
Progress in Studies of Disentangled Polymers and Composites

Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, 92-414 Lodz, Poland
*
Author to whom correspondence should be addressed.
J. Compos. Sci. 2023, 7(12), 521; https://doi.org/10.3390/jcs7120521
Submission received: 14 November 2023
/
Revised: 6 December 2023
/
Accepted: 14 December 2023
/
Published: 18 December 2023
(This article belongs to the Special Issue Progress in Polymer Composites, Volume III)
Abstract
:Macromolecule entanglements are common in polymers. The first part of this review describes their influence on the properties of entangled polymers. Then, methods for reducing the entanglement density of macromolecule chains are discussed. It has been shown that research on partially disentangled polymers has provided a lot of new information about the relationship between the entangled state and properties of polymers. This research concerns, among others, mechanical and thermal properties and the crystallization process. A special disentangled polymer case, ultra-high-molecular-weight polyethylene, is also discussed. The results of research on polymer composites in which macromolecules were disentangled via processing and composites were produced using already disentangled polymers are presented in particular detail. It has been indicated that such composites and blends of disentangled polymers are promising and will probably be intensively researched in the near future.
1. Entanglements in Polymers
The chains of macromolecules with carbon skeletons are flexible. The isolated chain easily takes the shape of a coil [1]. When a macromolecule is surrounded by other macromolecules, its coils interpenetrate, and entanglements arise between these macromolecules. The condition of their occurrence is that their molecular weight exceeds a certain limit. Entanglement can be defined as an obstacle to the movement of a macromolecule’s chain caused by the presence of another macromolecule. These obstacles may be topological (these are common) or cohesive.
Most often, topological entanglements take the form of a loop of one molecule around another. Cohesive entanglements are defined as the interactions of neighboring chain fragments, for example, through hydrogen bonds [2]. In this review, we focused on topological entanglements. These entanglements occur in concentrated polymer solutions, polymer blends, and also in the amorphous phase of the solid polymer [3]. Because these entanglements restrict the movement of macromolecules, they are thought to be responsible for properties that depend on macromolecular mobility. These include rheology in the melt or solution (polymer flow), mechanical properties (deformation of polymer network under the influence of force), crystallization (transport of macromolecules to growing crystals), and diffusion behaviors [4].
The presence of macromolecule entanglements was discovered during studies of the rheology of polymers in the molten state and the deformation of elastomers in the solid state. Earlier studies by Berry and Fox [5] showed that the zero shear viscosity of a low-molecular-weight polymer is proportional to the molecular weight, M, i.e., , whereas, for higher molecular weights, when entanglements are formed, this relationship changes to . In the polymer literature, it is customary to use the term “molecular weight” when the term “molecular mass” is more correct, as indicated by the unitary unit g/mol.
Elastic materials, such as rubbers or the amorphous phase in polymers above the glass transition temperature, exhibit large deformations and strong elastic responses, explained by the stretching of macromolecules forming a network of entanglements. The parameter characterizing the density of entanglements in a polymer is the molecular mass between entanglements, Me, usually determined from the rheology in the melt or the mechanical properties in the solid state.
According to the classical theory of rubber elasticity [6], the equilibrium shear modulus is equal to
where is the density of the rubber, R is the gas constant, T is the temperature, and Mr is the molecular mass between the network nodes [7,8,9]. Assuming that a similar approach is correct for polymers in the melt, the appropriate equation has the form of
where GN0 is the shear modulus in the plateau region, is the density of the polymer, and g is a coefficient equal to 1 [10] or 0.8 [11].
GN0 can be determined by measuring linear viscoelastic properties in an oscillatory shear experiment, performed over a wide range of frequencies and temperatures. GN0 is the value of the storage modulus, G′, at the frequency ωmin, in which the loss modulus, G″, reaches its minimum [12] (Figure 1). Since this minimum is not always visible, a number of methods have been developed to circumvent this limitation [12,13,14,15].
Any selected high-molecular-weight polymer is assumed to have an equilibrium entanglement density proportional to 1/Me. This density depends mainly on the architecture of the macromolecular chain. Calculations of the monodisperses polybutadiene, polyisoprene, and polystyrene have shown that Me is independent of Mw and is also independent of polydispersity (i.e., the Mw/Mn ratio) [12]. However, tests of mechanical properties (described below) show that, in semi-crystalline polymers, Me may depend on Mw. Me depends on the tacticity of the macromolecule [13,16]. The results obtained in many laboratories vary significantly, suggesting that the above statements are not generally true or that other factors are at play. For example, the measured Me values are as follows: polyethylene (PE), 830–2600 g/mol; polylactide (PLA), 4000–10,500 g/mol; isotactic polypropylene (PP), 5500–8100 g/mol. Me values for other polymers can be found in the literature [17,18,19]. Typically, entanglements are measured for bulk polymers. Wang et al. [20] showed that, in ultrathin polystyrene (PS) films, the entanglement density is lower than in bulk samples, and the increase in Me begins when the film thickness is below 3.3 gyration radii, i.e., 13–28 nm.
Much effort has been devoted to the theoretical description and modeling of chain motion confined by the presence of other macromolecules [11,21,22,23,24]. De Gennes [23] came up with the idea that the chain movement inside the entangled polymer could be described as snake-like, which is known as the reptation concept. To quantitatively describe the topological constraints caused by chain entanglement, Doi and Edwards [25] proposed the tube model. In the simplest version of this model, the polymer chain moves like a snake inside a virtual tube, the diameter of which depends on the constraints imposed by other macromolecules (Figure 2). The movement of the macromolecule perpendicular to the tube is restricted by the tube wall, but its motion along the tube is free. The time of diffusion through the tube is a parameter characterizing the dynamics of the chain [21,26]. The reptation model provides an interpretation of the observed dependence of viscosity on molecular weight. The improvement of the initial reptation model, taking into account contour length fluctuations and constraint release, has led to progress in approaching the experimental value, η0~M3.4, instead of the value predicted by the original model, η0~M3 [11,22,27,28]. Recently, Ransom et al. [29], while studying polyisobutylene, suggested that the rheology of polymers with a high number of entanglements per macromolecule (e.g., 430) is closer to the η0~M3 relationship predicted by the original tube theory than to that observed for the lower Mw polymer relationship of η0~M3.4.
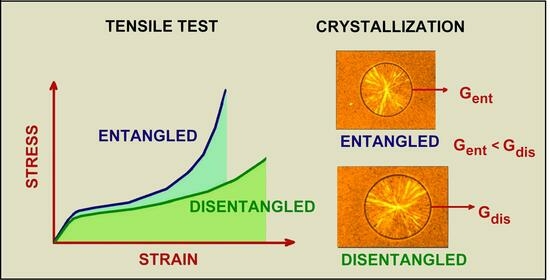
Many properties of polymers depend on the presence of entanglements. Entanglements determine the mechanical properties of polymers [30]. In amorphous polymers, the macromolecular network is responsible for maintaining the continuity of material under the action of forces. The entanglements act as physical cross-links, equivalent to chemical cross-links in rubber [31]. The reasons for the mechanical properties of semi-crystalline polymers are more complicated. It is assumed that entanglements and tie molecules are responsible for the connections of lamellar crystals [32]. When deforming such a polymer, both phases, i.e., crystalline and amorphous, participate in the deformation stages in different ways. When the polymer is ductile, the interconnection of crystals caused by macromolecules causes lamella fragmentation, occurring beyond the yield point, which results in the orientation of the amorphous phase and the formation of fibrillar crystals.
In semi-crystalline polymers, the role of entanglements is best visible in the last phase of tensile deformation, during so-called strain hardening. After exceeding the yield point, the change in true stress, σ, due to longitudinal stretch, λ, can be described by the following equation [33]:
where σy is the yield stress, N is the number of entanglements per unit volume, and n is the number of flexible units between entanglements. When n is large or λ is small, Equation (3) reduces to the Gaussian equation:
where Gp is the strain-hardening modulus [30], proportional to the entanglement density [34]. The Gp measurement is an alternative possibility for calculating Me for semi-crystalline polymers, using an equation analogous to Equation (1) [33]:
σ = σy + λ(NkTn0.5)/3[λ(3 − λ2/n)/(1 − λ2/n) − 1/λ2(3 − 1/λn)(1 − 1/λn)]
Experimental studies of the influence of the macromolecular network on deformation [35,36,37] have shown that strain hardening occurs earlier and is stronger as the density of chain entanglements in the amorphous component increases. Kennedy et al. [35] performed a tensile test on a number of differently crystallized polyethylenes and concluded that the entanglement density in the interlamellar region increases with molecular weight (Figure 3). Similar observations were presented by Schrauwen et al. [37] for poly(ethylene terephthalate) (PET), polyethylene, and polypropylene. Slowly crystallized samples exhibited lower strain hardening, which resulted from lower entangling caused by reeling in the molecular chains during the crystallization process. Bartczak et al. [36], examining polyethylene with Mw in a range of 5 × 104 to 5 × 106 g/mol, found that Me decreases from 1020 to 414 g/mol with increasing Mw. This dependence results from the two-phase structure of the semicrystalline polymer, but it does not apply to fully amorphous polymer, where Me should be independent of Mw. When using Equations (3)–(5) to describe mechanical properties, it is usually assumed that the density of entanglements is constant. However, according to Bartczak et al. [38], during plastic deformation, some disentangling of the chains occurs, which is the reason for the non-reversibility of strains. The entanglement network affects not only the shape of the stress-strain relationship but also the micro-mechanisms of deformation, such as crazing and/or shear yielding, and the failure of polymers [39,40,41].
The presence of macromolecular entanglements influences the crystallization process in at least two aspects: nucleation and the transport of macromolecules to growing crystals. Macromolecules diffuse through the environment from other macromolecules to the crystallization site. The rate of diffusion is controlled by obstacles in the path of the macromolecular chain, including entanglements [42]. The fragment of the chain adjacent to the crystal surface must be disentangled because, in the crystal structure, the macromolecules are ordered and do not have entanglements. It is debatable whether this means local disentangling or just pushing the entanglements some distance from the surface [36,43,44]. For rapidly crystallized higher-molecular-weight polymers, the second option is preferred [45,46].
Another issue is whether the length of the formed stem, equal to the thickness of the crystal, depends on the distance between entanglements, i.e., the length of the untangled fragment capable of crystallization. Crystallization can be accelerated when the polymer melt is sheared, which is known as flow-induced crystallization [47]. The orientation and stretching of macromolecules during shear cause some disentangling of the chains and thus accelerate the crystallization kinetics [48].
The entanglement density of miscible blends can be determined using the same rheological procedure as for homopolymers. The entanglement in the blend depends on the composition and entanglement density of each of the components [49,50]. It is usually assumed that, in immiscible blends and polymer composites, the level of entanglements is the same as in the polymers that compose them.
The properties of blends in terms of the influence of entanglements were examined by Song et al. [49] and Hao et al. [51]. In their opinion, dissimilar chains entangle more often than similar chains. Song et al. [52] studied the deformation of polyphenylene oxide/polystyrene (PPO/PS) blends toughened by HIPS (high-impact polystyrene) and PB-g-PS (polybutadiene-graft-polystyrene) copolymer. They assumed that the blends had “natural”, i.e., equilibrium, density of entanglements proportional to the composition. It was observed that the impact strength and elongation at the break gradually increased with the entanglement density of the matrix chains. The deformation mechanism changed from multiple crazing, through crazing together with shear yielding, to the shear yielding only for the highest entanglement density. Xie et al. [53], after blending ultra-high-molecular-weight polyethylene (UHMWPE) with PP and poly(ethylene glycol) (PEO), attained a significant reduction in melt viscosity. The entanglement density of the UHMWPE/PP (80/20) blend calculated from the storage modulus at a rubbery plateau was lower than that of UHMWPE. A further reduction in the modulus and the entanglement density of the blend was attained by adding PEO.
The accumulated knowledge about the interpenetration of macromolecular chains is impressive. However, all the conclusions of early research have been drawn from experiments on polymers with equilibrium entanglement densities. In order to obtain more information confirming or modifying the existing background, it has been necessary to perform studies on polymers with non-equilibrium-entangled macromolecular chains. This is the main reason for developing methods of disentangling polymers.
2. Disentangling of Polymers
It is difficult to find a way to increase equilibrium entanglement, but it is possible to reduce entanglement. Three approaches are proposed for disentangling macromolecules: during polymerization, via dissolving, and via shear in the melt. The effectiveness of each of these methods depends on its characteristic parameters, such as temperature, the molecular mass of the polymer, or the type of solvent. Roles are described for each method. Typically, when the literature mentions “disentangling”, the authors mean only a partial, not complete, reduction in entanglements. A standard confirmation of reduced contacts between macromolecules is a change in the rheological modulus and viscosity.
Entanglements can be reduced during polymerization [54,55]. For this purpose, the freshly polymerized macromolecules should immediately form crystals, thus limiting the amorphous phase content and the possibility of entanglement. In traditional Ziegler–Natta (Z-N) polymerization, the polymerization rate is usually faster than the crystallization rate; the active sites are close to each other, so adjacent chains easily form entanglements. In homogeneous synthesis, the metallocene catalyst and co-catalyst are dispersed in the polymerization medium, and it may be possible to control the polymerization and crystallization rates and the separation of polymerization sites. As a result, a disentangled polymer can be obtained [56,57,58]. The reactor powder is usually called nascent. Polymers that disentangle during the polymerization process include PE, PP, and poly(tetrafluoroethylene)(PTFE). The disadvantages of this method are low output and a limitation on synthesizable polymers [59].
The most common disentangling method used In laboratories is dissolving the polymer, followed by freezing and drying [60,61,62,63,64,65,66,67]. This is called the freeze-drying method. In a concentrated solution, the chains of macromolecules are still entangled, but as the concentration decreases, the so-called critical concentration, c*, is reached, below which, the macromolecular coils are separated. The critical overlap concentration is usually less than a few percent. If the dissolved polymer is quickly frozen using liquid nitrogen, and the solvent is then removed, for example, via sublimation, a solid polymer with limited entanglements can be obtained. Instead of freezing, disentanglement stabilization can be achieved via crystallization in solution. A long list of polymers disentangled using the solvent method can be found in Pawlak’s review [19]. Unfortunately, freeze-drying diluted polymer solutions is not suitable for large-scale production given the small amount of material obtained in a single process [68] and the harmfulness of the solvent to the environment.
Recently, much attention has been paid to the development of a method of disentangling polymer via shearing. It has been known for many years that the application of shear to the polymer melt causes the partial orientation of macromolecules, which is responsible, among other things, for the faster crystallization of the polymer. However, the nature of the process is often not recognized or discussed in terms of the disentangling necessary to achieve the high orientation of macromolecules. During processing, the orientation and disentangling of the chains lead to the rheological phenomenon of shear-thinning, i.e., a reduction in steady-state viscosity. This happens when the shear rate exceeds the reciprocal of the linear relaxation time [68].
The shearing of molten polymer can be performed using laboratory equipment. Examples are shearing stages or rheometers, in which the polymer is placed between parallel plates, one of which rotates or oscillates in a controlled manner. Shearing changes the rheological properties of the polymer, which allows the effectiveness of the method to be assessed. One variant of shear application is large amplitude oscillatory shear (LAOS) flow, which can be realized in a plate–plate configuration for the rheometer. Examples of test conditions used for PP are as follows: strain amplitude, 50–100%; frequency, 1 Hz; T = 180 °C; time, 200 s [59,69]. The disentanglement of polymer using LAOS can be controlled by adjusting the shear conditions. For example, a larger amplitude and a longer time promote disentangling. LAOS flow produces a less disentangled melt state with respect to steady-shear flow, which is another disentangling possibility. In a steady-shear experiment, the shear rate and time are constant. For example, Liu et al. [70] used a steady-state shear of 5 s−1 for 360 s to disentangle linear PP. LAOS, as an experimental method, simulates the large and rapid deformations experienced by polymers during industrial processing and subsequent use well [71].
The steady-state shear has been compared with another variant of shear using rheometer, a ramping-up shear, which is a deformation with an increasing shear rate during an experiment. A comparison of the steady-state shear at a rate of 5 s−1 and a ramping-up shear (0–10 s−1), both within 360 s, for PP disentanglement was performed by [70]. The measured Me masses were 11,442 g/mol for “initial”, 18,974 g/mol for “steady”, and 67,181 g/mol for “ramping-up” PP. The reason for the ramp-up shear efficiency is that, with a slow rate of ramp-up, shear banding, which is typical of steady shear, was avoided; thus, the bulk sample was sheared efficiently, and the more tangled network could undergo structural breakdown because of the buildup of retractive force [72]. In one study, to vary the degree of high-density polyethylene (HDPE) disentanglement, shear was used in a rotational rheometer with a linearly increasing rate from 1 s−1 to a final rate ranging from 2 s−1 to 12 s−1 [68] (Figure 4). The higher shear rate led to better disentanglement, confirmed by a change in Me from 2840 g/mol to 17,230 g/mol.
Attempts have been made to achieve the effective disentangling of polymers by shearing them in processing machines such as extruders, mixers, or injection molding machines. The difficulty in this case is that not only must the polymer be disentangled inside the machine but it must also be retained while forming the product outside the machine. One of the first to attempt the large-scale production of “in-pellet” materials from disentangled polymers was Ibar [73]. The concept was based on an additional, simultaneous simple shearing treatment for the polymer in the machine. As pointed out by Ibar, the orientation of an entangled polymer, which causes it to flow more easily, has often been incorrectly interpreted as “disentangling” the polymer.
In processing equipment, the shear process can be assisted by applying a vibration field or adding a pressure field. A vibrational force field added to polymer extrusion affects the molecular motion and rheological behavior of the polymer melt [74]. An example of a system with vibrations was proposed by An et al. [75]. The polymer was sheared by two rods vibrating in the opposite direction. Vibration support improved the rheological properties of molten PP. Both the tensile strength and elongation at the break of the PP increased after applying a shear vibration field, as did the melting temperature and crystallinity. A similar device was previously built by Isayev [76]. Often, these systems constitute a modified end part of the extruder. Another solution, involving the use of a vibration field in the entire extruder, was proposed by Chen [75].
Lin et al. [77] designed an eccentric rotor extruder that could generate an elongation flow to promote a good mixing of two polyolefines and reduce chain entanglement. The partially disentangled state can also be realized directly in injection molding using a vibration field [78] (Figure 5). The designed machine can impose a complex shear field, i.e., rotational shear + oscillatory shear. A comparison of different shearing methods, i.e., vibratory, rotational, or combined, showed that the latter solution is the most effective in disentangling polycarbonate (PC). The same machine was used for disentangling PLA [79].
Please note that the disentangled polymers obtained with the three methods above do not have exactly the same internal structure because, in the case of polymerization, the amount of the amorphous phase is smaller than usual; in the case of the freeze-drying methods; a solvent may still be present; and in the case of disentangling via shear, one can expect at least a local orientation of macromolecules.
3. Change in Properties Due to Polymer Disentangling
Studies on partially disentangled materials have provided further information on the impact of entanglements on polymer properties. Previous research up to 2016 was described in detail in Pawlak’s review [19]. The latest results, which clearly illustrate the current state of knowledge, are summarized below. This research has focused on crystallization, mechanical properties, and glass transition.
Crystallization from disentangled melt has been examined for isotactic PP [70,80,81,82,83], syndiotactic PP [65], PLA [61,79], PS [66], PEO [62], and UHMWPE [63,84]. Cold crystallization was studied for PC in [85] and PET in [86]. Generally, these studies confirmed conclusions from studies of equilibrium-entangled polymers, i.e., that both crystal nucleation and crystal growth may depend on reducing entanglement. Romano et al. [87] showed that the degree of polymer disentanglement can be evaluated based on differences in the melting kinetics of entangled and disentangled crystals. Wang et al. [78] crystallized PP after a LAOS process and concluded that the nucleation density and growth rate of spherulites increase as the entanglement density decreases. According to Fu et al. [68], less entanglement in sheared HDPE samples results in thicker lamellae in the non-isothermally crystallized polymer. Wang et al. [88] studied the crystallization kinetics of freeze-extracted PLA as a function of the concentration in solution below and above critical concentrations for a chain-overlapping c*, which was calculated to be 11 g/L. It was found that crystallization was faster and lamellae were thicker when the concentration of the precursor solution decreased, but only when it was lower than the c*. The crystallization of PLA was also studied by Hu et al. [79] in shear-disentangled samples. They concluded that, under isothermal and non-isothermal conditions, crystallization was faster and the degree of crystallinity was higher when the degree of entanglement was lower.
In another study, similar results were obtained from a comparison of fully entangled PET with another PET prepared from a trifluoroacetic acid solution, called reorganized PET (RPET) [89]. Observations of the transition of crystallization regimes I-II were interesting. The temperature was 7 °C lower for RPET. Using Hoffman–Weeks extrapolation, the calculated equilibrium melting temperature was found to be several degrees higher for RPET.
Many polymers, such as isotactic polybutene-1 (PB), can crystallize in different crystallographic forms. Ni et al. [90] showed the role of chain entanglement in the kinetics of polymorphic crystallization and the solid–solid phase transition. The temperature of crystallization, the temperature of melting, and the crystallinity of form II were found to decrease with the degree of disentanglement. This was accompanied by lower nucleation density and unusually lower spherulite growth rate (Figure 6). The reasons for this behavior were unclear, although the loose packing of the crystal lattice or highly mobile side ethyl groups were indicated as probable causes. Form II in PB is metastable and spontaneously transforms at room temperature into thermodynamically stable form I. Disentangling the chain significantly increases the phase transition rate and final degree of transition.
The crystallization of poly(ε-caprolactone) and its blends with poly(styrene-co-acrylonitrile) was studied by Liu et al. [91]. The authors prepared disentangled materials using strong shear or the “self-nucleation” process. The second term means that disentangled polymer was created by melting crystals and holding them just above the melting temperature. The ease of disordering and re-entangling such material was studied after applying weak shear immediately after disentangling. The weak shear decelerated the crystallization of the partially disentangled melt because of chain re-entanglement. However, when the weak shear rate was higher, additional disentanglement was attained, and crystallization occurred more quickly.
Zhang et al. [92] increased the mobility of poly(L-lactide) (PLLA) macromolecules using a complex shear method. As a result of disentangling, the crystallization kinetics of PLlA was accelerated. Moreover, PLLA macromolecular chains are usually aligned along the flow direction under strong shear fields, resulting in thicker shear layers and the formation of denser and more perfect shish–kebab structures.
The crystallization of the sterocomplex of polylactide enantiomers is usually restricted to high-molecular-weight components. Sun et al. [93] demonstrated the crucial role of chain entanglement in regulating this kind of crystallization. PLLA/PDLA blends with various degrees of entanglement were prepared by freeze-drying a dioxane solution. The disentangling not only increased the crystallization rate but also the crystallinity under both non-isothermal and isothermal conditions. The less-entangled samples crystallized exclusively as high-crystallinity complexes, in contrast to the predominant homo-crystallization that occurred in the entangled samples.
The crystallization of PP with its entanglements halved was the subject of several works by Pawlak et al. [81,94]. It was found that, similar to other polymers, the crystallization temperature and degree of crystallinity are higher in the disentangled polymer. The thickness of the lamellae also increases. It was shown for the first time that, in polymers under the influence of disentangling, the transition temperature between the II and III crystallization regimes decreases by 4 °C (Figure 7) [81]. Krajenta et al. [95] studied the crystallization of PLA after limiting the density of entanglements to 20% of the initial density. The crystal growth was found to be 10% faster, and it was shown that the boundary temperatures of the crystallization regimes shift under the influence of disentangling.
Another polymer in which the course of isothermal and non-isothermal crystallization was studied was poly(ethylene oxide) (PEO) [96]. It turned out that with a decrease in entanglement density spherulitic nucleation increases, and the resulting spherulites grow faster. With fewer macromolecular entanglements, the temperature of transition between crystallization regimes I and II shifted in the direction of lower temperatures. Equilibrium melting temperature measurements using the Hoffman-Weeks method showed little difference between entangled and disentangled PEO. X-ray observations of the size of the formed lamellae have shown that slightly thicker and more perfect crystals grew from a partially disentangled melt. The macromolecules in the samples annealed in the melt gradually re-entangled, but this process was slow.
The experimental crystallization studies have been supported by simulations. For example, Zhai et al. [97] performed coarse-grained molecular dynamics simulations of the isothermal crystallization of bimodal and unimodal polymers with equivalent average Mw. They analyzed the evolution of entanglement during crystallization. With the onset of crystal growth, the entanglement concentration decreased rapidly, but at the end of crystallization, it saturated at a level lower than the initial one. The actual crystallinity and lamellae thickness were linearly proportional to the degree of disentanglement. The authors built a scenario out of the entire process of chain disentangling and lamellar thickening based on the chain-sliding diffusion mechanism. Compared with the unimodal system, the increase in the crystallinity of the bimodal system was faster than disentanglement. The reason was that the chains of the component with longer chains slid more slowly.
Peng et al. [98] performed molecular dynamics simulations to account for the role of entanglement in the nucleation of polymer melts. In highly entangled polymers like PC, nucleation is completely suppressed. The inhibition of nucleation may result from an entanglement-enhanced nucleation barrier or from constrained chain dynamics. Which of these is important is not clear. The determined nucleation-free energy barriers and critical nucleus size decreased at lower entanglement densities.
Still, the effect of entanglement on the glass transition temperature (Tg) is not entirely clear, even as new results are available for disentangled polymers. A decrease in Tg has been observed for disentangled polymers, mainly PS but also PC, PLLA, and PET [78,89,99,100,101,102,103,104,105,106] (Figure 8). The decrease in Tg is interpreted in the literature as the enhanced degree of freedom of the bond rotation due to a reduction in entanglements, affecting the chain relaxation process [107]. Another possible explanation for the decrease in the Tg is that, after freeze-drying, the proliferation of free-volume holes facilitates the movement of molecular chains [108]. Since a decrease in Tg has also been observed in non-entangled, low-Mw PS prepared from solution, Simon and co-workers [102,109] suggested that the reason for the glass transition shift is not disentanglement but the presence of residual solvent.
Research has also been carried out to better understand the relationship between the entanglements of macromolecules and the mechanical properties of polymers. Three of these experiments focused on impact behavior. Yue et al. [110] examined the Izod impact strength of two sintered UHMWPE, including one with reduced entanglements. Interestingly, the polymer in the weakly entangled state was characterized by significantly better impact strength (80 vs. 70 kJ/m2). The reason may be better sintering of fewer entangled chains.
In the experiment by Shan et al. [111], micro-sized spherical polystyrene projectiles made of three polymers with different Mw values (9300, 40,000, 270,000 g/mol) below and well above the Me (13,400 g/mol) were launched against a rigid substrate, causing deformation at an ultra-high strain rate (Figure 9). The bottom part of the projectiles heated up and deformed as a result of plastic flow. The upper part showed localized shear banding and brittle fracture for disentangled PS and extensive shear banding followed by crazing in the entangled samples. The absence of entanglements in PS with a molecular weight of 9300 g/mol was the reason for the non-forming of crazes and the rapid transition to fractures. Zhang et al. [112] compared the mechanical properties of two commercially available UHMWPE with similar Mw values and concluded that the less entangled UHMWPE had three times more Charpy impact strength, although the tensile and flexural properties were comparable.
More traditional were the studies on the tensile properties of PC conducted by Wang et al. [78]. The novelty of their approach was the use of a modified extruder, in which the disentangling of PC was achieved by applying different shear procedures (vibration, rotation, or together). Surprisingly, no significant changes were observed between the tensile properties of disentangled and raw polymers. While examining the tensile properties of PP, Pawlak et al. [113] noticed that the differences between the entangled and non-entangled polymers become visible when, instead of engineering parameters, the values of true stress and true strain were calculated. It was found that properties at yield were not changed by the reduction in entanglements. However, in the strain-hardening phase, the increase in stress was slower when the polypropylene was less entangled. Increased cavitation was observed in partially disentangled polypropylene.
The process of pore formation in amorphous polyethylene during uniaxial stretching was modeled using the molecular dynamics method by Logunova and Orekhova [114]. The results of the statistical analysis of pore sizes indicate that intermolecular entanglements slow down the processes of their growth and aggregation in the bulk of the polymer.
In recent years, publications have appeared concerning other studies of materials containing disentangled chains, e.g., those that are not electrically neutral [115,116]. For example, the idea of studying relaxation processes using broadband dielectric spectroscopy has been presented. Dielectric spectroscopy allows for the identification of polarization and conductivity phenomena, the measurement of charge transport, and other things. In the case of non-polar polymers, it may be necessary to introduce a substance with a constant dipole moment. In Drakopoulos et al.’s [115] research on disentangled UHMWPE, Al2O3 catalytic ash played such a role. Five relaxation processes were identified in the polymer, two of which were attributed to the disentangled and entangled amorphous phase. Based on the results of dielectric spectroscopy, the formation of entanglements was also analyzed, estimating the minimum temperature at which the re-entanglement begins (58 °C).
The role of entanglement on the rheological and mechanical properties of polymerized ionic liquids, a class of polyelectrolytes, was investigated by Liu et al. [116]. Samples with a wide range of molecular weights were tested. The high density of electrical charges in the polymer backbone of this material provides unique rheological properties, such as resistance to entanglement. The molecular weight of the entanglement of the tested polyelectrolyte was 1.6 × 105 g/mol, which was much higher than that of conventional polymers. Disentangling the polymerized ionic liquid under shear in the LAOS process was also observed, with re-entangling occurring within 6 min after the high shear had ceased.
4. Special Case: Ultra-High-Molecular-Weight Polyethylene
A special polymer whose properties are being tested, taking into account the entanglements of macromolecules, is ultra-high-molecular-weight polyethylene. It is a polymer with very good mechanical properties, often used in medical applications. Its processing is usually difficult because the macromolecules are very long and, therefore, highly entangled. UHMWPE does not form a conventional melt when heated above its melting point.
There are two possible ways to improve processability. One of them is blending with lower-molecular-weight polyethylene, e.g., HDPE [117,118,119,120] or linear low-density polyethylene (LLDPE) [121] (Figure 10). Compatible macromolecules penetrate UHMWPE during blending, reducing its entanglements. Often, the blended UHMWPE is partially disentangled [122,123,124], which increases the effectiveness of the procedure. The blend of disentangled UHMWPE with HDPE can be used for the melt-spinning process, leading to the formation of high-strength fibers [120]. The formation of fibers from disentangled polyethylenes was studied in the 1970s by the Lemstra group [125]. In order to improve processability, attempts have also been made to reduce contacts between macromolecules and, thus, viscosity in the melt by introducing nanoparticles into the polymer structure. This issue is discussed in the section devoted to composites.
The second possible way to improve processability is to disentangle UHMWPE during controlled polymerization. Progress in this area concerns the use of new catalysts, such as imido vanadium trichloride complexes [126], polyhedral oligomeric silsesquioxane (POSS)/MgCl2 nanoaggregates modifying Z-N catalysts [127], POSS-modified Z-N catalysts with judicious immobilization of TiCl4 [110], Z-N catalyst with TiCl4 anchored on PS-modified silica [128], nanodispersed MgO/MgCl2/TiCl4 catalyst [129], and POSS Z-N catalysts [130]. UHMWPE/HDPE wax blends have been produced directly during polymerization [122,131].
Some research on UHMWPE has examined the properties of the polymer after disentangling. For example, mechanisms of relaxation [132] and the physical network junctions have been studied [133]. Petrov et al. [134] performed large-scale molecular dynamics simulations concerning the uniaxial stretching of UHMWPE and found that there is an optimal number of entanglements per macromolecule necessary to maximize chain orientations in fibers formed from plastically damaged crystals.
Tao et al. [123] melt-blended three types of UHMWPE with different entanglement states and molecular weights with an HDPE matrix (1:4 by weight). The lower degree of entanglement and higher molecular weight of the UHMWPE promoted an increase in the thickness of the lamellae and the crystallinity of the produced blend. This led to the formation of more oriented shish structures and more kebab lamellae. Moreover, the molecular chains of weakly entangled UHMWPE acted as tie molecules, significantly improving the impact resistance. Chen et al. [130] obtained HDPE/UHMWPE blends containing 0–50 wt.% of disentangled or entangled UHMWPE by synthesis in a chemical reactor. The mechanical properties depended on the composition but were similar for entangled and disentangled UHMWPE. The Izod impact strength increased rapidly in the disentangled blends compared with the disentangled UHMWPE, and the effect of entangled blends was half that of disentangled blends.
5. Re-Entangling Macromolecules
A disentangled polymer is not in a state of equilibrium, so if the movement of macromolecules is possible, it tends to reach equilibrium. The rate of the process depends on the temperature, but it is usually not fast [135,136]. Re-entangling can be studied by measuring changes in the storage modulus over time. The modulus after re-entangling should return to its pre-disentanglement value, and there are some observations that confirm this [137].
However, it is often observed that the storage modulus increases quickly at first and then slower and slower, and after some time, a final plateau value is reached, well below the expected value representing the polymer before disentangling [63]. It is unclear what causes such a low plateau in polymers disentangled in solution [29]. Many authors have tried to avoid discussing the reasons for this behavior by showing the G′(t) data normalized to the G′ value for the plateau (i.e., to the maximum of the curve) and assuming that, when the ratio is one, the polymer is completely entangled again. In both ways of presenting rheological data, the re-entangling time is defined as the moment at which the modulus stops increasing [68,69,122,128,138] (see Figure 11). The second issue not discussed in the literature is that, sometimes, the curve describing the increase in G′ over time is not smooth but has “waves” (undulations) visible on it [68,121,128].
Many years ago, Barham and Sadler carried out an experiment by melting polyethylene mats containing single-chain crystals. During melting, the macromolecules that had previously formed crystals took the shape of a coil within a few seconds [139]. In real materials, crystals consist of many macromolecules, and there are also tie macromolecules that connect adjacent lamellae, which means that the macromolecules that form crystals are not completely separated from the others after melting. As a result, in a semi-crystalline polymer, melting the crystals creates areas with lower and higher densities of entanglements. As a consequence of this, two growth rates of G′ are observed in the re-entanglement process, i.e., the slow and fast return to the equilibrium state of the melt [133]. However, in Fu’s opinion [68], the entanglement process is a two-stage one given the type of entanglements that arise. At the early stage (a time of 100 s for HDPE), simple entanglements (twists, knots, links) are formed, and in the second phase, loops are formed. The second, slower phase of entanglement network formation is mainly controlled by reptation dynamics [70].
Experimental results have shown that the kinetics of re-entanglement is much slower than that predicted by tube theory [68], i.e., longer than the average reptation time [69]. The time of recovery depends on the polymer, temperature, and disentangling level [69]. Ni et al. [90] observed in PB that the recovery time was 32,100 s for PB obtained from the most diluted solution and 6000 s when the solution concentration was close to the critical c*. In the above example, the entanglement time was long, but in many cases, the recovery time is too short in terms of processing. So, there have been attempts to increase it. Liu et al. [138] effectively delayed the recovery of entanglements in HDPE by adding a small (3–5%) amount of UHMWPWE. The re-entangling time in the blend was twice as long as in the case of HDPE.
Rheological tests of PP have shown that, after annealing for 120 min, the initial viscosity level can still not be achieved. The mechanical properties become similar to those of entangled polymers after 2 h, but the higher the molecular weight, the more time is required for them to increase (Figure 12). The time after which the transition temperature between crystallization regimes returns to the initial state depends on the degree of disentanglement and the molecular weight of PP [94].
Chammingkwan et al. [129], examining disentangled and entangled UHMWPE, surprisingly found similar increases of the G′ modulus as a function of time. The reason was that voids initially present in the entangled samples were reduced by the prolonged heating at a temperature above the melting point, which changed the conditions of the rheological experiment, i.e., the contact of the polymer with the rheometer plates. This example shows how important it is to properly control the course of the experiment. It cannot be excluded that technical or procedural reasons not taken into account may cause the observed lower final values of G′ after re-entangling.
6. Composites Created Using Disentangled Polymers
Over the last ten years, interest in polymer nanocomposites has been steadily increasing. Classic composites contain a micrometer-sized dispersed phase (fillers, particles, plates) with a relatively high amount of reinforcing agent (10–50 wt.%). The progress in composites involves the use of smaller dispersed phase elements, nanometer-sized, which allow the content to be reduced to 1–5 wt.% while also improving the properties of the matrix. An additional aspect of introducing very small dispersed phase elements into the composite is that some new effects or behaviors may be revealed. An example is unexpected rheological behavior. It is known that in micro-composites the dispersed phase increases the viscosity of the melt, but when nanoparticles are used in a polymer composite, the behavior may be the opposite, i.e., a reduction in viscosity, as long as the size of the nanoparticles is smaller than the mesh size of the matrix [140,141,142,143,144].
For example, such an effect was observed for poly(methyl methacrylate) (PMMA) with 2 wt.% dispersed 11 nm grafted silicon nanoparticles [143]. A decrease in the storage modulus with grafted polyoxometalate particle content was observed by Chai et al. [140]. A decrease in the GNo modulus of a commercial UHMWPE composite with POSS compared with UHMWPE was observed by Zhang et al. [106]. The effect was best visible at a filler content of 0.5% by weight and less at higher contents. Sui et al. [145] formulated a UHMWPE composite with 0.1–0.5 wt.% TiO2 and observed that the viscosity of the solution in the presence of nanoparticles decreased by 91% compared with the pure UHMWPE solution. Also, the critical overlap concentration, i.e., the concentration in the solution at which the separation of the macromolecular coils occurs, increased from 1 wt.% in UHMWPE up to 1.4 wt.% in the composite. A determination of the plateau modulus with the DMTA method showed that the amount of 0.3% TiO2 was optimal, most effectively reducing the modulus, resulting in a twofold increase in Me, assuming that the application of Equation (2) is correct for composites. Waterborne acrylic coatings (butyl acrylate: methyl methacrylate: acrylic acid, 54:42:2 mol%) with the addition of 1–3 wt.% nano-TiO2 were studied by Romo-Uribe et al. [146]. A two-order reduction in G′ and an increase in the entanglement relaxation time were observed after the addition of TiO2, which was especially large at 1 wt.%. In this case, the nanoparticles also caused a partial disentangling of the chains.
The reason for the observed rheological behavior is that sufficiently small nanoparticles can increase the free volume in the melt, thereby reducing chain entanglement and accelerating the relaxation process. As a result of disentanglement, a lower melt viscosity (or modulus) can be observed compared with the polymer used as the matrix [140]. Table 1 shows the nanocomposites prepared in which the matrix polymer was disentangled.
Developed nanocomposites with partial disentanglement have been used to study their properties. In one study, in PMMA/SiO2 composites, the glass transition did not change in relation to the PMMA-to-SiO2 content below 10 wt.% but increased with higher amounts [143]. For PS/polyoxymetalate, a regular shift in Tg toward lower temperatures was observed with an increase in nanofiller content in [140]. Flash DSC studies of UHMWPE with 0.8 nm POSS particles showed that the composite had lower melting activation energy than pure UHWMPE, confirming the lower entanglement level, and the chain segment activity was improved with the addition of POSS. The POSS particles used were smaller than the theoretical tube size of PE (3.6 nm) [106] (Figure 13). Disentangling the UHMWPE/TiO2 composite made it processable, as demonstrated by measurements of the melting index. The obtained value of the melt index was low, only 0.069 g/10 min (at 21.6 kg and 230 °C), but it was measurable instead of UHMWPE [145]. The UHMWPE/TiO2 composite had much better tensile properties than UHMWPE. The elongation to break increased by six times, and the tensile strength also increased significantly. However, regardless of improvement, this composite was still highly entangled, and the increased elongation at the break may also be a result of reducing impurities in the polymer during solution preparation.
The second approach to obtaining disentangled composites is shear in melt, similar to the disentangling of homopolymers. Luo et al. [147] disentangled PP containing 0.1–4 wt.% graphene nanoplatelets (Figure 14). The subject of their studies was the rheology and crystallization of the composite. The re-entanglement process was investigated at a temperature of 200 °C. The disentangled-by-shear neat PP was fully entangled; i.e., it reached initial viscosity after 2000 s. In the composites, after 100 s, the increasing viscosity of the composite reached a constant value, but at a level much lower than the value expected for the equilibrium entangled composite. According to the authors of the article, this means it maintained a disentangled state. Given their strong interaction, graphene platelets limit the movement of chains. PP in the sheared graphene composite crystallized faster compared with the non-sheared composite.
The subject of other studies from Luo’s group was the properties of a PP micro-composite with a high amount (35 wt.%) of boron nitride [148]. The PP in the composite was disentangled via a steady-state shear, and the re-entangling was examined (Figure 15). The viscosities of the non-sheared composites were higher compared with PP, were stable during the experiment, and depended on the size of microparticles. After shearing, the order changed, and PP had a higher viscosity than the composites. This confirmed the disentanglement. After the cessation of shear, the viscosity of each materias increased rapidly during the first 30 s of the time sweep experiment and then remained stable (at least 300 s); however, it was far from the equilibrium value of PP. The authors interpreted this as maintaining a state of disentanglement in the case of the composites. In many experiments, reaching a viscosity plateau is interpreted as full entanglement. According to Luo, the attachment of disentangled molecular chains to the surface of particles and, thus, the limitation on the mobility of the macromolecular chains are the cause of the reduced plateau viscosity of composites. The crystallization of PP/boron nitride composites was slightly faster when the PP matrix was partially disentangled.
A third possible approach to obtaining disentangled composites is not formulating them by dissolving or shearing entangled polymers with filler but by using a previously disentangled polymer as the matrix. This was tested by Drakopoulos et al. [149], who obtained a UHMWPE/1 wt.% Au nanocomposite from a solution. UHMWPE was disentangled during polymerization. The obtained nanocomposite powder was calendered to obtain draw ratios of up to 100, or it was additionally stretched into higher draw ratios (100–200). An increase in crystallinity from 75% to 87% for the deformed composite and the occurrence of gold agglomeration (from 10.6 to 12.3 nm) with deformation were observed. The reason for the agglomeration was a reduction in the content of the amorphous phase, which contained gold particles.
The entanglement dynamics of the oriented UHMWPE/1 wt.% Au nanocomposite were the subject of another publication by the same authors [150]. Dielectric spectroscopy was used to study the transition of a composite into a molten state. A decrease in DC conduction was observed with the formation of entanglements.
Barangizi and Pawlak [151] investigated the crystallization of solution-disentangled PP in a composite with 1 wt.% Al2O3 nanopowder. For comparison, entangled nanocomposite, solution-disentangled PP, and entangled PP were examined. It was observed that isothermal crystallization occurred faster and non-isothermal crystallization occurred at a higher temperature if the composite had fewer entanglements (Figure 16). However, it was also observed that crystallization in the nanocomposite was slower compared with the homopolymer at the same entangling level. The dispersed alumina nanoparticles hindered the movement of macromolecules into the growing crystals. The re-entanglement of macromolecules as a result of annealing at 200 °C was also investigated. The time to equilibrium for the less-entangled composite and similarly entangled PP was around 80 min. More entangled samples reached equilibrium after 40–50 min.
Recently, Barangizi et al. [152] published the results of studies on the mechanical and thermal properties of PLA composites containing 0.1–1.0 wt.% multi-wall carbon nanotubes (MWCNT). The authors compared fully and partially entangled composites and PLA. The partial disentangling of macromolecules improved filler dispersion during composite fabrication via extrusion. The increased mobility of the less-entangled PLA macro-molecules accelerated crystallization in the nanocomposites, which had occurred already during the cooling of the melt and not only as a cold crystallization. Isothermal crystallization studies showed faster crystallization because of a favorable combination of matrix disentanglement and increased nucleation in the nanotubes. The reduction in entanglements influenced the mechanical properties of the composites (Figure 17). The initiation of plastic deformation was easier, i.e., at lower yield stress, and the increase in stress in the strain-hardening phase was slower for the disentangled homopolymers. In the composites, the strain-hardening was stronger with a higher concentration of nanotubes, but it also depended on their dispersion, which was better in the less-entangled polylactide matrix, resulting in the fastest increase in stress.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Nano- and micro-composites prepared for studies of the matrix disentangling effect.
| Polymer Matrix | Filler | Contents of Filler (wt.%) | Method of Dispersion | Disentangling of Matrix | Reference |
|---|---|---|---|---|---|
| PMMA | Grafted SiO2, 11 nm | 2 | Solution | During mixing | [143] |
| PS | Grafted polyoxometalate, 0.5–6 nm | 1–5 | Solution | During mixing | [140] |
| UHMWPE | POSS | 0.2–3 | During polymerization | Previously disentangled | [63] |
| UHMWPE | POSS, 0.8 nm | 0.1–1 | Melt mixing | During mixing | [106] |
| UHMWPE | TiO2 | 0.1–0.5 | Solution | During mixing | [145] |
| UHMWPE | Gold, nano | 1 | Solution | Previously disentangled | [149] |
| PP | Graphene | 0.1–4 | Shear in melt | During mixing | [147] |
| PP | Boron nitride, 1, 5, 27 µm | 35 | Steady-state shear | During shear | [148] |
| PP | Al2O3, 78 nm | 1 | Melt mixing | Previously disentangled | [151] |
| Waterborne acrylic coatings | TiO2 | 1–3 | Solution | During mixing | [146] |
| PLA | MWCNT | 0.1–1 | Melt mixing | Previously disentangled | [152] |
A specific approach to formulating composites is the polymerization of UHMWPE in the presence of POSS nanoparticles, such as in [63]. In that study, extending the polymerization time resulted in a decrease in the POSS content in the composite, which was 0.74–2.31%. Rheological tests showed that the highest G′ modulus value (higher than UHMWPE) was obtained for the polymer composite with the highest molecular weight and the lowest POSS content, which resulted from the longest polymerization process (Figure 18). During the time sweep test, the composites gradually reached the equilibrium G′ values, which, however, were not the same as for the homopolymer. The times to reach equilibrium were 103–104 min and were the longest for UHMWPE. The crystallinity and lamella thickness of the resulting UHMWPE increased with increasing POSS loading. This suggested that the POSS particles were nucleating agents.
Disentangled polymers can also be used to produce special types of composites called all-polymer composites. These are composites in which not only the matrix but also the reinforcement is polymer. Smith and Lemstra [125] showed that very strong fibers can be obtained by spinning from solution. At that time, it was not known that this was possible by limiting entanglements. A similar concept was later used by Galeski [83,153]. It is known that the plastic deformation of a semi-crystalline polymer is limited by the network of macromolecules in the amorphous phase. It was assumed that if the network was less dense, it would be possible to obtain much greater deformations and transform the lamellar structure of the polymer into a fibrous one. Such a transformation can be achieved by blending a solid, partially disentangled polymer powder with a second molten polymer. The concept was experimentally verified via extrusion, and all-polymer composites were created from HDPE/PP and PS/PP pairs [83] (Figure 19). Microscopic observations confirmed that long, flexible micro- and nanofibers were obtained, well dispersed in the second polymer matrix. Tests of mechanical properties showed effective strengthening in the polymer matrix.
7. Concludings Remarks and Perspectives
Polymers with partially disentangled chains may be an interesting alternative in the production of materials dedicated to special applications. They have properties that are different from their fully entangled counterparts. The facilitation of the crystallization process and changes in mechanical properties with the goal of obtaining large deformations is worth mentioning here. It is important that attempts are being made to develop methods of industrial production for such polymers and the materials using them. Modifications to the extrusion process can be mentioned here. In the future, the observation that very small nanoparticles can participate in the disentangling of polymers may also be important. While the literature on disentangled polymers is quite extensive, the number of reports on composites does not exceed twenty, and these are reports from the last 2–3 years. This indicates the direction of future research, which, in addition to nanocomposites, will probably also involve blends of disentangled polymers.
Author Contributions
Both authors participated in collecting information and writing the text. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre, Poland, on the basis of decision 2022/06/X/ST5/00654. A statutory fund from the Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, is acknowledged.
Data Availability Statement
No new data was created.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of the data; in the writing of the manuscript; or in the decision to publish the results.
References
- Watanabe, H. Viscoelasticity and dynamics of entangled polymers. Prog. Polym. Sci. 1999, 24, 1253–1403. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, C.; Zhang, Y.; Zhao, J. Study on the chain entanglement of polyvinyl alcohol fiber during the dry-jet wet spinning process. Fibers Polym. 2015, 16, 345–353. [Google Scholar] [CrossRef]
- Wool, R.P. Polymer entanglements. Macromolecules 1993, 26, 1564–1569. [Google Scholar] [CrossRef]
- Pearson, D.S.; Ver Strate, G.; Von Meerwall, E.; Schilling, F.C. Viscosity and self-diffusion coefficient of linear polyethylene. Macromolecules 1987, 20, 1133–1141. [Google Scholar] [CrossRef]
- Berry, G.C.; Fox, T.G. The viscosity of polymers and their concentrated solutions. Adv. Polym. Sci. 1968, 5, 261–357. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity, 3rd ed.; Clarendon Press: Oxford, UK, 1975; pp. 64–65. [Google Scholar]
- Ward, M.; Sweeney, J. Mechanical Properties of Solid Polymers; John Wiley & Sons Ltd.: Chichester, UK, 2013; p. 72. [Google Scholar]
- Haward, R.N. Strain hardening of thermoplastics. Macromolecules 1993, 26, 5860–5869. [Google Scholar] [CrossRef]
- Deplancke, T.; Lame, O.; Rousset, F.; Seguela, F.; Vigier, G. Mechanisms of Chain Reentanglement During the Sintering of UHMWPE Nascent Powder: Effect of Molecular Weight. Macromolecules 2015, 48, 5328–5338. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; Wiley: New York, NY, USA, 1980; p. 372. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon: Oxford, UK, 1986. [Google Scholar]
- Liu, C.; He, J.; van Ruymbeke, E.; Keunings, R.; Bailly, C. Evaluation of different methods for the determination of the plateau modulus and the entanglement molecular weight. Polymer 2006, 47, 4461–4479. [Google Scholar] [CrossRef]
- Eckstein, A.; Suhm, J.; Friedrich, C.; Maier, R.; Sassmannshausen, J.; Bochmann, M.; Mulhaupt, R. Determination of Plateau Moduli and Entanglement Molecular Weights of Isotactic, Syndiotactic, and Atactic Polypropylenes Synthesized with Metallocene Catalysts. Macromolecules 1998, 31, 1335–1340. [Google Scholar] [CrossRef]
- Kong, D.-H.; Yang, M.-H.; Zhang, X.-S.; Du, Z.-C.; Fu, Q.; Gao, X.-Q.; Gong, J.W. Control of Polymer Properties by Entanglement: A Review. Macromol. Mater. Eng. 2021, 306, 2100536. [Google Scholar] [CrossRef]
- Wu, S. Chain structure an entanglement. J. Polym. Sci. Polym. Phys. 1989, 27, 723–741. [Google Scholar] [CrossRef]
- Wu, S.; Beckerbauer, R. Effect of Tacticity on Chain Entanglement in Poly(methyl methacrylate). Polym. J. 1992, 24, 1437–1442. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. The connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Colby, R.H. Physical Properties of Polymers Handbook, 2nd ed.; Mark, J.E., Ed.; Springer Science: New York, NY, USA, 2007; pp. 447–454. [Google Scholar]
- Pawlak, A. The Entanglements of Macromolecules and Their Influence on the Properties of Polymers. Macromol. Chem. Phys. 2019, 220, 1900043. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, Z.; Lin, X.; Zhang, C.; Tanaka, K.; Zuo, B.; Zhang, W.; Wang, X. Suppressed Chain Entanglement Induced by Thickness of Ultrathin Polystyrene Films. Macromolecules 2021, 54, 3735–3743. [Google Scholar] [CrossRef]
- Doi, M. Viscoelastic and rheological properties. In Materials Science and Technology. A Comprehensive Treatment. Volume 12. Structure and Properties of Polymers; Cahn, R.W., Haasen, P., Kramer, E.J., Thomas, E.L., Eds.; VCH Verlag: Weinheim, 1993; pp. 389–425. [Google Scholar]
- Zulli, F.; Giordano, M.; Andreozzi, L. Onset of entanglement and reptation in melts of linear homopolymers: Consistent rheological simulations of experiments from oligomers to high polymers. Rheol. Acta 2015, 54, 185–205. [Google Scholar] [CrossRef]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. 4. Rheological properties. J. Chem. Soc. Faraday Trans. II 1979, 75, 38–54. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. Part 1—Brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1789–1801. [Google Scholar] [CrossRef]
- Talebi, S. Disentangled Polyethylene with Sharp Molar Mass Distribution: Implications for Sintering. Ph.D. Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2008. [Google Scholar]
- Robertson, C.G.; Warren, S.; Plazek, D.J.; Roland, C.M. Reentanglement kinetics in sheared polybutadiene solutions. Macromolecules 2004, 37, 10018–10022. [Google Scholar] [CrossRef]
- Costanzo, S.; Huang, Q.; Ianniruberto, G.; Marrucci, G.; Hassager, O.; Vlassopoulos, D. Shear and extensional rheology of polystyrene melts and solutions with the same number of entanglements. Macromolecules 2016, 49, 3925–3935. [Google Scholar] [CrossRef]
- Ransom, T.C.; Debjani, R.; Puskas, J.E.; Kaszas, G.; Roland, M.C. Molecular Weight Dependence of the Viscosity of Highly Entangled Polyisobutylene. Macromolecules 2019, 52, 5177–5182. [Google Scholar] [CrossRef]
- Hoy, R.; Robbins, M. Strain hardening of polymer glasses: Effect of entanglement density, temperature, and rate. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 3487–3500. [Google Scholar] [CrossRef]
- Hiss, R.; Hobeika, S.; Lynn, C.; Strobl, G. Network stretching, slip processes, and fragmentation of crystallites during uniaxial drawing of polyethylene and related copolymers. A comparative study. Macromolecules 1999, 32, 4390–4403. [Google Scholar] [CrossRef]
- Zuo, F.; Keum, J.K.; Chen, X.; Hsiao, B.S.; Chen, H.; Lai, S.Y.; Wevers, R.; Li, J. The role of interlamellar chain entanglement in deformation-induced structure changes during uniaxial stretching of isotactic polypropylene. Polymer 2007, 48, 6867–6880. [Google Scholar] [CrossRef]
- Haward, R.N. Strain Hardening of High Density Polyethylene. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 1090–1099. [Google Scholar] [CrossRef]
- van Melick, H.G.H.; Govaert, L.E.; Meijer, H.E.H. On the origin of strain hardening in glassy polymers. Polymer 2003, 44, 2493–2502. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Peacock, A.J.; Mandelkern, L. Tensile properties of crystalline polymers: Linear polyethylene. Macromolecules 1994, 27, 5297–5310. [Google Scholar] [CrossRef]
- Bartczak, Z.; Kozanecki, M. Influence of molecular parameters on high-strain deformation of polyethylene in the plane-strain compression. Part I. Stress-strain behavior. Polymer 2005, 46, 8210–8221. [Google Scholar] [CrossRef]
- Schrauwen, B.A.G.; Janssen, R.P.M.; Govaert, L.E.; Meijer, H.E.H. Intrinsic deformation behavior of semicrystalline polymers. Macromolecules 2004, 37, 6069–6078. [Google Scholar] [CrossRef]
- Bartczak, Z.; Grala, M.; Richaud, E.; Gadzinowska, K. Erosion of the molecular network in the amorphous layers of polyethylene upon high-strain deformation. Polymer 2016, 99, 552–565. [Google Scholar] [CrossRef]
- Donald, A.M.; Kramer, E.J. Effects of molecular entanglements on craze microstructure in glassy polymers. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 899–909. [Google Scholar] [CrossRef]
- Donald, A.M.; Kramer, E.J. The competition between shear deformation and crazing in glassy polymers. J. Mater. Sci. 1982, 17, 1871–1879. [Google Scholar] [CrossRef]
- Garcıa-Franco, C.A.; Harrington, B.A.; Lohse, D.J. On the rheology of ethylene–octene copolymers. Rheol. Acta. 2005, 44, 591–599. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hikosaka, M.; Gu, F.; Ghosh, S.K.; Arakaki, M.; Toda, A. Effect of entanglement on nucleation rate of polyethylene. Polymer J. 2001, 33, 906–908. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Zhong, L.W.; Yang, S.; Liang, D.H.; Chen, E.Q. Memory effect on solution crystallization of high molecular weight poly(ethylene oxide). Polymer 2012, 53, 3621–3628. [Google Scholar] [CrossRef]
- Robelin-Souffache, E.; Rault, J. Origin of the long period and crystallinity in quenched semicrystalline polymers. Macromoelcules 1989, 22, 3581–3594. [Google Scholar] [CrossRef]
- Flory, P.J.; Yoon, D.Y. Molecular morphology in semicrystalline polymers. Nature 1978, 272, 226–229. [Google Scholar] [CrossRef]
- Jani, F.; Sepahi, A.; Afzali, S.K.; Moyad, S.H. Experimental study on the effect of molecular weight and chemical composition distribution on the mechanical response of high-density polyethylene. Polym. Eng. Sci. 2023, 63, 176–188. [Google Scholar] [CrossRef]
- Peters, G.W.M.; Balzano, L.; Steenbakkers, R.J.A. Flow-induced Crystallization. In Handbook of Polymer Crystallization; Piorkowska, E., Rutledge, G.C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 399–431, Chapter 14. [Google Scholar]
- Martins, J.A.; Zhang, W.; Brito, A.M. Origin of the melt memory effect in polymer crystallization. Polymer 2010, 51, 4185–4194. [Google Scholar] [CrossRef]
- Song, L. Effect of Entanglement Density on Mechanical Properties and Deformation Behavior of Rubber-Modified PVC/α-MSAN Blends. Ind. Eng. Chem. Res. 2013, 52, 12567–12573. [Google Scholar] [CrossRef]
- Prest, W.M.; Porter, R.S. Rheological properties of poly(2, 6-dimethylphenylene oxide)-polystyrene blends. J. Polym. Sci. (A-2) 1972, 10, 1639–1655. [Google Scholar] [CrossRef]
- Hao, X.; Kaschta, J.; Liu, X.; Pan, Y.; Schubert, D.W. Entanglement network formed in miscible PLA/PMMA blends and its role in rheological and thermo-mechanical properties of the blends. Polymer 2015, 80, 38–45. [Google Scholar] [CrossRef]
- Song, L.; Zhang, Y.; Ren, J.; Li, Y.; Yang, B.; Xing, E.; Wang, Y.; Shi, Y. Effect of Entanglement Density on Mechanical Properties and the Deformation Mechanism of Rubber-Modified PPO/PS Blends. Macromol. Mater. Eng. 2022, 307, 2200325. [Google Scholar] [CrossRef]
- Xie, M.; Li, M. Viscosity reduction and disentanglement in ultrahigh molecular weight polyethylene melt: Effect of blending with polypropylene and poly(ethylene glycol). Eur. Polym. J. 2007, 43, 3480–3487. [Google Scholar] [CrossRef]
- Huang, Y.F.; Xu, J.Z.; Zhang, Z.C.; Xu, L.; Li, L.B.; Li, J.F.; Li, Z.M. Melt processing and structural manipulation of highly linear disentangled ultrahigh molecular weight polyethylene. Chem. Eng. J. 2017, 315, 132–141. [Google Scholar] [CrossRef]
- Pandey, A.; Champouret, Y.; Rastogi, S. Heterogeneity in the Distribution of Entanglement Density during Polymerization in Disentangled Ultrahigh Molecular Weight Polyethylene. Macromolecules 2011, 44, 4952–4960. [Google Scholar] [CrossRef]
- Rastogi, S.; Kurelec, L.; Cuijpers, J.; Lippits, D.; Wimmer, M.; Lemstra, P.J. Disentangled state in polymer melts; a route to ultimate physical and mechanical properties. Macromol. Mater. Eng. 2003, 288, 964–970. [Google Scholar] [CrossRef]
- Westfahl, H., Jr.; Cardoso, M.B. Accessing the hidden lamellar nanostructure of semi-crystalline nascent polymers by small-angle X-ray scattering contrast variation. J. Appl. Crystall. 2011, 44, 1123–1126. [Google Scholar] [CrossRef]
- Yamazaki, S.; Gu, F.; Watanabe, K.; Okada, K.; Toda, A.; Hikosaka, M. Two-step formation of entanglement from disentangled polymer melt detected by using nucleation rate. Polymer 2006, 47, 6422–6428. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Zhang, X.; Zhang, B.; Chen, J. Evolution of chain entanglements under large amplitude oscillatory shear flow and its effect on crystallization of isotactic polypropylene. Polymer 2020, 186, 121899. [Google Scholar] [CrossRef]
- Bu, H.S.; Gu, F.M.; Bao, L.; Chen, M. Influence of entanglements on crystallization of macromolecules. Macromolecules 1998, 31, 7108–7110. [Google Scholar] [CrossRef]
- Hao, H.; Liu, R.J.; Zhao, Y.L. Concentration Dependence of Crystalline Poly(L-lactide) Prepared by Freeze-drying Solutions. Polym. Polym. Compos. 2009, 17, 31–35. [Google Scholar] [CrossRef]
- Gu, F.M.; Bu, H.S.; Zhang, Z. A unique morphology of freeze-dried poly(ethylene oxide) and its transformation. Polymer 2000, 41, 7605–7609. [Google Scholar] [CrossRef]
- Li, W.; Guan, C.; Xu, J.; Mu, J.; Gong, D.; Chen, Z.R.; Zhou, Q. Disentangled UHMWPE/POSS nanocomposites prepared by ethylene in situ polymerization. Polymer 2014, 55, 1792–1798. [Google Scholar] [CrossRef]
- Huang, B.; Ito, M.; Kanamoto, T. Deformation mechanism of amorphous poly(ethylene terephthalate) as function of molecular weight and entanglements. Polymer 1994, 35, 1210–1216. [Google Scholar] [CrossRef]
- Sun, Q.; Fu, Q.; Xue, G.; Chen, W. Crystallization Behavior of Syndiotactic Poly(propylene) Freeze-Dried from Toluene at very Dilute Concentration. Macromol. Rapid Comm. 2001, 22, 1182–1185. [Google Scholar] [CrossRef]
- Xue, G.; Wang, Y.; Liu, S.; Liao, Y.T. FT-IR Study of Concentration Dependence for Crystallization of Isotactic Polystyrene Arising from Freeze-Drying Dilute Solutions. Macromolecules 1995, 28, 4344–4346. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ohta, T. The influence of chain entanglement density on ultra-drawing behavior of ultra-high-molecular-weight polypropylene in the gel-casting method. Polymer 2008, 49, 621–627. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Y.; Shen, K.; Fu, Q.; Zhang, J. Insight into Shear-Induced Modification for Improving Processability of Polymers: Effect of Shear Rate on the Evolution of Entanglement State. J. Polym. Sci. B Polym. Phys. 2019, 57, 598–606. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Chen, J. Delay of re-entanglement kinetics by shear-induced nucleation precursors in isotactic polypropylene melt. Polymer 2020, 210, 123000. [Google Scholar] [CrossRef]
- Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Investigating the disentanglement of long chain branched polypropylene under different shear fields. J. Appl. Polym. Sci. 2022, 139, 51642. [Google Scholar] [CrossRef]
- Kamkar, M.; Salehiyan, R.; Goudoulas, T.B.; Abbasi, M.; Saengow, C.; Erfanian, E.; Sadeghi, S.; Natale, G.; Rogers, S.A.; Giacomini, A.J.; et al. Large amplitude oscillatory shear flow: Microstructural assessment of polymeric systems. Prog. Polym. Sci. 2022, 132, 101580. [Google Scholar] [CrossRef]
- Wang, S.-Q.; Ravindranath, S.; Wang, Y.; Boukany, P. New theoretical considerations in polymer rheology: Elastic breakdown of chain entanglement network. J. Chem. Phys. 2007, 127, 064903. [Google Scholar] [CrossRef] [PubMed]
- Ibar, J.P. Processing Polymer Melts under Rheo-Fluidification Flow Conditions, Part 1: Boosting Shear-Thinning by Adding Low Frequency Nonlinear Vibration to Induce Strain Softening. J. Macromol. Sci. B Phys. 2013, 52, 407–441. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Zhou, N.-Q.; Liu, B.; Jin, G. Improved Mechanical Propertes and Structure of Polypropylene Pipe Prepared Under Vibration Force Field. J. Appl. Polym. Sci. 2009, 114, 3612–3620. [Google Scholar] [CrossRef]
- An, F.Z.; Gao, X.Q.; Lei, J.; Deng, C.; Li, Z.M.; Shen, K.Z. Vibration assisted extrusion of polypropylene. Chin. J. Polym. Sci. 2015, 33, 688–696. [Google Scholar] [CrossRef]
- Isayev, A.I.; Wong, C.M.; Zeng, X. Effect of oscillations during extrusion on rheology and mechanical properties of polymers. Adv. Polym. Technol. 1990, 10, 31–45. [Google Scholar] [CrossRef]
- Lin, W.; Yang, Z.T.; Qu, J.-P. Short-time fabrication of well-mixed high-density polyethylene/ultrahigh-molecular-weight polyethylene blends under elongational flow: Morphology, mechanical properties and mechanism. Polym. Int. 2019, 68, 904–914. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Efficient disentanglement of polycarbonate melts under complex shear field. Polymer 2020, 201, 122610. [Google Scholar] [CrossRef]
- Hu, H.; Chen, J.; Yang, T.; Wang, P.; Min, J.; Fu, Q.; Zhang, J. Regulation of Entanglement Networks under Different Shear Fields and Its Effect on the Properties of Poly(L-lactide). Ind. Eng. Chem. Res. 2023, 62, 7434–7446. [Google Scholar] [CrossRef]
- Wang, X.H.; Liu, R.; Wu, M.; Wang, Z.; Huang, Y. Effect of chain disentanglement on melt crystallization behavior of isotactic polypropylene. Polymer 2009, 50, 5824–5827. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. The crystallization of polypropylene with reduced density of entanglements. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 748–756. [Google Scholar] [CrossRef]
- Xiao, Z.G.; Sun, Q.; Xue, G.; Yuan, Z.; Dai, Q.; Hu, Y.L. Thermal behavior of isotactic polypropylene freeze-extracted from solutions with varying concentrations. Europ. Polym. J. 2003, 39, 927–931. [Google Scholar] [CrossRef]
- Krajenta, J.; Pawlak, A.; Galeski, A. Deformation of disentangled polypropylene crystalline grains into nanofibers. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1983–1994. [Google Scholar] [CrossRef]
- Pandey, A.; Toda, A.; Rastogi, S. Influence of Amorphous Component on Melting of Semicrystalline Polymers. Macromolecules 2011, 44, 8042–8055. [Google Scholar] [CrossRef]
- Ji, G.; Xue, G.; Ma, J.; Dong, C.; Gu, X. Concentration dependence of crystallinity of polycarbonate by shock-cooling and subsequent freeze-drying of its various solutions. Polymer 1996, 37, 3255–3258. [Google Scholar] [CrossRef]
- Xie, Z.P.; Liu, D.; Zhu, P.P.; Yang, H.Y. Crystallization behavior of chain-disentangled poly(ethylene terephthalate). Acta Polym. Sinica 2010, 5, 522–529. [Google Scholar] [CrossRef]
- Romano, D.; Tops, N.; Andablo-Reyes, E.; Ronca, S.; Rastogi, S. Influence of Polymerization Conditions on Melting Kinetics of Low Entangled UHMWPE and Its Implications on Mechanical Properties. Macromolecules 2014, 47, 4750–4760. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, J.; Liu, M.; Fu, Q.; Zhang, J. Understanding the effect of chain entanglement state on melt crystallization of the polymer freeze-extracted from solution: The role of critical overlap concentration. Polymer 2019, 178, 121588. [Google Scholar] [CrossRef]
- Gaonkar, A.A.; Murudkar, V.V.; Deshpande, V.D. Comparison of crystallization kinetics of polyethylene terephthalate (PET) and reorganized PET. Thermochim. Acta 2020, 683, 178472. [Google Scholar] [CrossRef]
- Ni, L.; Xu, S.; Sun, C.; Qin, Y.; Zheng, Y.; Zhou, J.; Yu, C.; Pan, P. Retarded Crystallization and Promoted Phase Transition of Freeze-Dried Polybutene-1: Direct Evidence for the Critical Role of Chain Entanglement. ACS Macro Lett. 2022, 11, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, W. Weak Shear-Induced Slowdown in Crystallization of Less-Entangled Poly(ε-caprolactone). Macromolecules 2021, 54, 3347–3357. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Fu, Q.; Zhang, J. Novel Strategy to Improve the Performance of Poly(L-lactide): The Synergistic Effect of Disentanglement and Strong Shear Field. ACS Sustain. Chem. Eng. 2023, 11, 9630–9642. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, Y.; Xu, S.; Ni, L.; Li, X.; Shan, G.; Bao, Y.; Pan, P. Role of Chain Entanglements in the Stereocomplex Crystallization between Poly(lactic acid) Enantiomers. ACS Macro. Lett. 2021, 10, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Krajenta, J.; Safandowska, M.; Pawlak, A. The re-entangling of macromolecules in polypropylene. Polymer 2019, 175, 215–226. [Google Scholar] [CrossRef]
- Krajenta, J.; Safandowska, M.; Pawlak, A.; Galeski, A. All-polymer composites—A new approach with the use of disentangled semi-crystalline polymers. Part 1. Disentangling and properties of disentangled polylactide. Polimery 2020, 65, 167–173. [Google Scholar] [CrossRef]
- Krajenta, J.; Polinska, M.; Lapienis, G.; Pawlak, A. The crystallization of poly(ethylene oxide) with limited density of macromolecular entanglements. Polymer 2020, 197, 122500. [Google Scholar] [CrossRef]
- Zhai, Z.; Fusco, C.; Morthomas, J.; Perez, M.; Lame, O. Disentangling and Lamellar Thickening of Linear Polymers during Crystallization: Simulation of Bimodal and Unimodal Molecular Weight Distribution Systems. ACS Nano. 2019, 13, 11310–11319. [Google Scholar] [CrossRef]
- Peng, F.; Nie, C.; Xu, T.Y.; Sheng, J.F.; Chen, W.; Yu, W.C.; Li, L.B. Entanglement on nucleation barrier of polymer crystal. Chin. J. Polym. Sci. 2022, 40, 1640–1650. [Google Scholar] [CrossRef]
- Dai, Q.; Lu, Y.; Xue, G.; Liao, Y.T. Glass transition of atactic polystyrene with less chain entanglements. Polym. Bull. 1995, 35, 209–214. [Google Scholar] [CrossRef]
- Sasaki, T.; Yamauchi, N.; Irie, S.; Sakurai, K. Differential scanning calorimetry study on thermal behaviors of freeze-dried poly(L-lactide) from dilute solutions. J. Polym. Sci. B Polym. Phys. 2005, 43, 115–124. [Google Scholar] [CrossRef]
- Huang, D.H.; Yang, Y.; Zhuang, G.; Li, B. Influence of Entanglements on the Glass Transition and Structural Relaxation Behaviors of Macromolecules. 1. Polycarbonate. Macromolecules 1999, 32, 6675–6678. [Google Scholar] [CrossRef]
- Bernazzani, P.; Simon, S.L.; Plazek, D.J.; Ngai, K.L. Effects of entanglement concentration on Tg and local segmental motions. Europ. Phys. J. E 2002, 8, 201–207. [Google Scholar] [CrossRef]
- Rong, W.; Fan, Z.; Yu, Y.; Bu, H.; Wang, M. Influence of entanglements on glass transition of atactic polystyrene. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2243–2251. [Google Scholar] [CrossRef]
- Huang, D.H.; Yang, Y.; Zhuang, G.; Li, B. Influence of Intermolecular Entanglements on the Glass Transition and Structural Relaxation Behaviors of Macromolecules. 2. Polystyrene and Phenolphthalein Poly(ether sulfone). Macromolecules 2000, 33, 461–464. [Google Scholar] [CrossRef]
- Singh, M.K.; Hu, M.; Cang, Y.; Hsu, H.P.; Therien-Aubin, H.; Koynov, K.; Fytas, G.; Landfester, K.; Kremer, K. Glass Transition of Disentangled and Entangled Polymer Melts: Single-Chain-Nanoparticles Approach. Macromolecules 2020, 53, 7312–7321. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, S.; Xin, Z. The chain dis-entanglement effect of polyhedral oligomeric silsesquioxanes (POSS) on ultra-high molecular weight polyethylene (UHMWPE). Polymer 2020, 202, 122631. [Google Scholar] [CrossRef]
- Lu, X.L.; Xue, G.; Mi, Y.L. Understanding the effect of chain entanglement on the glass transition of a hydrophilic polymer. J. Appl. Polym. Sci. 2011, 119, 2310–2317. [Google Scholar] [CrossRef]
- Li, N.; Yang, Q.; Huang, Y.; Zhang, Q.; Zhao, W. Entropy reduction phenomenon in the non-equilibrium state of freeze-dried polymethyl methacrylate samples. J. Polym. Res. 2014, 21, 392. [Google Scholar] [CrossRef]
- Zheng, W.; Simon, S.L. Polystyrene freeze-dried from dilute solution: Tg depression and residual solvent effects. Polymer 2006, 47, 3520–3527. [Google Scholar] [CrossRef]
- Yue, Z.; Wang, N.; Cao, Y.; Li, W.; Dong, C.-D. Reduced Entanglement Density of Ultrahigh-Molecular-Weight Polyethylene Favored by the Isolated Immobilization on the MgCl2 (110) Plane. Ind. Eng. Chem. Res. 2020, 59, 3351–3358. [Google Scholar] [CrossRef]
- Shan, W.; Xiao, K.; Thomas, E.L. Influence of Entanglements on Ultrahigh Strain Rate Deformation of Polystyrene Micro projectiles. Macromolecules 2022, 55, 9594–9600. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, S.; Yu, X.; Xin, Z.; Ye, C.; Li, Z.; Xia, J. Nascent Particle Sizes and Degrees of Entanglement Are Responsible for the Significant Differences in Impact Strength of Ultrahigh Molecular Weight Polyethylene. J. Polym. Sci. B Polym. Phys. 2019, 57, 632–641. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. Cavitation phenomenon and mechanical properties of partially disentangled polypropylene. Polymer 2018, 151, 15–26. [Google Scholar] [CrossRef]
- Logunova, M.A.; Orekhov, N.D. The Role of Intermolecular Entanglements in the Formation of Nanosized Pores during Deformation of Polyethylene: Atomistic Modeling. Polym. Sci. Series A 2021, 63, 591–599. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Psarras, G.C.; Forte, G.; Martin-Fabiani, I.; Ronca, S. Entanglement dynamics in ultra-high molecular weight polyethylene as revealed by dielectric spectroscopy. Polymer 2018, 50, 35–43. [Google Scholar] [CrossRef]
- Liu, G.; Larson, R.G.; Li, L.; Luo, H.; He, X.; Niu, Y.; Li, G. Influence of Chain Entanglement on Rheological and Mechanical Behaviors of Polymerized Ionic Liquids. Macromolecules 2023, 56, 2719–2728. [Google Scholar] [CrossRef]
- Wu, B.; Cai, Y.; Zhao, X.; Ye, L. Polymer Testing, Fabrication of well-miscible and highly enhanced polyethylene/ultrahigh molecular weight polyethylene blends by facile construction of interfacial intermolecular entanglement. Polym. Test. 2021, 93, 106973. [Google Scholar] [CrossRef]
- Tanaka, H.; Saijo, S.; Kakiage, M.; Yamanobe, T.; Uehara, H. In-situ analysis for melt-drawing behavior of ultra-high molecular weight polyethylene/normal molecular weight polyethylene blend films. Polymer 2021, 213, 123213. [Google Scholar] [CrossRef]
- Takazawa, A.; Kakiage, M.; Yamanobe, T.; Uehara, H.; Shimizu, Y.; Ohnishi, T.; Wakabayashi, Y.; Inatomi, K.; Abe, S.; Aoyama, K. Effect of blending small amount of high-density polyethylene on molecular entanglements during melt-drawing of ultrahigh-molecular-weight polyethylene. Polymer 2022, 241, 124528. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, Y.; Li, W.; Wang, J.; Yang, Y. Melt spinning of polyethylene composites fibers containing disentangled ultra-high molecular weight polyethylene. In Proceedings of the World Textile Conference AUTEX 2022, Lodz, Poland, 7–10 June 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Di, Y.; Chunlin Ye, C.; Zhang, L.; Tang, X.; Shu, B.; Yan, X.; Li, W.; Wang, J.; Yang, Y. Morphology evolution and mechanical property enhancement of linear low-density polyethylene by adding disentangled ultra high molecular weight polyethylene. Polym. Adv. Technol. 2022, 33, 1047–1056. [Google Scholar] [CrossRef]
- Schirmeister, C.G.; Hees, T.; Dolynchuk, O.; Licht, E.H.; Thurn-Albrecht, T.; Muelhaupt, R. Digitally Tuned Multidirectional All-Polyethylene Composites via Controlled 1D Nanostructure Formation during Extrusion-Based 3D Printing. ACS Appl. Polym. Mater. 2021, 3, 1675–1686. [Google Scholar] [CrossRef]
- Tao, G.; Chen, Y.; Mu, J.; Zhang, L.; Ye, C.; Li, W. Exploring the entangled state and molecular weight of UHMWPE on the microstructure and mechanical properties of HDPE/UHMWPE blends. J. Appl. Polym. Sci. 2021, 138, 50741. [Google Scholar] [CrossRef]
- Tang, X.; Xing, J.; Yan, X.; Ye, C.; Zhang, L.; Zhang, Y.; Shu, B.; Mu, J.; Wei, L.; Wang, J.; et al. Metallocene Polyolefins Reinforced by Low-Entanglement UHMWPE through Interfacial Entanglements. Adv. Polym. Technol. 2022, 2022, 9344096. [Google Scholar] [CrossRef]
- Smith, P.; Lemstra, P.J. Ultra-drawing of high molecular weight polyethylene cast from solution. Coll. Polym. Sci. 1980, 258, 891–894. [Google Scholar] [CrossRef]
- Spronck, M.; Klein, A.; Blom, B.; Romano, D. Synthesis of Disentangled Ultra-High Molecular Weight Polyethylene using Vanadium(V)-Based Catalysts. Z. Anorg. Allg. Chem. 2018, 644, 993–998. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, P.; Yue, Z.; Li, W.; Dong, C.; Jiang, B.; Wang, J.; Yang, Y. Entanglement Formation Mechanism in the POSS Modified Heterogeneous Ziegler−Natta Catalysts. Macromolecules 2019, 52, 7593–7602. [Google Scholar] [CrossRef]
- Cao, Y.; Wu, Y.; Tang, X.; Zhou, Q.; Stapf, S.; Mattea, C.; Li, W. Long-term efficiency for reducing entanglements of nascent polyethylene by a polystyrene-modified Ziegler-Natta catalyst. J. Appl. Polym. Sci. 2022, 39, 51790. [Google Scholar] [CrossRef]
- Chammingkwan, P.; Bando, Y.; Mai, L.T.T.; Wada, T.; Thakur, A.; Terano, M.; Sinthusai, L.; Taniike, T. Less Entangled Ultrahigh-Molecular-Weight Polyethylene Produced by Nano-Dispersed Ziegler−Natta Catalyst. Ind. Eng. Chem. Res. 2021, 60, 2818–2827. [Google Scholar] [CrossRef]
- Chen, Y.; Li, W.; Zhang, L.; Ye, C.; Tao, G.; Ren, C.; Jiang, B.; Wang, J.; Yang, Y. In Situ Synthesized Self-Reinforced HDPE/UHMWPE Composites with High Content of Less Entangled UHMWPE and High Gradient- Distributed Oriented Structures. ACS Appl. Polym. Mater. 2023, 5, 88–98. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, G.; Li, W.; Zhou, Q.; Shu, B.; Jiang, B.; Wang, J.; Yang, Y. The in situ synthesis of weakly entangled ultrahigh-molecular weight polyethylene/polyethylene wax blends and its synergetic disentanglement effect on LLDPE reinforcement. Polym. Adv. Technol. 2022, 33, 3728–3739. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Forte, G.; Ronca, S. Relaxation Dynamics in Disentangled Ultrahigh Molecular Weight Polyethylene via Torsional Rheology. Ind. Eng. Chem. Res. 2020, 59, 4515–4523. [Google Scholar] [CrossRef]
- Christakopoulos, F.; Bersenev, E.; Grigorian, S.; Brem, A.; Ivanow, D.; Tervoort, T.O.; Litvinov, V. Melting-Induced Evolution of Morphology, Entanglement Density, and Ultradrawability of Solution-Crystallized Ultrahigh-Molecular-Weight Polyethylene. Macromolecules 2021, 54, 5683–5693. [Google Scholar] [CrossRef]
- Petrov, A.; Rudyak, V.Y.; Chertovich, A. Optimal. Entanglement of Polymers Promotes the Formation of Highly Oriented Fibers. Macromolecules 2022, 55, 6493–6504. [Google Scholar] [CrossRef]
- Li, W.; Guan, C.; Xu, J.; Chen, Z.; Jiang, B.; Wang, J.; Yang, Y. Bimodal/broad polyethylene prepared in a disentangled state. Industr. Eng. Chem. Res. 2014, 53, 1088–1096. [Google Scholar] [CrossRef]
- Lippits, D.R.; Rastogi, S.; Talebi, S.; Bailly, C. Formation of entanglements in initially disentangled polymer melts. Macromolecules 2006, 39, 8882–8885. [Google Scholar] [CrossRef]
- Talebi, S.; Duchateau, R.; Rastogi, S.; Kaschta, J.; Peters, G.; Lemstra, P.J. Molar mass and molecular weight distribution determination of UHMWPE synthesized using a living homogeneous catalyst. Macromolecules 2010, 43, 2780–2788. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Y.; Chen, J.; Luo, J.; Fu, Q.; Zhang, J. The retarded recovery of disentangled state by blending HDPE with ultra-high molecular weight polyethylene. Polymer 2020, 192, 122329. [Google Scholar] [CrossRef]
- Barham, P.J.; Sadler, D.M. A neutron scattering study of the melting behavior of polyethylene single crystals. Polymer 1991, 32, 393–395. [Google Scholar] [CrossRef]
- Chai, S.-C.; Xu, T.-Y.; Cao, X.; Wang, G.; Chen, Q.; Li, H.-L. Ultrasmall Nanoparticles Diluted Chain Entanglement in Polymer Nanocomposites. Chin. J. Polym. Sci. 2019, 37, 797–805. [Google Scholar] [CrossRef]
- Mackay, M.E.; Dao, T.T.; Tuteja, A.; Ho, D.L.; Horn, B.V.; Kim, H.C.; Hawker, C.J. Nanoscale effects leading to non-Einstein-like decrease in viscosity. Nat. Mater. 2003, 2, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Nusser, K.; Schneider, G.J.; Pyckhout-Hintzen, W.; Richter, D. Viscosity decrease and reinforcement in polymer-silsesquioxane composites. Macromolecules 2011, 44, 7820–7830. [Google Scholar] [CrossRef]
- Mangal, R.; Srivastava, S.; Archer, L.A. Phase stability and dynamics of entangled polymer-nanoparticle composites. Nat. Commun. 2015, 6, 7198. [Google Scholar] [CrossRef] [PubMed]
- Senses, E.; Ansar, S.M.; Kitchens, C.L.; Mao, Y.; Narayanan, S.; Natarajan, B.; Faraone, A. Small Particle Driven Chain Disentanglements in Polymer Nanocomposites. Phys. Rev. Lett. 2017, 118, 147801. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Yui, Y.; Wei, P.; Cong, C.; Meng, X.; Ye, H.-M.; Zhou, Q. Nanoscale effects of TiO2 nanoparticles on the rheological behaviors of ultra-high molecular weight polyethylene (UHMWPE). Soft Matter 2023, 19, 5459–5467. [Google Scholar] [CrossRef]
- Romo-Uribe, A. Dispersion at Single Unit TiO2 Nanoparticles Reduced Tg, Induced Chain Disentanglement and Reduced Tensile Modulus in Waterborne Acrylic Coatings. Macromol. Mater. Eng. 2021, 306, 200059. [Google Scholar] [CrossRef]
- Luo, J.; Liu, M.; Chen, J.; Min, J.; Fu, Q.; Zhang, J. Effectively maintaining the disentangled state of isotactic polypropylene in the presence of graphene nanoplatelet. Polymer 2021, 226, 23806. [Google Scholar] [CrossRef]
- Luo, J.; Chen, J.; Liu, M.; Min, J.; Fu, Q.; Zhang, J. Investigating the Influence of Incorporation of Boron Nitride on the Kinetics of Isotactic Polypropylene Entanglement Recovery. Ind. Eng. Chem. Res. 2021, 60, 12901–12910. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Tarallo, O.; Guan, L.; Martin-Fabiani, I.; Ronca, S. Nanocomposites of Au/Disentangled UHMWPE: A Combined Optical and Structural Study. Molecules 2020, 25, 3225. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Psarras, G.C.; Ronca, S. Oriented ultra-high molecular weight polyethylene/gold nanocomposites: Electrical conductivity and chain entanglement dynamics. Express Polym. Lett. 2021, 15, 492–502. [Google Scholar] [CrossRef]
- Barangizi, H.; Pawlak, A. Crystallization of partially disentangled polypropylene in nanocomposites with aluminum oxide. Polymer 2022, 254, 125049. [Google Scholar] [CrossRef]
- Barangizi, H.; Krajenta, J.; Pawlak, A. The influence of entanglements of macromolecules on the mechanical and thermal properties of polylactide composites with carbon nanotubes. Express Polym. Lett. 2023, 17, 738–758. [Google Scholar] [CrossRef]
- Jurczuk, K.; Galeski, A.; Piorkowska, E. All-polymer nanocomposites with nanofibrillar inclusions generated in situ during compounding. Polymer 2013, 54, 4617. [Google Scholar] [CrossRef]
Figure 1.
Storage (G′) and loss (G″) moduli for a monodisperse long-chain polymer in the function of frequency. The plateau modulus, GN0, is also shown.
Figure 1.
Storage (G′) and loss (G″) moduli for a monodisperse long-chain polymer in the function of frequency. The plateau modulus, GN0, is also shown.
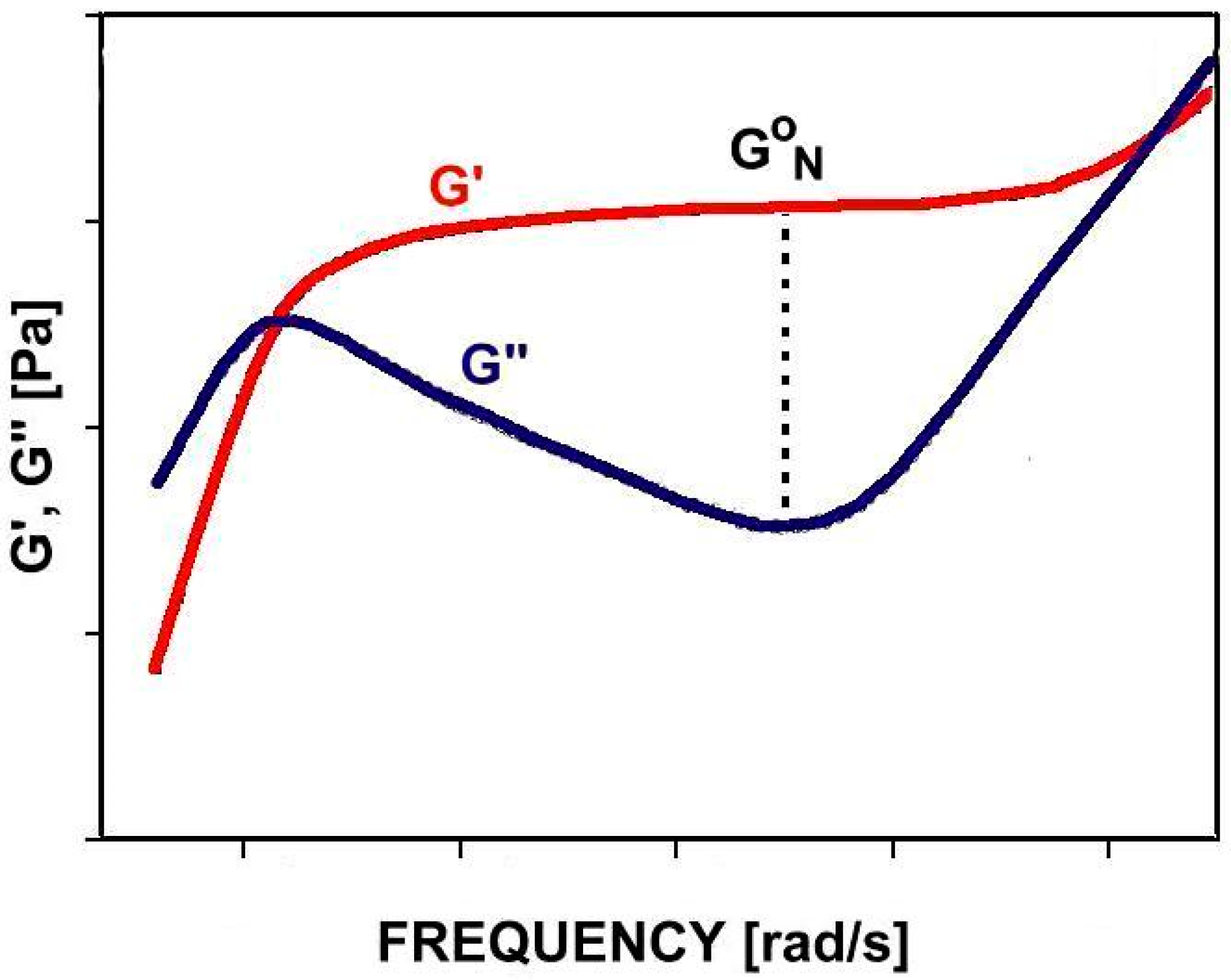
Figure 2.
The idea of a macromolecule moving inside a virtual tube caused by the entanglement of other macromolecules.
Figure 2.
The idea of a macromolecule moving inside a virtual tube caused by the entanglement of other macromolecules.

Figure 3.
Plot of nominal stress against nominal strain for a series of rapidly quenched linear PEs with the most probable molecular weight distributions. The values of Mw are indicated. A strain-hardening phase is clearly visible [35]. Reproduced with permission: 1994, American Chemical Society.
Figure 3.
Plot of nominal stress against nominal strain for a series of rapidly quenched linear PEs with the most probable molecular weight distributions. The values of Mw are indicated. A strain-hardening phase is clearly visible [35]. Reproduced with permission: 1994, American Chemical Society.

Figure 4.
Variation in viscosity of HDPE with shear time for different end shear rates in a range of 2–12 s−1 [68]. Reproduced with permission: John Wiley and Sons.
Figure 4.
Variation in viscosity of HDPE with shear time for different end shear rates in a range of 2–12 s−1 [68]. Reproduced with permission: John Wiley and Sons.

Figure 5.
Schematic of polymer melt disentangling machine [78]. Reproduced with permission: Elsevier.
Figure 5.
Schematic of polymer melt disentangling machine [78]. Reproduced with permission: Elsevier.
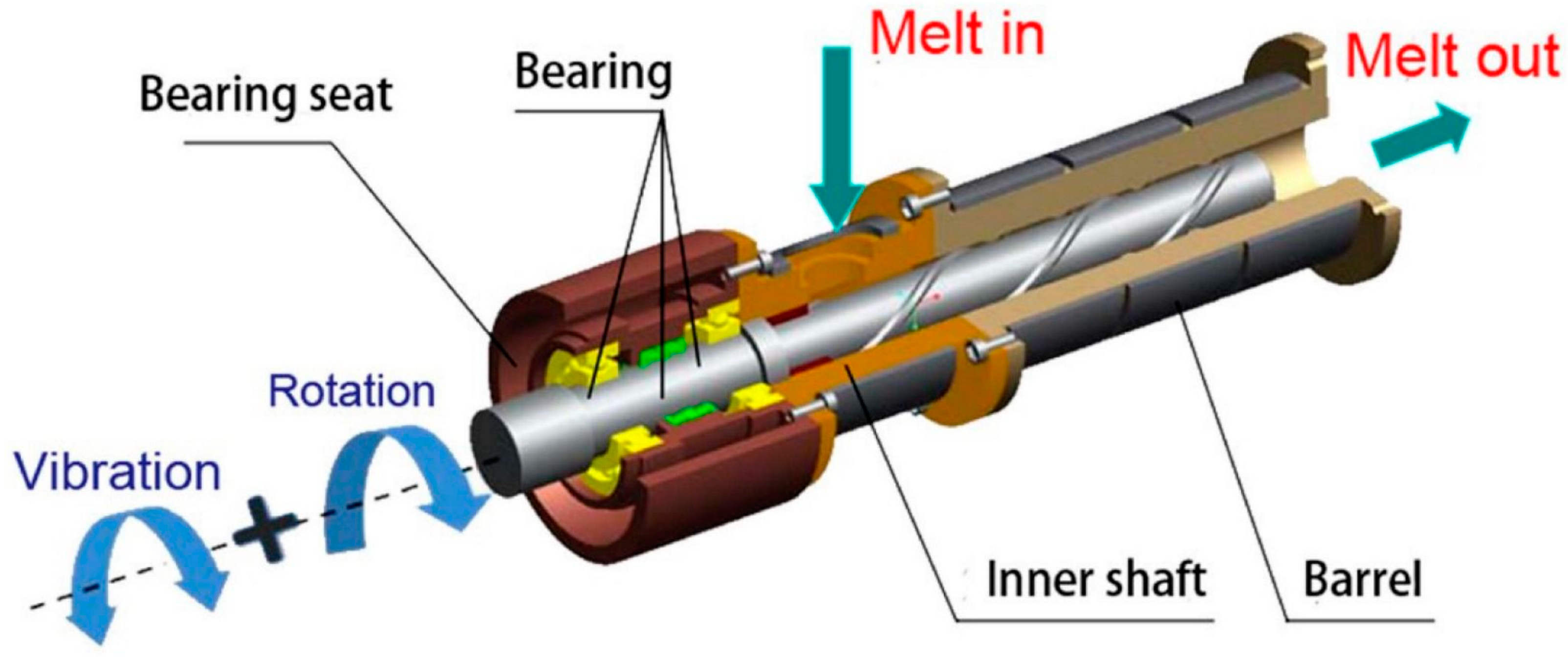
Figure 6.
Spherulite growth rate in PB disentangled by dissolving it in p-xylene, with a concentration of c = 0.1−2 wt.%. The critical overlap concentration, c*, was estimated to be ~0.6 wt.%. The growth rate for bulk, entangled polymer is also shown [90]. Reproduced with permission: American Chemical Society.
Figure 6.
Spherulite growth rate in PB disentangled by dissolving it in p-xylene, with a concentration of c = 0.1−2 wt.%. The critical overlap concentration, c*, was estimated to be ~0.6 wt.%. The growth rate for bulk, entangled polymer is also shown [90]. Reproduced with permission: American Chemical Society.
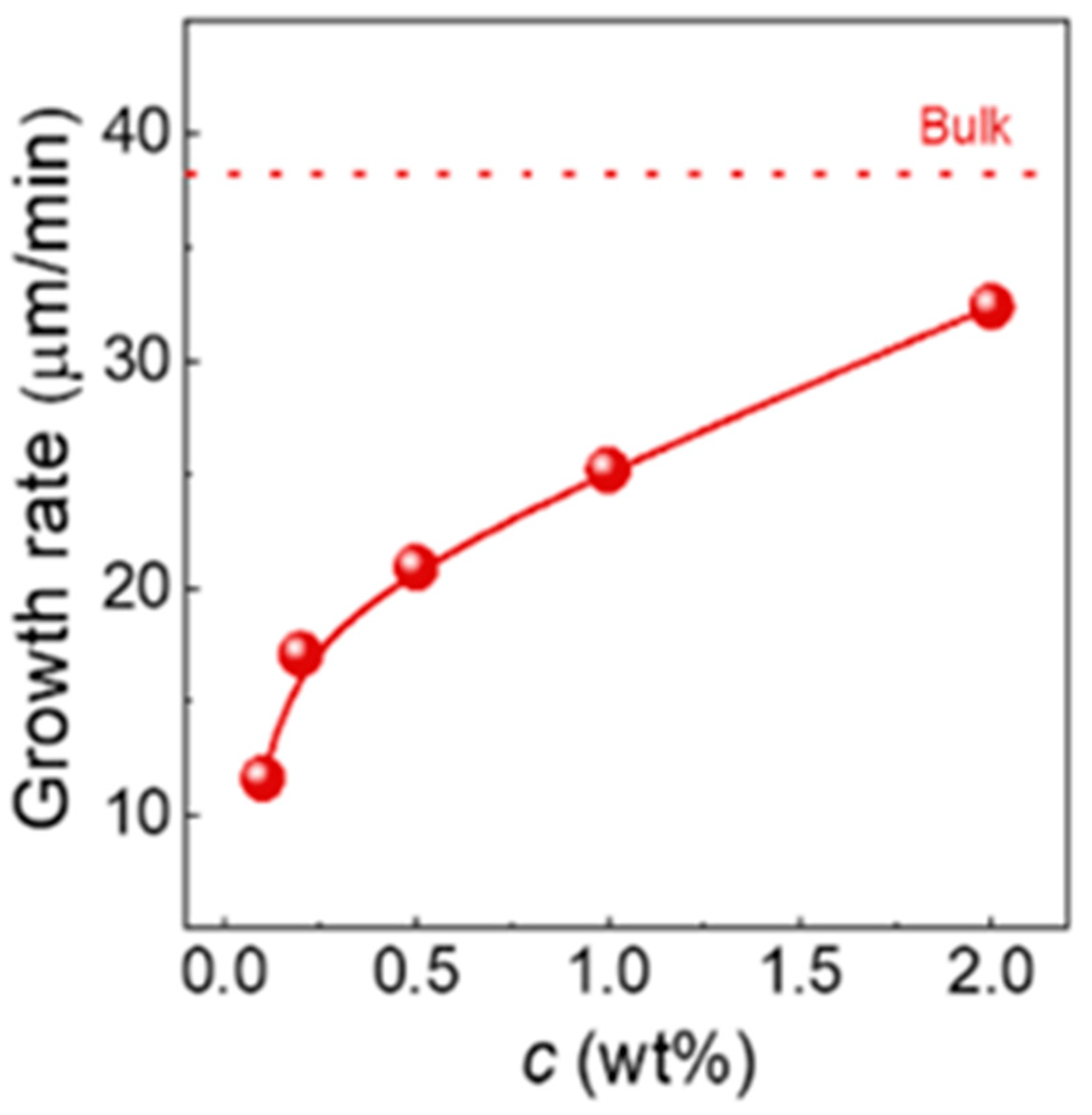
Figure 7.
A dependence log of G + ∆E/[2.3 R(Tc − T∞)] versus 1/(Tc∆T) drawn for the initial PP (PPi) and two disentangled PPs (PPo, PPx). The temperatures of the transition between crystallization regime II (on the right side) and regime III (on the left side) are also shown [81]. Reproduced with permission: John Wiley and Sons.
Figure 7.
A dependence log of G + ∆E/[2.3 R(Tc − T∞)] versus 1/(Tc∆T) drawn for the initial PP (PPi) and two disentangled PPs (PPo, PPx). The temperatures of the transition between crystallization regime II (on the right side) and regime III (on the left side) are also shown [81]. Reproduced with permission: John Wiley and Sons.
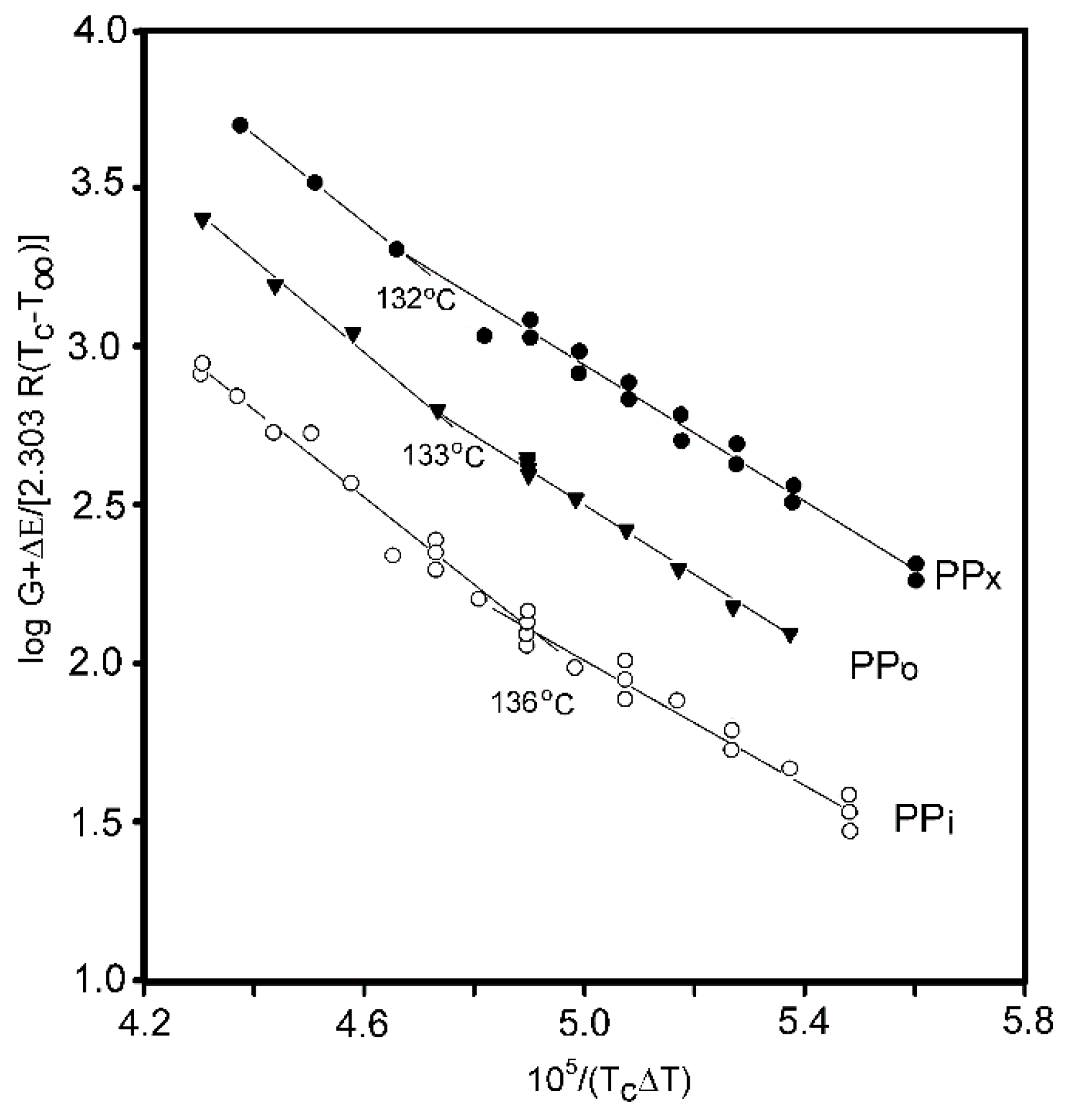
Figure 8.
Comparison of the glass transition temperature, Tg, of entangled and disentangled PS. NPs—single-polymer chain nanoparticles, NFs—nanofibers [105]. Reproduced with permission: American Chemical Society.
Figure 8.
Comparison of the glass transition temperature, Tg, of entangled and disentangled PS. NPs—single-polymer chain nanoparticles, NFs—nanofibers [105]. Reproduced with permission: American Chemical Society.
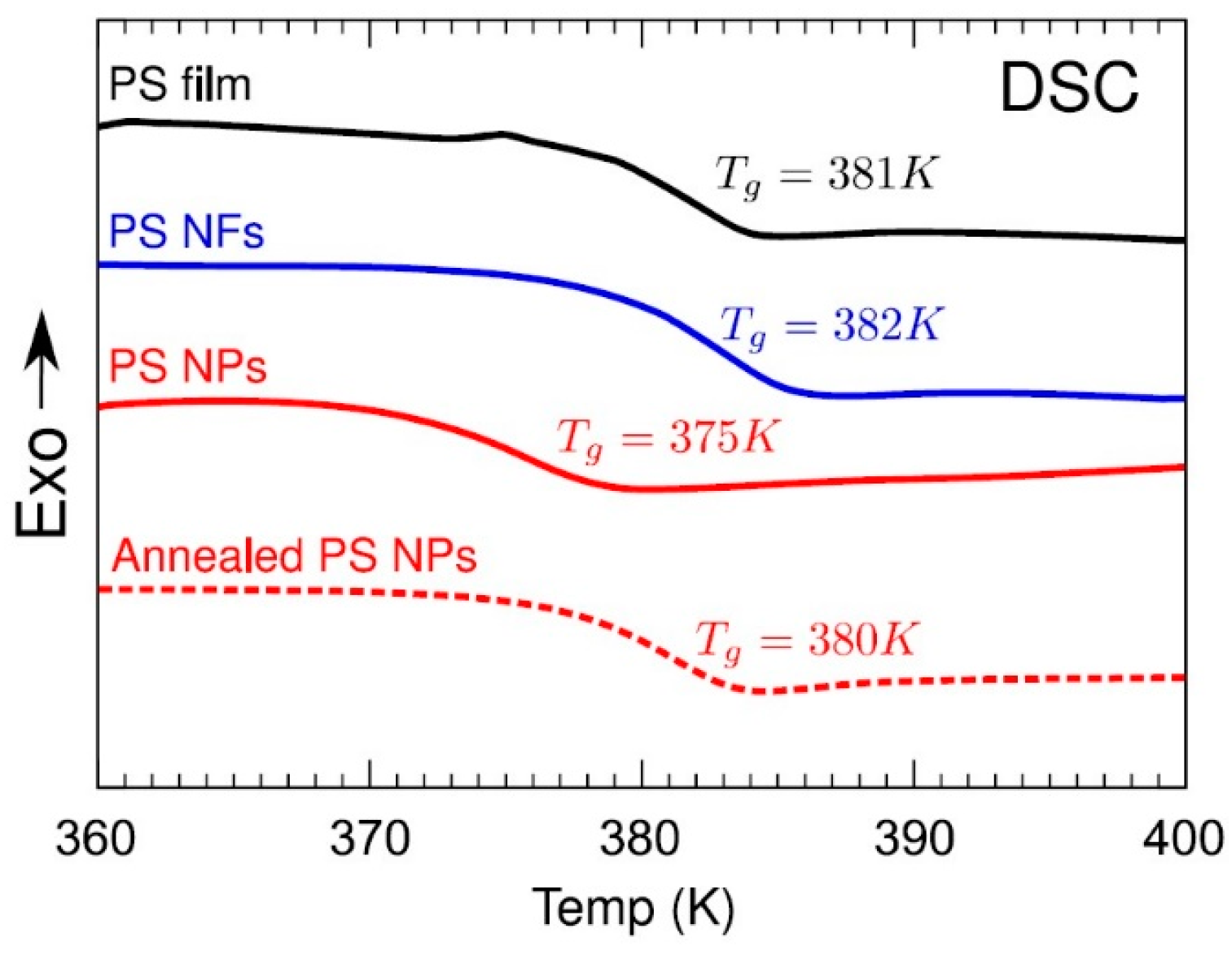
Figure 9.
Schematic of the high-rate deformation processes for disentangled (a) and entangled (b) glassy PS [111]. Reproduced with permission: American Chemical Society.
Figure 9.
Schematic of the high-rate deformation processes for disentangled (a) and entangled (b) glassy PS [111]. Reproduced with permission: American Chemical Society.
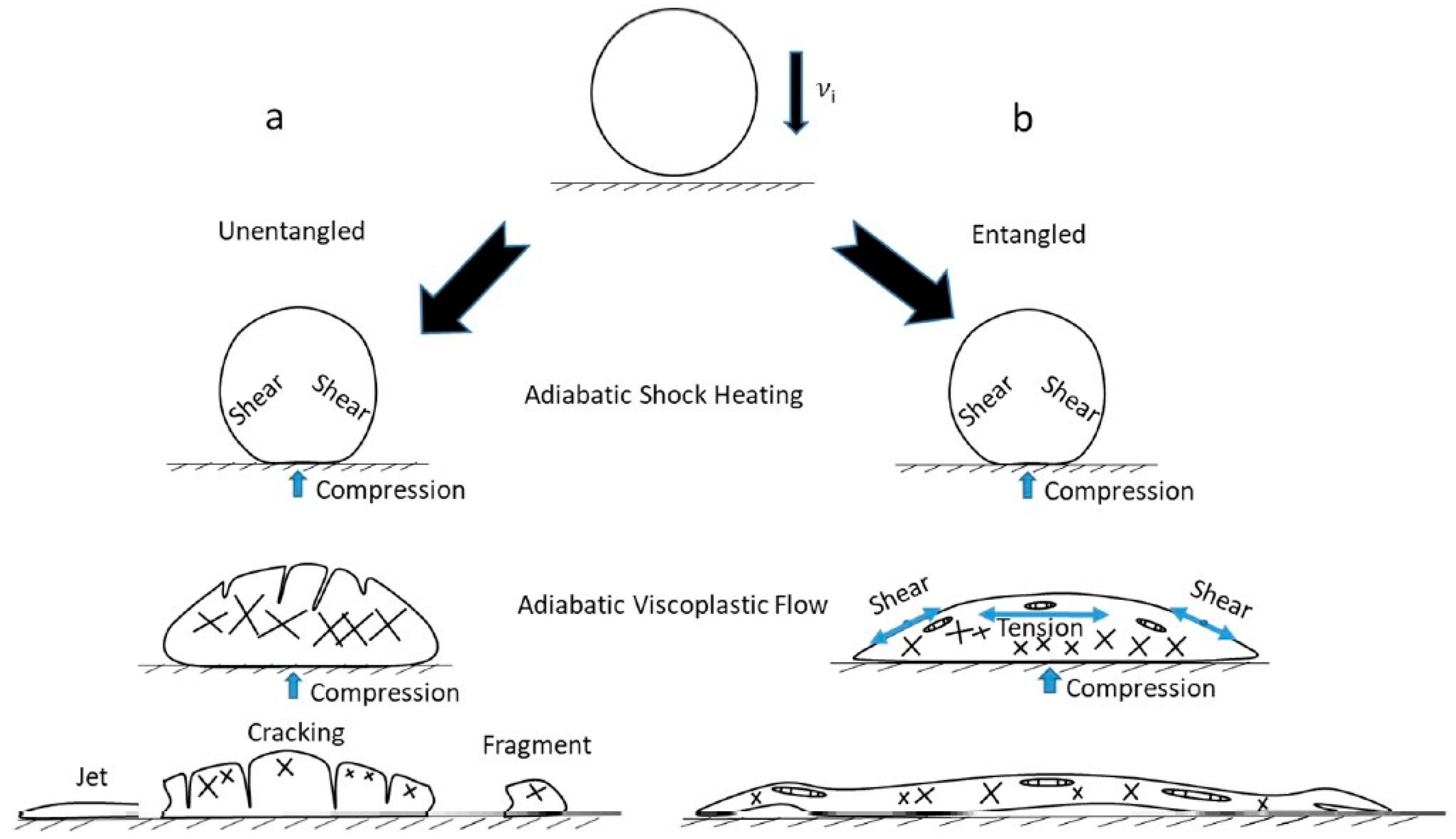
Figure 10.
(A–D) The mechanical properties of UHMWPE/LLDPE blends. The numbers in blend names indicate the weight fraction of UHMWPE [121]. Reproduced with permission: John Wiley and Sons.
Figure 10.
(A–D) The mechanical properties of UHMWPE/LLDPE blends. The numbers in blend names indicate the weight fraction of UHMWPE [121]. Reproduced with permission: John Wiley and Sons.
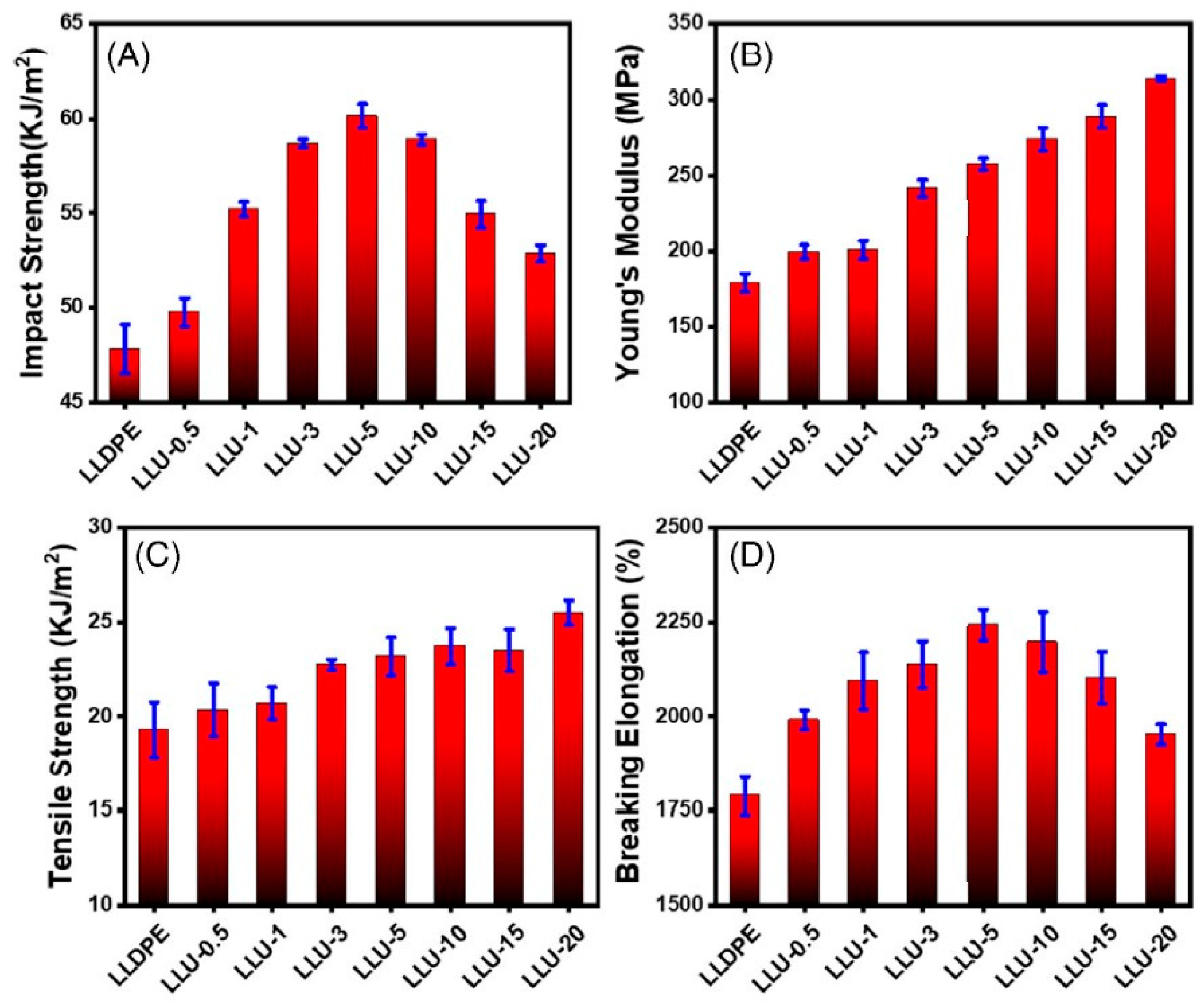
Figure 11.
(a) Changes in the G′ modulus over time for rheological measurements, at 180 °C, of entangled and disentangled PP [80]. (b) Buildup of normalized storage modulus for HDPE/UHMWPE blends at 200 °C. The H/U5-18 designation means HDPE with a 5% UHMWPE addition with a shear rate of 18 s−1 [138]. Reproduced with permission: Elsevier.
Figure 11.
(a) Changes in the G′ modulus over time for rheological measurements, at 180 °C, of entangled and disentangled PP [80]. (b) Buildup of normalized storage modulus for HDPE/UHMWPE blends at 200 °C. The H/U5-18 designation means HDPE with a 5% UHMWPE addition with a shear rate of 18 s−1 [138]. Reproduced with permission: Elsevier.
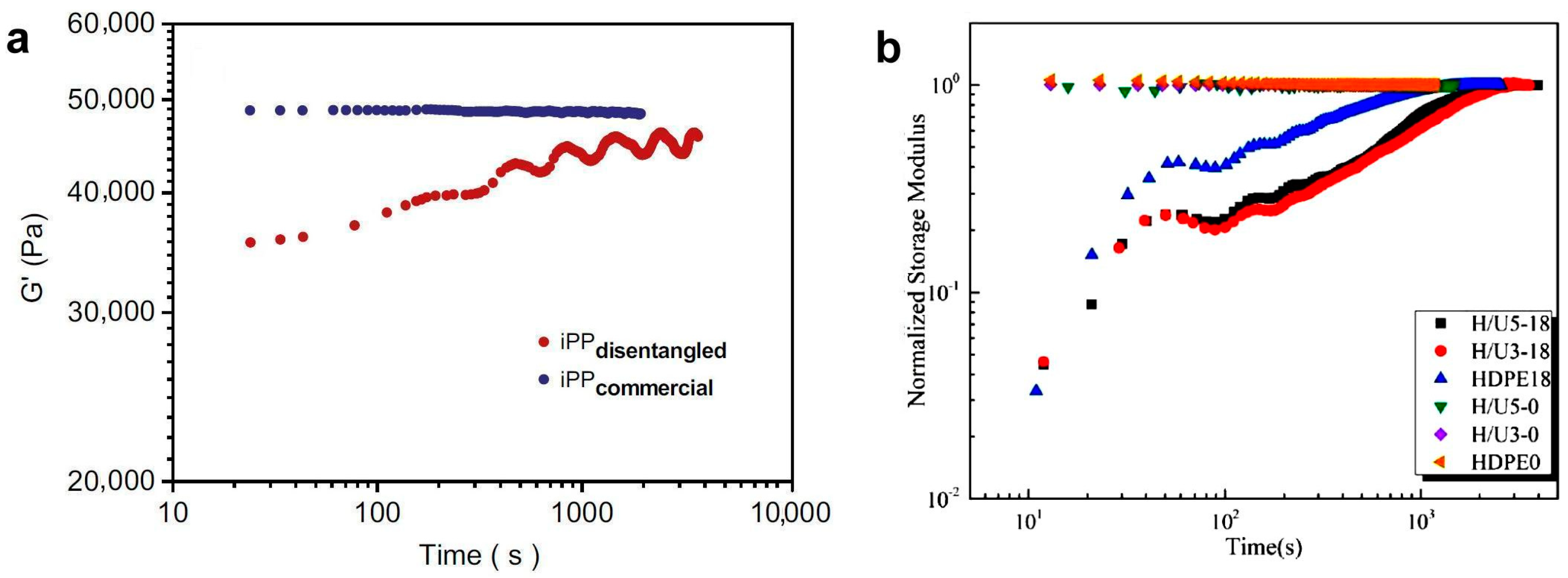
Figure 12.
Dependence of stress on the degree of compression of samples made of entangled (PPi) and partially disentangled (PP2%) polypropylene. The effect of heating in the melt is also shown, leading to the regeneration of entanglements after 2 h [94]. Reproduced with permission: Elsevier.
Figure 12.
Dependence of stress on the degree of compression of samples made of entangled (PPi) and partially disentangled (PP2%) polypropylene. The effect of heating in the melt is also shown, leading to the regeneration of entanglements after 2 h [94]. Reproduced with permission: Elsevier.

Figure 13.
(a) Complex viscosity of UHMWPE and POSS (0.1–1 wt.%) composite measured with torque rheometer; (b) tensile properties of composite regarding the function of POSS content [106]. Reproduced with permission: Elsevier.
Figure 13.
(a) Complex viscosity of UHMWPE and POSS (0.1–1 wt.%) composite measured with torque rheometer; (b) tensile properties of composite regarding the function of POSS content [106]. Reproduced with permission: Elsevier.

Figure 14.
(a) The storage modulus regarding the function of frequency for PP/graphene nanocomposites. Interlayer slipping in the composite decreased the G′ value, but for higher graphene (GnP) content, the opposite effect of chain movement restriction due to nanoplatelets is visible; (b) buildup of normalized viscosity with time at 200 °C during recovery of entanglement [147]. Reproduced with permission: Elsevier.
Figure 14.
(a) The storage modulus regarding the function of frequency for PP/graphene nanocomposites. Interlayer slipping in the composite decreased the G′ value, but for higher graphene (GnP) content, the opposite effect of chain movement restriction due to nanoplatelets is visible; (b) buildup of normalized viscosity with time at 200 °C during recovery of entanglement [147]. Reproduced with permission: Elsevier.
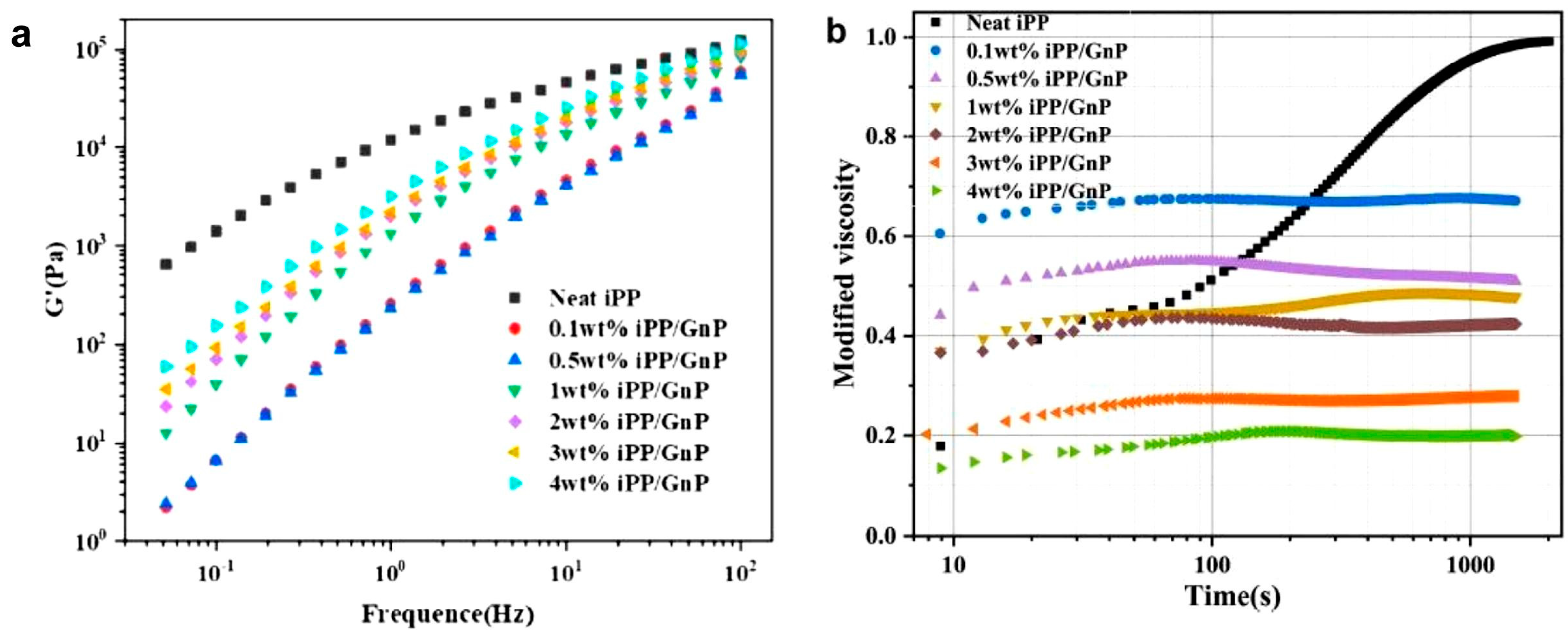
Figure 15.
Schematic showing changes in the structure of PP and a PP/boron nitride composite during disentanglement and recovery in the melt [148]. Reproduced with permission: American Chemical Society.
Figure 15.
Schematic showing changes in the structure of PP and a PP/boron nitride composite during disentanglement and recovery in the melt [148]. Reproduced with permission: American Chemical Society.
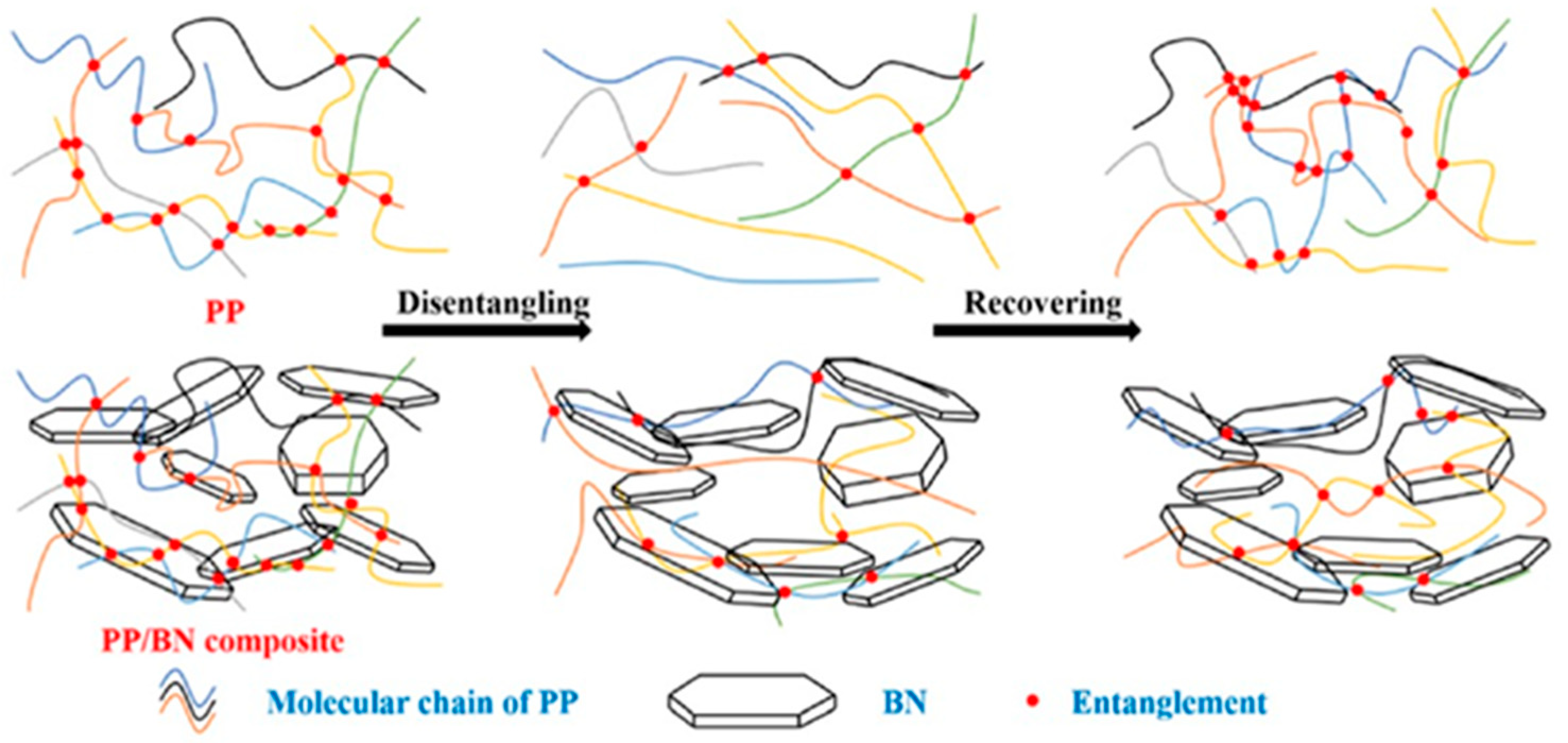
Figure 16.
Conversion of the melt into crystals during non-isothermal crystallization of PP and PP/Al2O3 composites. PP2 is a disentangled polymer, and PP2c is its composite with 1 wt.% Al2O3; PP10 is an entangled polymer, and PP10c is its composite with 1 wt.% Al2O3 [151]. Reproduced with permission: Elsevier.
Figure 16.
Conversion of the melt into crystals during non-isothermal crystallization of PP and PP/Al2O3 composites. PP2 is a disentangled polymer, and PP2c is its composite with 1 wt.% Al2O3; PP10 is an entangled polymer, and PP10c is its composite with 1 wt.% Al2O3 [151]. Reproduced with permission: Elsevier.
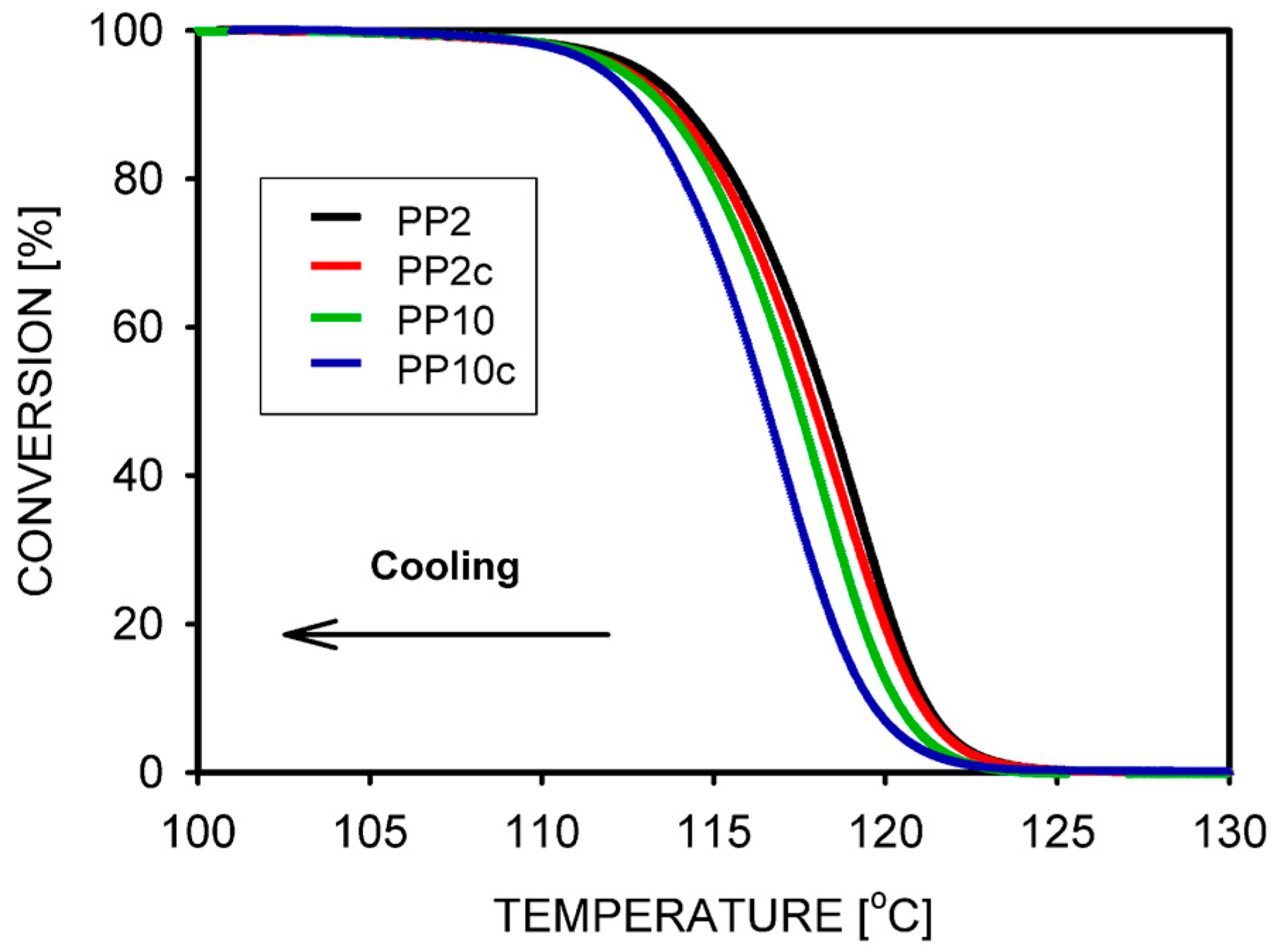
Figure 17.
Strain–stress curves showing the mechanical properties of PLA and PLA/MWCNT composite samples in a tensile test at 70 °C. PLAi—entangled polymer; PLAd—disentangled polymer; PLAi 0.1 and PLAi 1.0—composites of entangled polymers with 0.1 and 1.0 wt.% MWCNT, respectively; PLAd 0.1 and PLAd 1.0—composites of entangled polymers with 0.1 and 1.0 wt.% MWCNT, respectively [152].
Figure 17.
Strain–stress curves showing the mechanical properties of PLA and PLA/MWCNT composite samples in a tensile test at 70 °C. PLAi—entangled polymer; PLAd—disentangled polymer; PLAi 0.1 and PLAi 1.0—composites of entangled polymers with 0.1 and 1.0 wt.% MWCNT, respectively; PLAd 0.1 and PLAd 1.0—composites of entangled polymers with 0.1 and 1.0 wt.% MWCNT, respectively [152].
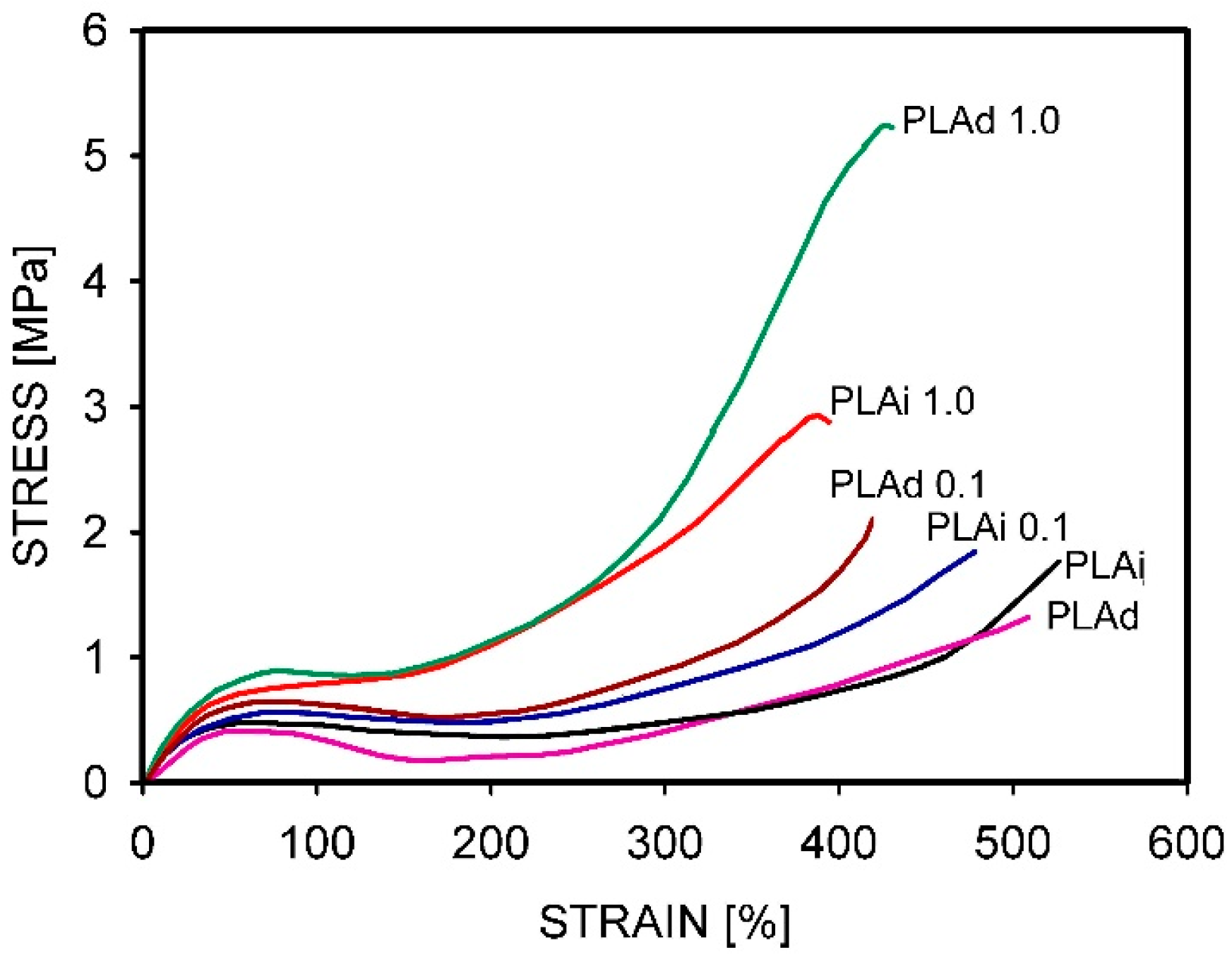
Figure 18.
Changes in storage modulus over time for UHMWPE/POSS composites [63]. Reproduced with permission: Elsevier.
Figure 18.
Changes in storage modulus over time for UHMWPE/POSS composites [63]. Reproduced with permission: Elsevier.
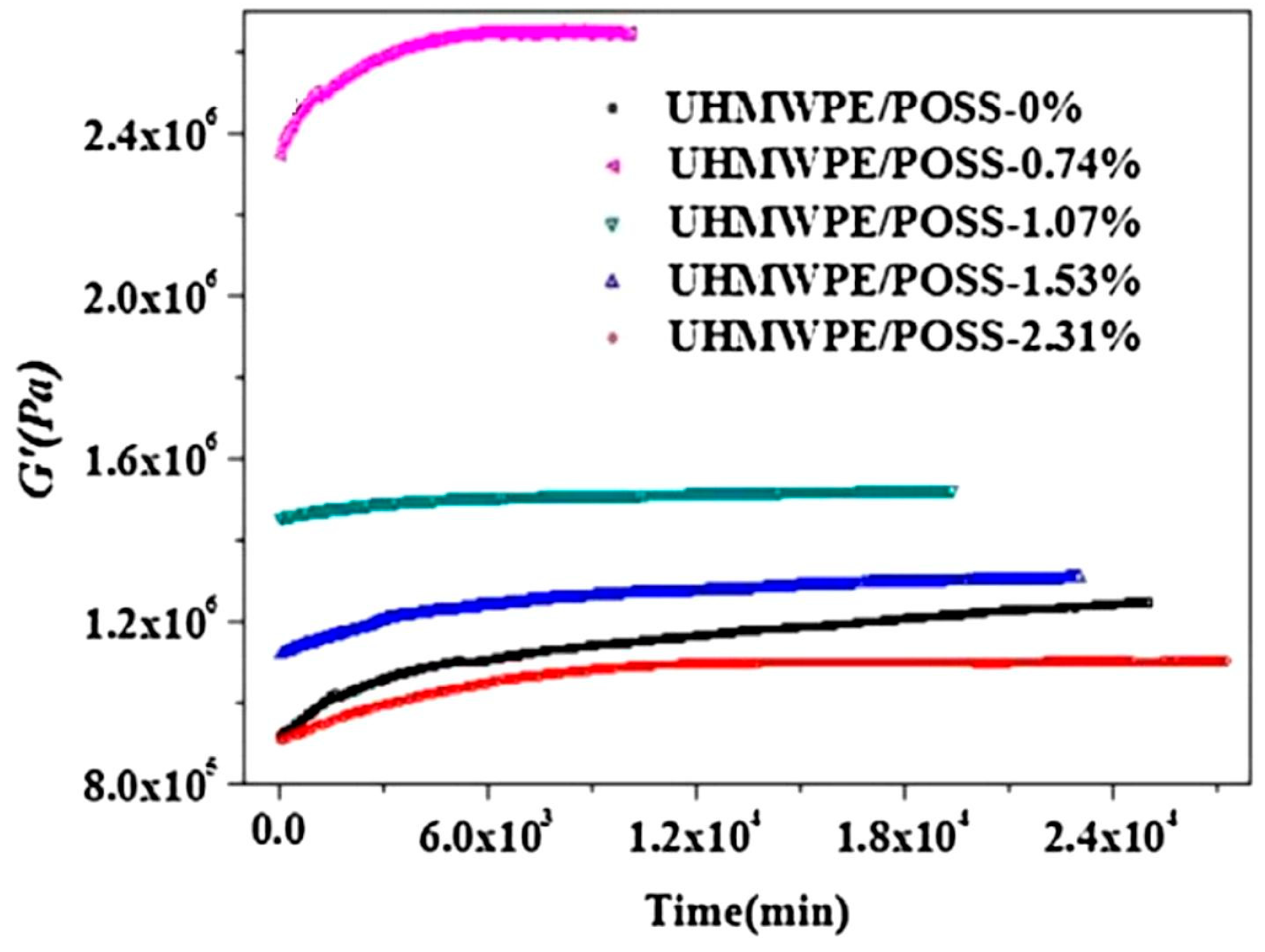
Figure 19.
SEM image of PP fibers formed via shear deformation during compounding with PS. The PS matrix was removed here via dissolution in toluene [83]. Reproduced with permission: John Wiley and Sons.
Figure 19.
SEM image of PP fibers formed via shear deformation during compounding with PS. The PS matrix was removed here via dissolution in toluene [83]. Reproduced with permission: John Wiley and Sons.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pawlak, A.; Krajenta, J. Progress in Studies of Disentangled Polymers and Composites. J. Compos. Sci. 2023, 7, 521. https://doi.org/10.3390/jcs7120521
AMA Style
Pawlak A, Krajenta J. Progress in Studies of Disentangled Polymers and Composites. Journal of Composites Science. 2023; 7(12):521. https://doi.org/10.3390/jcs7120521
Chicago/Turabian StylePawlak, Andrzej, and Justyna Krajenta. 2023. "Progress in Studies of Disentangled Polymers and Composites" Journal of Composites Science 7, no. 12: 521. https://doi.org/10.3390/jcs7120521