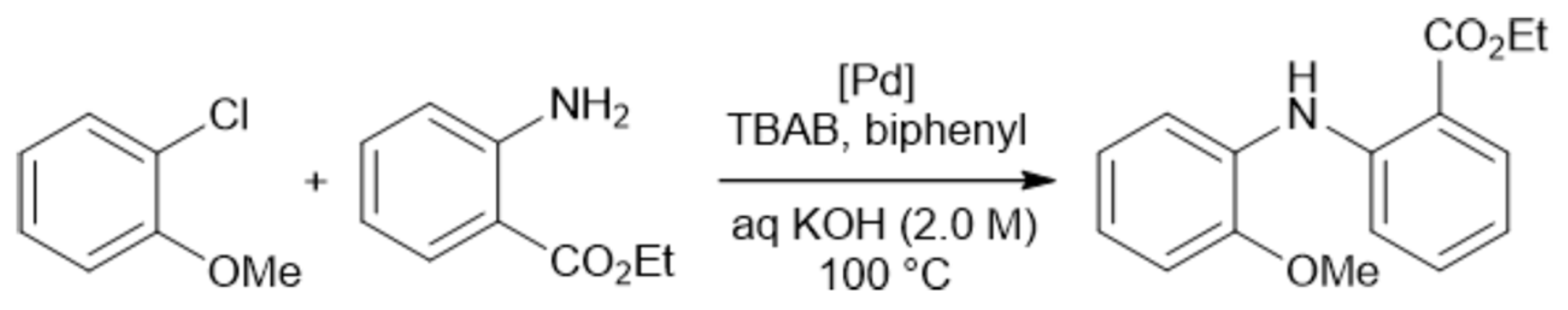
Figure 6.
An example of the crucial role of mixing efficiency: the reaction (see Scheme 1) proceeds in high yield only when packed bed capillary is used for mixing reagents. Time indicated designates the residence time. Adapted with permission from [120]. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 6.
An example of the crucial role of mixing efficiency: the reaction (see Scheme 1) proceeds in high yield only when packed bed capillary is used for mixing reagents. Time indicated designates the residence time. Adapted with permission from [120]. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.

Scheme 2.
C–N cross-coupling palladium-catalyzed reaction performed in flow with various packed bed volumes (see Figure 7) [120].

Figure 7.
Comparison of the packed beds differing in size: the reaction (see Scheme 2) proceeds in high yield if the large packed bed capillary is used for the reaction proceeds (see also Figure 6). Time indicated designates the residence time. Adapted with permission from [120]. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 7.
Comparison of the packed beds differing in size: the reaction (see Scheme 2) proceeds in high yield if the large packed bed capillary is used for the reaction proceeds (see also Figure 6). Time indicated designates the residence time. Adapted with permission from [120]. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8.
Three micromixers used for performing alkylation reaction (see Scheme 3) featured by different mixing efficiency. (a)—simple channel micromixer without additional loops (“no loop”); (b)—abacus-like micromixer with some additional loops (“abacus”); (c)—micromixer with maximum additional loops (“full loop”). Adapted with permission from [121].
Figure 8.
Three micromixers used for performing alkylation reaction (see Scheme 3) featured by different mixing efficiency. (a)—simple channel micromixer without additional loops (“no loop”); (b)—abacus-like micromixer with some additional loops (“abacus”); (c)—micromixer with maximum additional loops (“full loop”). Adapted with permission from [121].
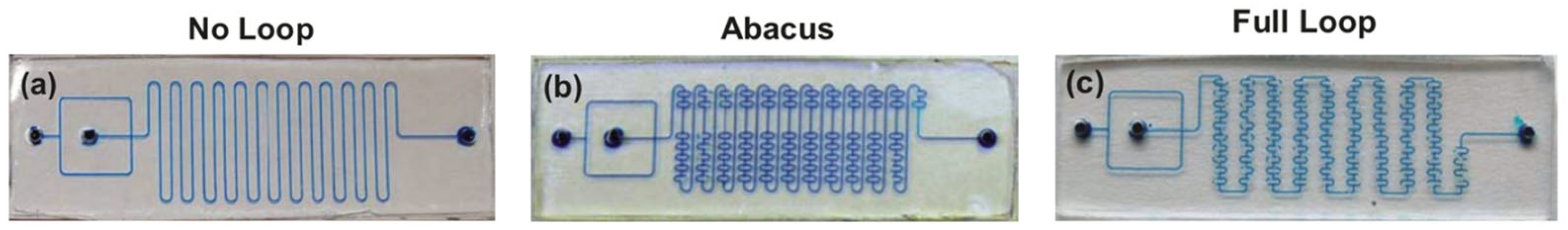
Figure 9.
Mixer design for performing oxidation reaction under flow conditions. (a)—illustration of various modes of contact area between gas and liquid phases; (b)—schematic illustration of the dual-channel system. Adapted with permission from [122].
Figure 9.
Mixer design for performing oxidation reaction under flow conditions. (a)—illustration of various modes of contact area between gas and liquid phases; (b)—schematic illustration of the dual-channel system. Adapted with permission from [122].
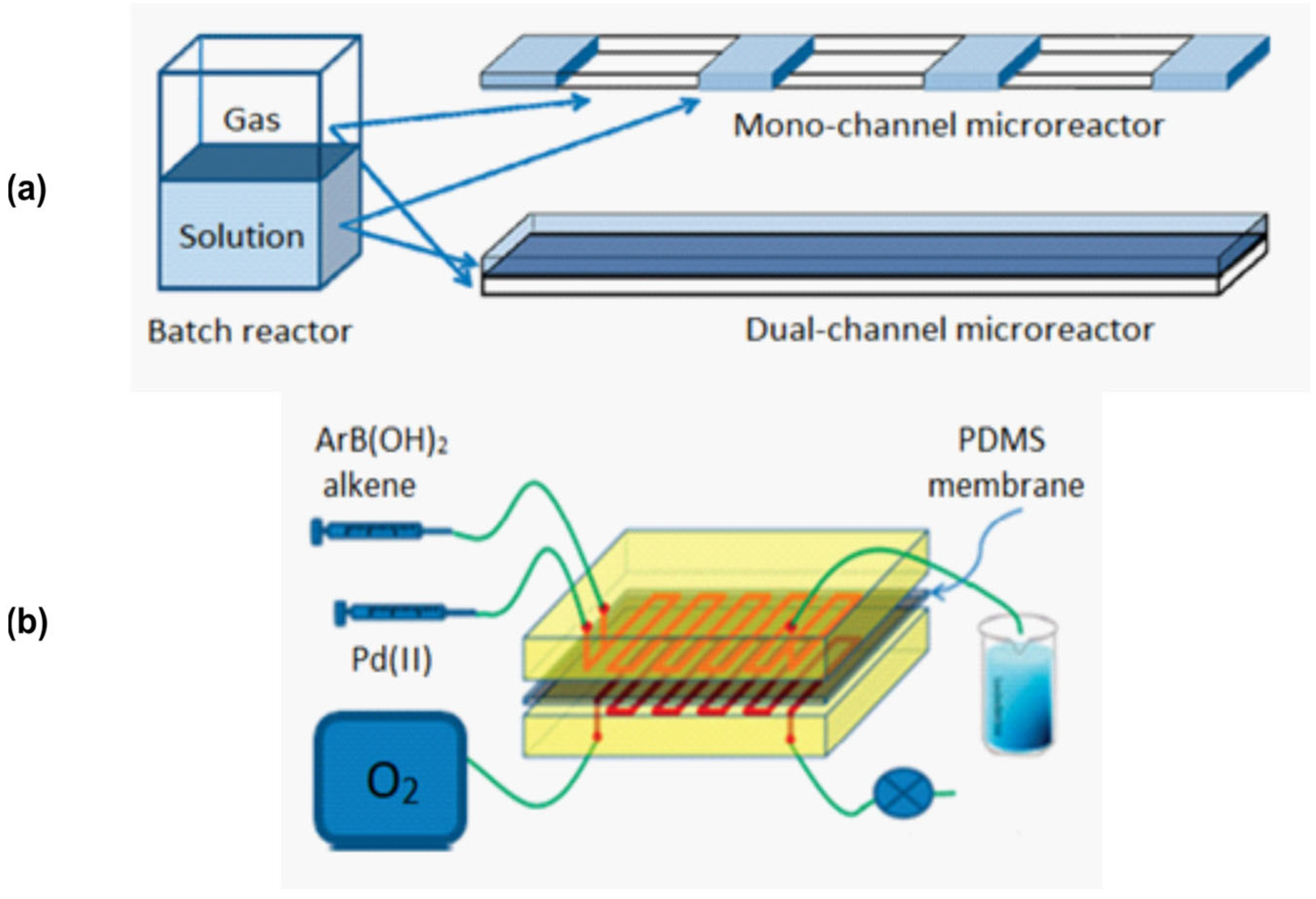
Figure 10.
Investigation of mixing performance for the metalation step as a function of flow rate of aryl bromide (1) and n-BuLi: (a)—schematic illustration of the flow system. (b)—the yield of 3 as a function of the total flow rate. Adapted with permission from [118]. Copyright 2016 American Chemical Society.
Figure 10.
Investigation of mixing performance for the metalation step as a function of flow rate of aryl bromide (1) and n-BuLi: (a)—schematic illustration of the flow system. (b)—the yield of 3 as a function of the total flow rate. Adapted with permission from [118]. Copyright 2016 American Chemical Society.

Scheme 4.
Friedel–Crafts reaction performed in flow using three types of micromixers: T-shaped, YM-1, and IMM [123].
Scheme 4.
Friedel–Crafts reaction performed in flow using three types of micromixers: T-shaped, YM-1, and IMM [123].
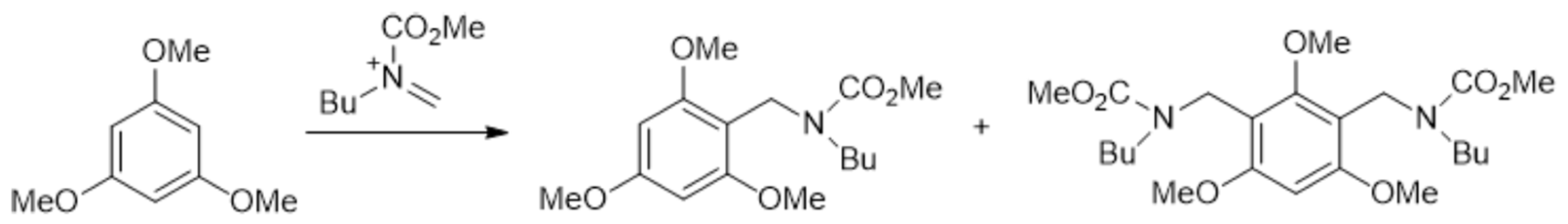
Figure 11.
A model [124] of the course of a liquid two-step reaction (like in Scheme 4) where diffusion of reagents is main limitation of reaction rate. A, B—initial reagents, P1—monosubstituted product, P2—disubstituted product. Adapted with permission from [123].
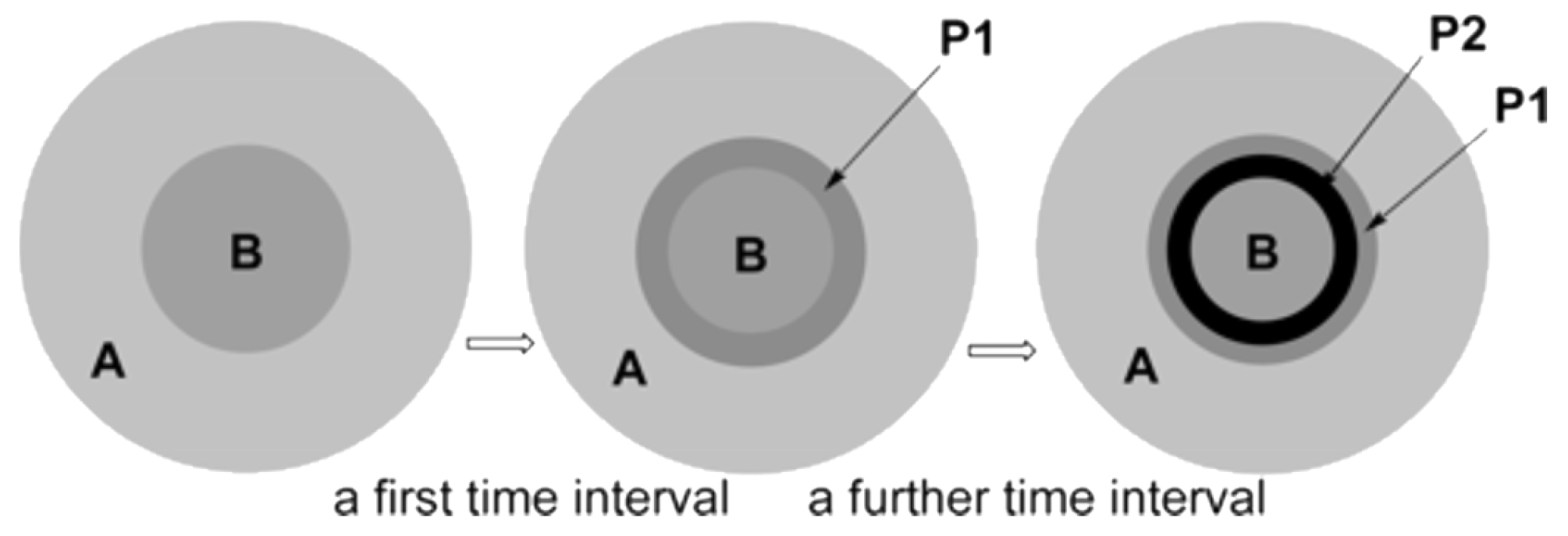
Scheme 5.
Sialylation reaction under phase transfer conditions performed in flow. CPP-OH—4-(3-chloropropoxy)phenol [113,114].

Figure 12.
Presentation of glycosyl cation on supramer surface: (a,b)—α- and β-selective reactions, respectively, (c)—nonselective reaction, (d)—no reaction. Adapted with permission from [114].
Figure 12.
Presentation of glycosyl cation on supramer surface: (a,b)—α- and β-selective reactions, respectively, (c)—nonselective reaction, (d)—no reaction. Adapted with permission from [114].
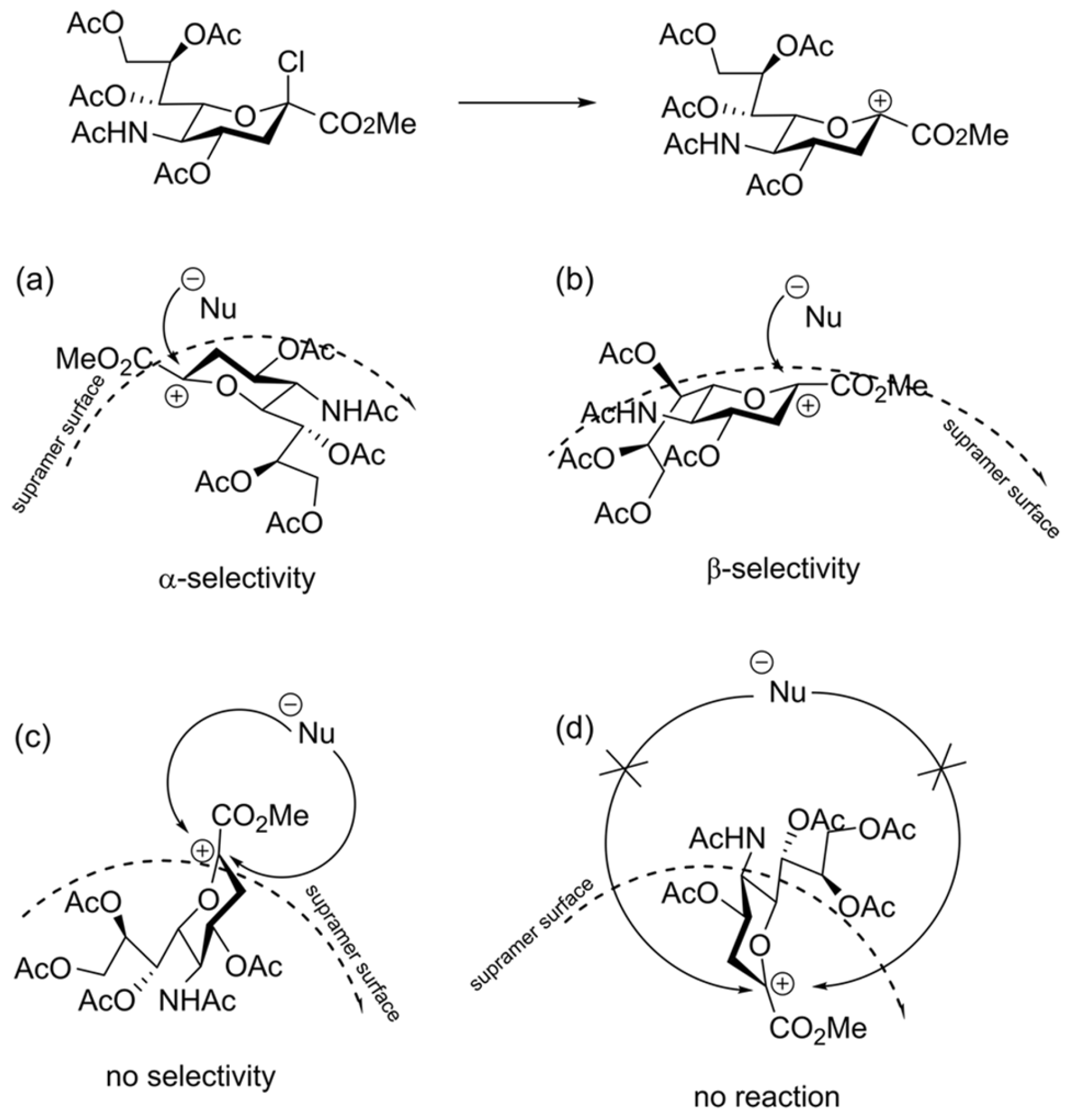
Figure 13.
Reagent molecules inside solution domains (supramers) [R1] and [R2] cannot react with each other. Efficient destruction of domains is required for all molecules of reagents R1 and R2 to react—a clue for increasing reactivity of reagents and achieving high yield of product P. Supramers may react on their surfaces, in this case asymmetric environment of the surface molecules (presentation, see Figure 12) may lead to stereoselective reactions. Reproduced from [27] with permission from the Royal Society of Chemistry.
Figure 13.
Reagent molecules inside solution domains (supramers) [R1] and [R2] cannot react with each other. Efficient destruction of domains is required for all molecules of reagents R1 and R2 to react—a clue for increasing reactivity of reagents and achieving high yield of product P. Supramers may react on their surfaces, in this case asymmetric environment of the surface molecules (presentation, see Figure 12) may lead to stereoselective reactions. Reproduced from [27] with permission from the Royal Society of Chemistry.
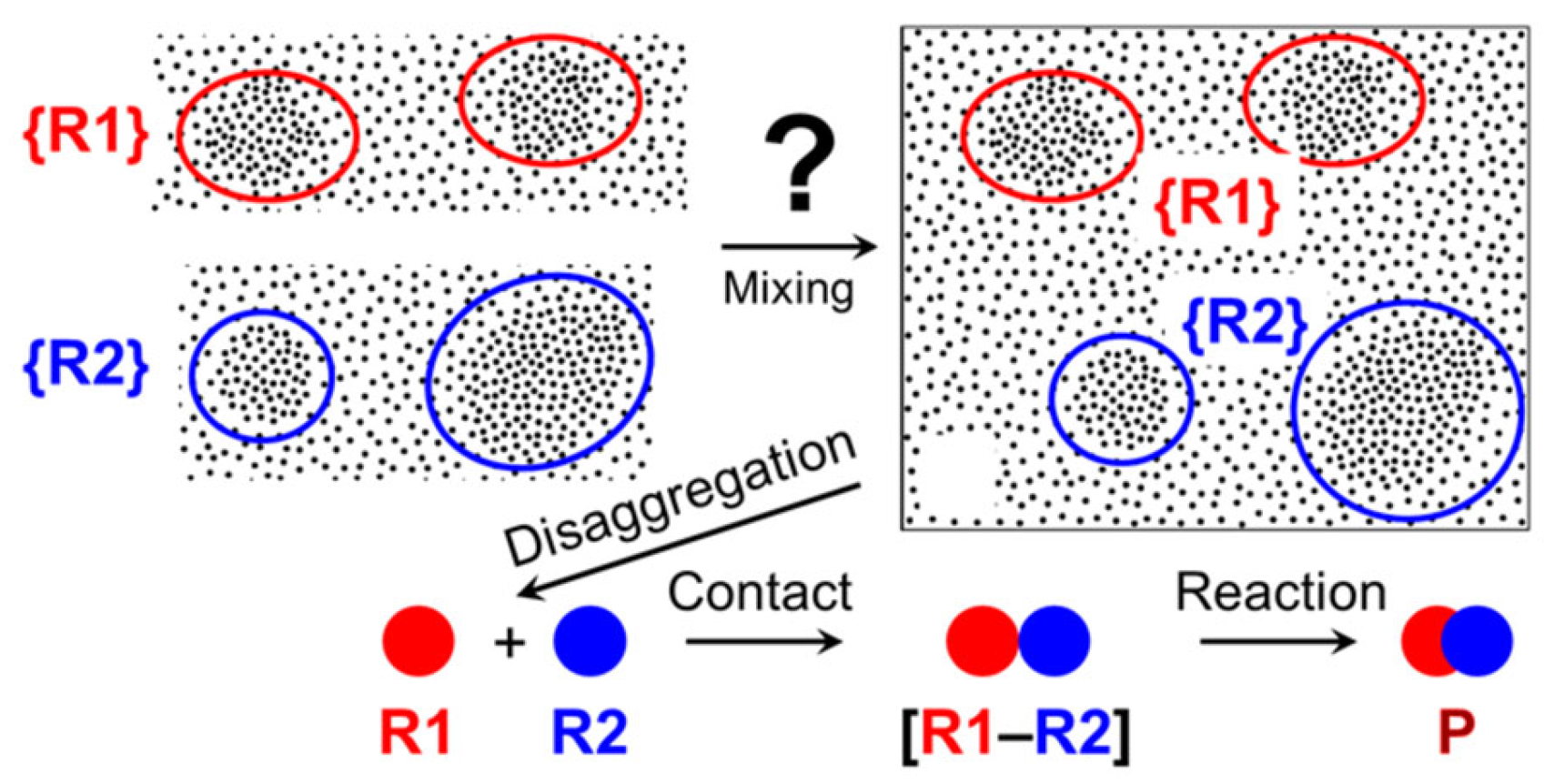
Figure 14.
A model of supramers of sialyl chloride in the reaction mixture. In batch reactor the supramers are visualized as a whole intact “apple” (left panel). The spatial orientation of molecules of sialyl chloride in such supramers (presentation, see Figure 12) allows glycosylation from only one face within SN2-like pathway leading to formation of only α-isomer. During “efficient mixing” of reaction mixture in Comet X-01 micromixer (M) the “apple” is chopped allowing the molecules of sialyl chloride to show both faces on the edge of the apple allowing SN1-like mechanism which leads to formation of both α- and β-isomers (right panel). Reproduced with permission from [113]. Copyright © 2022 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 14.
A model of supramers of sialyl chloride in the reaction mixture. In batch reactor the supramers are visualized as a whole intact “apple” (left panel). The spatial orientation of molecules of sialyl chloride in such supramers (presentation, see Figure 12) allows glycosylation from only one face within SN2-like pathway leading to formation of only α-isomer. During “efficient mixing” of reaction mixture in Comet X-01 micromixer (M) the “apple” is chopped allowing the molecules of sialyl chloride to show both faces on the edge of the apple allowing SN1-like mechanism which leads to formation of both α- and β-isomers (right panel). Reproduced with permission from [113]. Copyright © 2022 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.

Scheme 6.
Glycosylation reaction of isopropyl alcohol by glyco-oxazoline. CSA—camphorsulfonic acid.

Figure 15.
Formation of products in glycosylation reaction performed in flow at different flow rates (Scheme 6) using Comet X-01 micromixer. Relative amounts (ω) of products: dashed line—isopropyl glycoside, solid line—glycal (side product). f—flow rate, mL/h. No reaction was observed at low flow rates (≤43 μL/h). Adapted with permission from [110]. Copyright (2019), Springer Nature.
Figure 15.
Formation of products in glycosylation reaction performed in flow at different flow rates (Scheme 6) using Comet X-01 micromixer. Relative amounts (ω) of products: dashed line—isopropyl glycoside, solid line—glycal (side product). f—flow rate, mL/h. No reaction was observed at low flow rates (≤43 μL/h). Adapted with permission from [110]. Copyright (2019), Springer Nature.

Figure 16.
Observation of the change in flow regime during alkylation of benzoic acid by CH3I in DMF in the presence of DBU in the Low Flow reactor. Reproduced with permission from [159].
Figure 16.
Observation of the change in flow regime during alkylation of benzoic acid by CH3I in DMF in the presence of DBU in the Low Flow reactor. Reproduced with permission from [159].
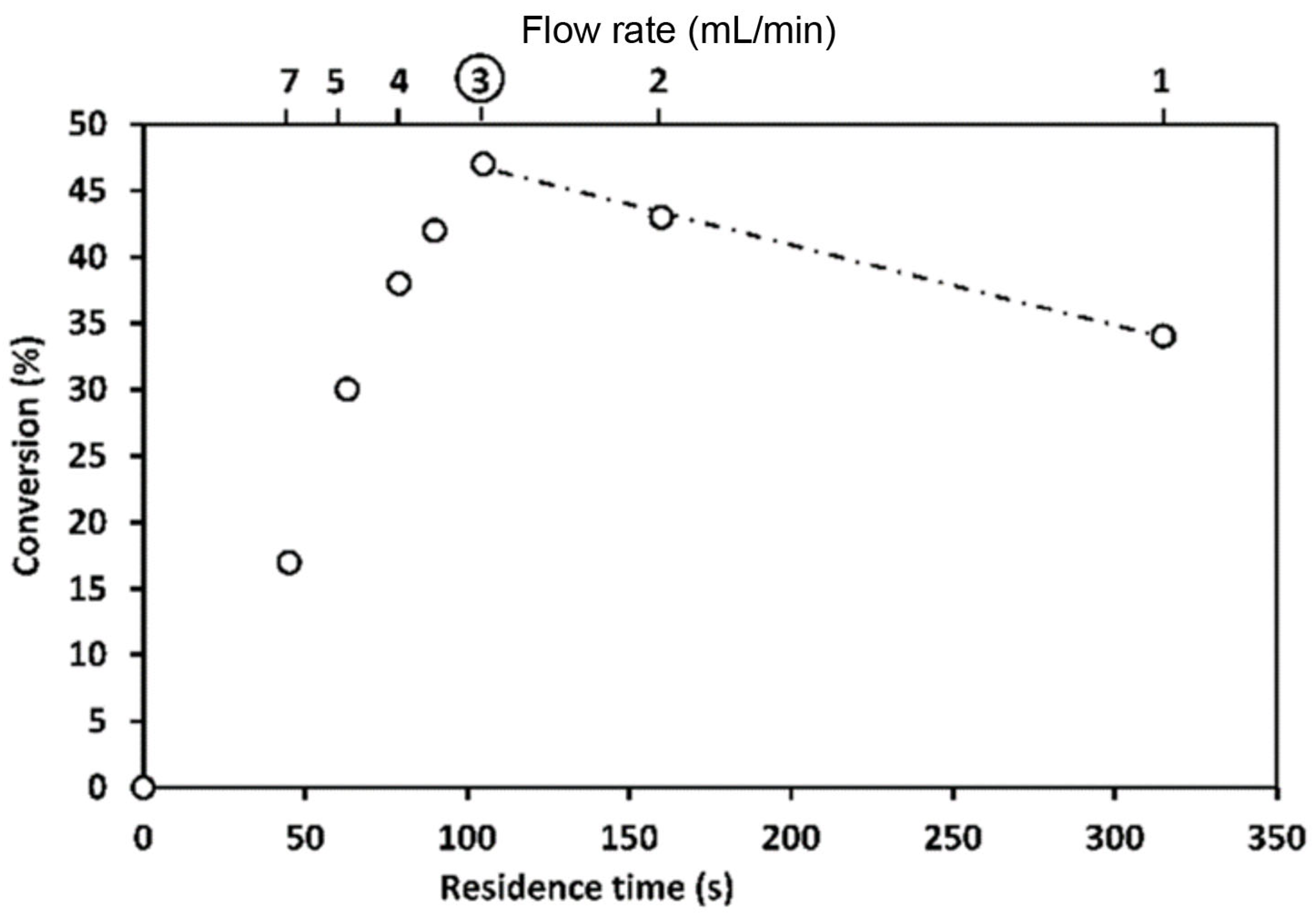
Figure 17.
A switch between kinetic and thermodynamic control by changing the residence time of the Br-Li exchange reaction followed by the reaction of the lithiated species with PriCHO as electrophile. ◆—5-substitued kinetically formed product, ■—3-substitued thermodynamically preferred product, ▲ —initial aromatic reagent. Reproduced with permission from [162]. Copyright © 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 17.
A switch between kinetic and thermodynamic control by changing the residence time of the Br-Li exchange reaction followed by the reaction of the lithiated species with PriCHO as electrophile. ◆—5-substitued kinetically formed product, ■—3-substitued thermodynamically preferred product, ▲ —initial aromatic reagent. Reproduced with permission from [162]. Copyright © 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
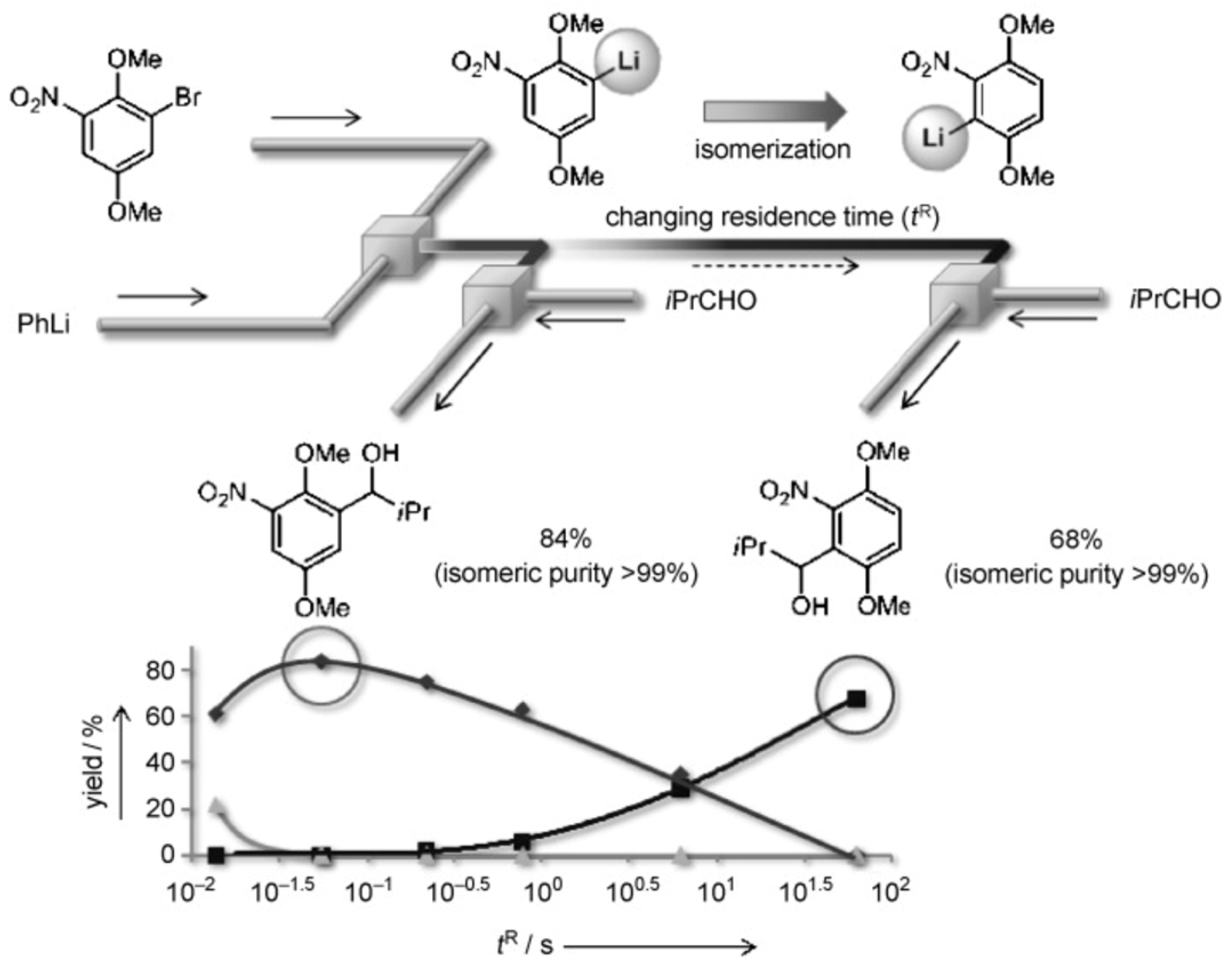
Figure 18.
Dependence of the yield on the residence time of the liquid-liquid reaction at 50 °C (●) and 20 °C (■) in the microreactor. Reproduced with permission from [40]. Copyright © 2001 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 18.
Dependence of the yield on the residence time of the liquid-liquid reaction at 50 °C (●) and 20 °C (■) in the microreactor. Reproduced with permission from [40]. Copyright © 2001 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
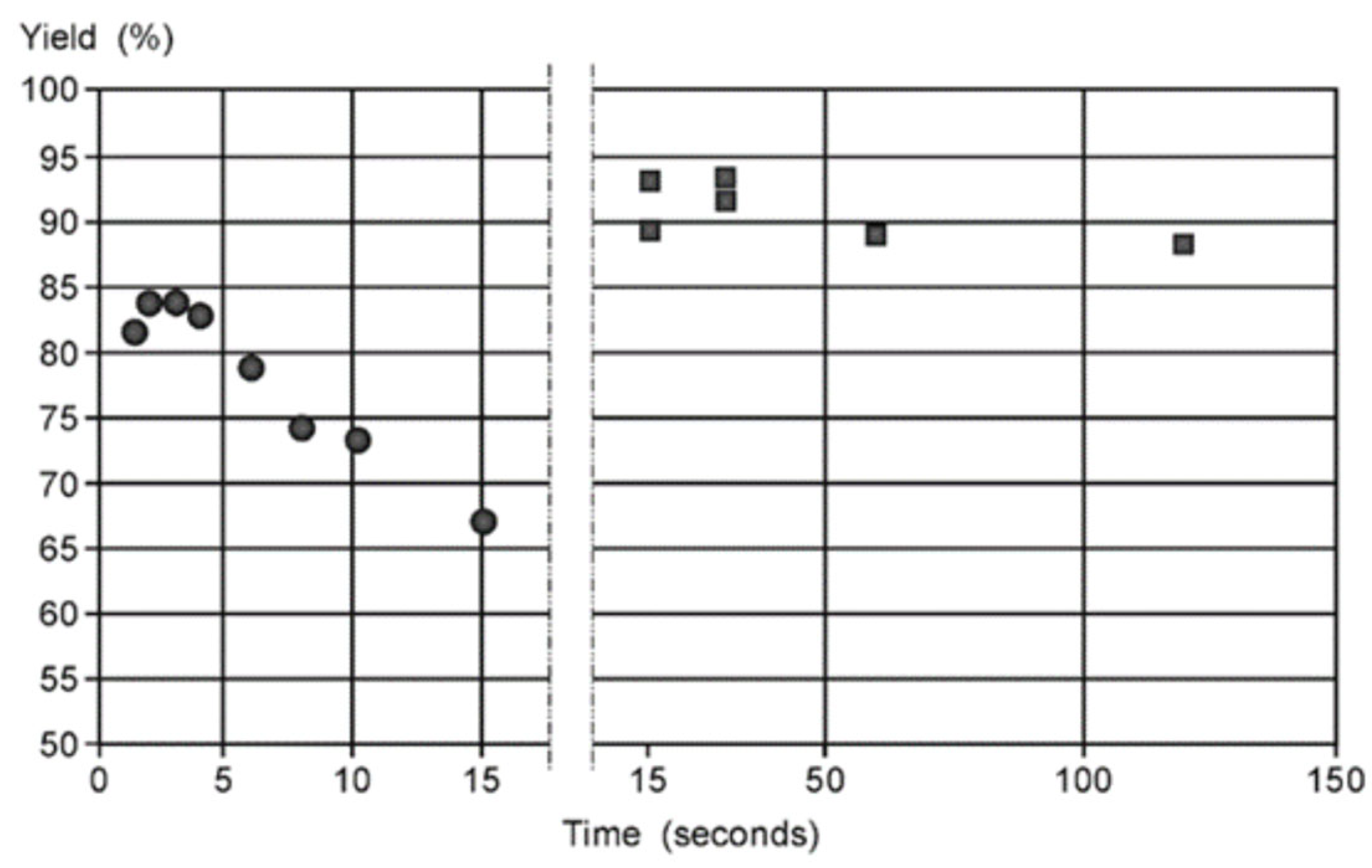
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Myachin, I.V.; Kononov, L.O. Mixer Design and Flow Rate as Critical Variables in Flow Chemistry Affecting the Outcome of a Chemical Reaction: A Review. Inventions 2023, 8, 128. https://doi.org/10.3390/inventions8050128
AMA Style
Myachin IV, Kononov LO. Mixer Design and Flow Rate as Critical Variables in Flow Chemistry Affecting the Outcome of a Chemical Reaction: A Review. Inventions. 2023; 8(5):128. https://doi.org/10.3390/inventions8050128
Chicago/Turabian StyleMyachin, Ilya V., and Leonid O. Kononov. 2023. "Mixer Design and Flow Rate as Critical Variables in Flow Chemistry Affecting the Outcome of a Chemical Reaction: A Review" Inventions 8, no. 5: 128. https://doi.org/10.3390/inventions8050128