

Temperature-Dependent Elastic Properties of B4C from First-Principles Calculations and Phonon Modeling

Department of Mechanical Engineering, University of Alberta, Edmonton, AB T6G 2R3, Canada
*
Author to whom correspondence should be addressed.
Ceramics 2024, 7(1), 235-249; https://doi.org/10.3390/ceramics7010015
Submission received: 14 December 2023
/
Revised: 12 February 2024
/
Accepted: 19 February 2024
/
Published: 21 February 2024
(This article belongs to the Special Issue Advances in Ceramics, 2nd Edition)
Abstract
:Boron carbide plays a crucial role in various extreme environment applications, including thermal barrier coatings, aerospace applications, and neutron absorbers, because of its high thermal and chemical stability. In this study, the temperature-dependent elastic stiffness constants, thermal expansion coefficient, Helmholtz free energy, entropy, and heat capacity at a constant volume () of rhombohedral B4C have been predicted using a quasi-harmonic approach. A combination of volume-dependent first-principles calculations (density functional theory) and first-principles phonon calculations in the supercell framework has been performed. Good agreement between the elastic constants and structural parameters from static calculations is observed. The calculated thermodynamic properties from phonon calculations show trends that align with the literature. As the temperature rises, the predicted free energy follows a decreasing trend, while entropy and follow increasing trends with temperature. Comparisons between the predicted room temperature thermal expansion coefficient (TEC) ( K−1) and bulk modulus (228 GPa) from the quasi-harmonic approach and literature results from experiments and models are performed, revealing that the calculated TEC and bulk modulus fall within the established range from the limited set of data from the literature (TEC = 5.73–9.50 × K−1, B = 221–246 GPa). Temperature-dependent are predicted, enabling stress analysis at elevated temperatures. Overall, the outcomes of this study can be used when performing mechanical and thermal stress analysis (e.g., space shielding applications) and optimizing the design of boron carbide materials for elevated temperature applications.
1. Introduction
Advanced ceramics, such as boron carbide, exhibit low density (∼2.5 [1]), high strength (∼3 to 5 GPa [2]), high hardness (∼25 to 38 GPa [3]), high melting temperature (2723 K [4]), and high chemical and thermal stability [5]. These desirable properties make boron carbide suitable for applications involving extreme environments, including aerospace [6], body armor [7], wear-resistant components [8], abrasive powder [9], and neutron absorbers [10]. Given the significance of advanced ceramics in various industries (e.g., medical devices [11], and aircraft and satellite components [12]) and broad research interests toward understanding their behavior under extreme conditions, accurately predicting their behavior and mechanical properties is crucial for designing and optimizing boron carbide-based materials. Computational methods like molecular dynamics and density functional theory provide efficient tools to investigate the behavior of materials under different loading conditions, with reduced cost and time compared to experimental approaches [13,14]; these tools are used here to study the effects of temperature on the elastic and thermal properties of boron carbide. The primitive cell of boron carbide is a rhombohedral lattice composed of 15 atoms with 12-atom boron icosahedra and a 3-atom chain in the middle of the lattice with the configuration of B12C3 or B4C [15]. The covalent bonds between B and C atoms contribute to the overall thermal and structural stability of the material [9,16].
Traditional methods for predicting the elastic properties of materials rely on empirical relationships, such as Hooke’s law, which are based on experimental data and generally provide reasonable estimates for well-established materials [17]. However, these methods can be limited when applied to extreme conditions (e.g., very low or high temperatures [18], high pressure [19], and corrosive environments [20]), as well as for complex materials (e.g., advanced ceramics [21,22] and composites [23,24]). The elastic properties reported for boron carbide typically originate from room-temperature experimental setups. For example, the Young’s modulus of B4C, measured using acoustic methods, falls within the range of 460–470 GPa [25,26]. Several studies have predicted the bulk modulus of boron carbide at room temperature through experiments [27] and atomistic calculations [28]. These studies suggest a range of 220–250 GPa for the bulk modulus of B4C at 298 K. Although the elastic properties of boron carbide have been extensively studied at room temperature [27,28,29,30,31,32], there is still a gap in the knowledge regarding how these properties vary at elevated temperatures, where boron carbide has numerous applications. Challenges arise from the need for extensive experimental testing and the difficulty of capturing the full complexity of material behavior. Traditional methods often overlook factors like microstructural effects [23], anisotropy [33], and temperature dependence [34]. To address these challenges, advanced computational techniques, including first-principles methods [28,35,36], molecular dynamics simulations [21,37], and machine learning approaches [14,38], have been developed. These methods not only enable more accurate predictions of elastic properties, but also, when appropriately validated, can overcome the limitations of traditional approaches based solely on empirical relationships and experimental data [17,39]. Significant advancements in computer resources and first-principles methodologies have enabled the prediction of single crystal static elastic stiffness constants (s) at 0 K using the strain energy and stress–strain methods. As an example, the elastic constants of single crystal materials such as B4C [28], Al2O3 [40], Ti3B4 [41], and Fe3C [42] have been successfully predicted through first-principles calculations. In addition to this, density functional theory and molecular dynamics simulations have been used for investigating the microstructure and mechanical properties [43], phase transformation and amorphization [44], and semiconductor behavior of boron carbide [45]. Despite these achievements, the exploration of the pressure dependence of elastic constants has received more attention, while the estimation of s at elevated temperatures remains largely unexplored, except for molecular dynamics simulations, which can lack sufficient accuracy at different temperatures [34,46]. While MD simulations offer a detailed understanding of dynamic behavior, their computational intensity and the accuracy of interatomic potentials may limit their feasibility for extensive exploration of high-temperature regimes, especially in the context of single crystal calculations for materials like boron carbide [34,39,46]. To address this gap, this article focuses on the calculation of the temperature-dependent elastic properties of single crystal B4C boron carbide, through the use of a quasi-harmonic approach involving density functional theory (DFT) and phonon calculations. Since first-principles methods typically provide calculations at zero temperature, the quasi-harmonic approach [47] is employed to obtain temperature-dependent elastic properties. The quasi-harmonic approach incorporates the effects of temperature by considering the Helmholtz free energy, which has thermal electronic and vibrational contributions to the material’s properties [35,47]. This allows for a more comprehensive understanding of the temperature-dependent elastic behavior of boron carbide. This approach has been previously applied to other materials such as alumina (Al2O3) [34], nickel aluminide (Ni3Al) [35], and lead titanate (PbTiO3) [47].
In the present work, we organize the article as follows. In Section 2, we present the theory and methodology employed to find the temperature-dependent elastic constants, including the Helmholtz free energy, static calculations and equation of state (EOS) for total energy versus cell volume (E-V) results, vibrational contributions based on phonon calculations, and the details of our first-principles calculations. In Section 3, the details of the DFT model validation, the properties calculated from the EOS, and the results of phonon calculations are provided. Results are discussed in the context of comparing structural parameters and elastic properties with literature data, static calculations and the equation of state, phonon calculations, temperature-dependent thermal and elastic properties, and the thermal expansion coefficient. The limitations of the current work are also provided in this section. In Section 4, conclusions of the present work are given.
2. Theory and Methodology
Density functional theory provides calculations at zero temperature [48]. To obtain the temperature-dependent elastic properties in this study, a quasi-harmonic approach [49] within the Helmholtz free energy framework is applied. The quasi-harmonic approach accurately captures temperature-dependent lattice variations, allowing a comprehensive analysis of the material’s elastic behavior across a wider temperature range [47,49,50,51].
The Helmholtz free energy at volume V and temperature T is approximated by
where is the static energy at 0 K and volume V determined by DFT calculations. The vibrational contribution to the free energy at volume V and temperature T is defined by , estimated by first-principles phonon calculations. The thermal electronic contribution to free energy is represented by , which is zero in the case of semiconductors and insulators due to the absence of electrons at the Fermi level [34,35,52]. Given that boron carbide exhibits semiconductor behavior [53], the term is disregarded in this study. In the following, the details of the equations and methods for static energy calculation (Section 2.1), vibrational contribution to the Helmholtz free energy (Section 2.2), and DFT and phonon calculations (Section 2.3) are provided.
2.1. Static Energy and E-V Equation of State
Various equations of state (EOS) for E-V relationships have been established in the literature. The most widely used EOS is the Birch–Murnaghan EOS (BM) [54]. The 4-parameter equation of the Birch–Murnaghan EOS has the following form:
where a, b, c, and d are the fitting parameters. In this study, we have adopted the modified Birch–Murnaghan (mBM) EOS proposed by Teter et al. [55], for which , to fit the E-V data of B4C, due to its ability to minimize fitting errors [35].
The bulk modulus corresponding to the mBM EOS is
where is the equilibrium volume. According to the first-principles methods of EOS fitting, a single-phase region with a volume range of approximately ±10% around the equilibrium volume should be considered. Additionally, it is recommended to use a minimum of five data points for EOS fitting [35].
2.2. Vibrational Contribution to the Helmholtz Free Energy
The vibrational contribution to the Helmholtz free energy as a function of volume (V) and temperature (T) is
where is the Boltzmann constant, ℏ is the reduced Planck constant, is the frequency, and is the phonon density of states (DOS). To obtain the vibrational contribution to the Helmholtz free energy, a systematic approach is undertaken. After finding the ground state properties of the initial structures from DFT calculations, the supercell method [56] is used to calculate the phonon dispersion relations. These relations describe the vibrational frequencies as a function of wave vector in the Brillouin zone. The force constants, which quantify how the potential energy changes with atomic displacement are then obtained from the DFT results. By analyzing the force-sets and phonon dispersion relations [56], the phonon DOS is calculated, which gives information about the distribution of vibrational frequencies. Utilizing force-sets and DOS and statistical mechanics, the thermodynamic properties are calculated as a function of temperature [56]. By repeating this process for different volumes of the initial structure, the thermodynamic properties are obtained as a function of temperature and volume. Then the T-V relationship is obtained by minimizing the F(V) for each temperature.
2.3. DFT and Phonon Calculations
DFT calculations were performed using the Vienna ab initio simulation package (VASP) [57,58]. The Projector Augmented Wave with Perdew–Burke–Ernzerhof (PAW-PBE) pseudopotential [59] with a cut-off energy of 520 eV for plane waves was employed. The PAW-PBE has been widely used in the literature within DFT simulations for electronic structure calculations because of its high accuracy and reliability [59]. For k-point sampling, the Gamma scheme with fine accuracy ( k-mesh for the unit cell with equilibrium volume as shown in Figure 1a) was used, with a careful assessment of k-point convergence to ensure reliable results. Next, the integration of band structure energy across the Brillouin zone was accomplished using the tetrahedron method with Blöch corrections [60]. This method effectively accounts for the total energy, ion forces, and stresses in semiconductors and insulators (e.g., Si [61], Al2O3 [35], and SiO2 [62]), and is also suitable for simulating the current structure because boron carbide exhibits semiconductor properties [63,64]. The Generalized Gradient Approximation (GGA) [65] was used for the exchange–correlation functional. During the structural optimization, the ionic relaxation scheme with a conjugate gradient algorithm was employed. Additionally, the relaxation of both the cell shape and atomic positions throughout the optimization process was ensured. The incorporation of these two parameters ensures a more precise representation of the equilibrium structure of the system [66]. The VASPkit code [67] was used for k-point generation and post-processing of the VASP calculated data.
Phonon calculations within the supercell framework [56] with supercells of cells and 135 atoms as shown in Figure 1b were employed to determine the vibrational contribution to the Helmholtz free energy (Equation (1)). The acting forces on atoms for each supercell of B4C were calculated with DFT by VASP (v.5.4.4) with a k-mesh. Phonopy software (v.2.19.1), a package for phonon calculations at harmonic and quasi-harmonic levels [68], was used for frozen phonon calculations and post-processing of the results. The supercell sizes of and were studied in phonon calculations. The phonon calculation results and thermal properties for both cells matched and the size was chosen as the supercell size to reduce the computational expenses. The studied structures in this study are perfect crystal boron carbides and they do not have any defects. Seven different B4C cells with varying cell volumes were considered to find the relationship between V and T. Phonon dispersion curves are obtained from the self-consistent field calculations of the supercells by DFT. The force constants for each volume are calculated from the force-sets derived from self-consistent field (SCF) calculations by DFT. Phonon DOSs are then calculated for each of the volumes from force constants and principles of statistical mechanics [56] resulting in temperature-dependent properties of boron carbide. The crystal structures and visualizations presented in this study were generated using VESTA software (v.3.5.7) [69].
Figure 1.
Atomistic visualization (by VESTA software (v.3.5.7) [69]) of the rhombohedral boron carbide (a) unit cell with an arrangement of B4C and (b) supercell with the same atomic configuration.
Figure 1.
Atomistic visualization (by VESTA software (v.3.5.7) [69]) of the rhombohedral boron carbide (a) unit cell with an arrangement of B4C and (b) supercell with the same atomic configuration.

In the following, the validation of the DFT model and outcomes of static and phonon calculations including the temperature-dependent thermal and elastic properties are presented and discussed.
3. Results and Discussion
In this section, the DFT calculations are validated (Section 3.1). Then, we present studies of the EOS and static energy contribution (Section 3.2), along with the vibrational contribution (Section 3.3) to the Helmholtz free energy for B4C. Using the properties derived from the static calculations and insights obtained from phonon calculations, we provide the temperature-dependent thermal and elastic properties of B4C.
3.1. First-Principles Calculations’ Validation
To validate our DFT calculations for boron carbide, we compared the relaxed lattice parameters, cell volume, energy per atom, and elastic parameters with experimental [29,30,70,71] and modeling [28,72,73,74] literature data.
Table 1 presents a detailed comparison of the lattice constants, cell volume, and total energy per atom for B4C obtained from different literature sources. This agreement (less than 2% relative error) indicates the robustness of our computational approach in accurately predicting the atomic structure of B4C. Table 2 and Table 3 show the calculated elastic constants and properties for B4C with DFT in this work and previous works. By comparing the DFT-calculated elastic properties with data available in the literature and from experimental measurements, a comprehensive assessment of the accuracy and reliability of the DFT results can be achieved. Understanding elastic properties, including Young’s modulus (E), shear modulus (G), bulk modulus (B), and Poisson’s ratio (), is crucial for unraveling the behavior of materials, guiding predictions of failure mechanisms [33,75], understanding dynamic loading in ceramics [21,37], and recognizing temperature dependency [26,34] under extreme conditions.
Figure 2 illustrates the DFT total energies calculated for B4C at various volumes (including the equilibrium volume), alongside the fitted E-V mBM EOS curve. The volumes were adjusted through uniform deformation applied to each direction while maintaining a consistent rhombohedral lattice shape, as is suggested in the literature [34,76]. This approach facilitates the comparison of independent elastic constants, ensuring uniformity across different structures. The fitting demonstrates the effectiveness of the chosen EOS in representing the material’s behavior across different volumes.
The fitted EOS is also validated through a comparison of the bulk modulus. The bulk modulus calculated from Equation (3) is compared with the results of DFT for the equilibrium cell of B4C, other calculations [28,31,32], and experiments [27,29,30]. Table 4 presents a validation of the bulk modulus for single crystal B4C. The comparison between the bulk modulus values obtained from the fitted EOS, our DFT calculations, experimental data [27,29,30], and other calculations [28,31,32] confirms the accuracy and reliability of our computational approach. The bulk modulus of B4C computed from mBM EOS (239 GPa) and DFT (225 GPa) both fall into the range of 220–246 GPa that was established for B4C [28,29,30].
3.2. Properties from Static Calculations
In addition to the total energy, the elastic stiffness constants of B4C were calculated with DFT simulations for seven different cell volumes. In this study, the DFT calculations provided valuable insights into these elastic properties. Figure 3 shows the six independent elastic constants of the single crystal B4C structures from DFT simulation for each volume (shown as square data points). The volume-dependent s are fitted by exponential functions, which will be used in combination with phonon calculation results to predict temperature-dependent s. The circle data points represent the data from previous DFT calculations performed by Taylor et al. [28] showing elastic constants at the equilibrium volume of a B4C unit cell. The small difference between these results arises from variations in structural relaxation (as shown in Table 1) and elastic constant calculation processes. The elastic constant versus volume curves for boron carbide exhibit dissimilar trends, reflecting its complex anisotropic behavior. Specifically, and show decreasing trends as the volume increases, indicating a weakening stiffness and reduced resistance to elongation along specific crystallographic directions. This behavior can be attributed to structural softening, lattice expansion, and anisotropic bonding [9]. In contrast, demonstrates an increasing trend, suggesting a higher resistance to shear deformation along certain crystallographic planes as the volume expands. These findings highlight the complexity of boron carbide’s mechanical response, which is a consequence of its unique crystal structure and the arrangement of boron and carbon atoms within the lattice [9,28].
3.3. Phonon Calculation and Thermal Properties
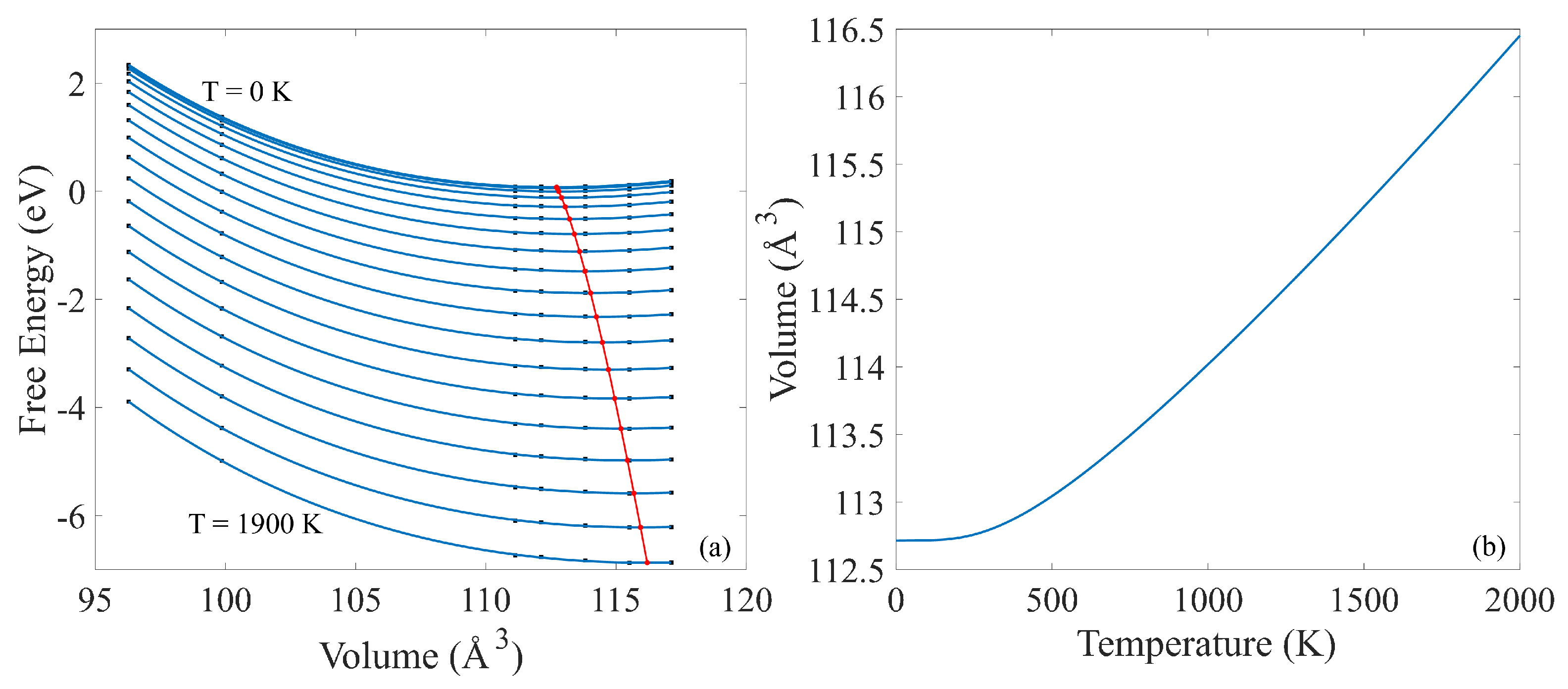
Combining the E-V results from static calculations at different volumes (Figure 2) with temperature-dependent thermal properties from phonon calculations (provided in Supplementary Materials) results in free energy vs. volume curves for different temperatures and establishes the relationship between volume and temperature (V-T), as shown in Figure 4b. Specifically, Figure 4a depicts the free energy as a function of volume for different temperatures starting from 0 to 1900 K.
Substituting the from Figure 4b in the exponential functions fitted to the static elastic stiffness constants () from Figure 3 results in temperature-dependent elastic stiffness constants (). Figure 5 illustrates the six independent elastic constants of the single crystal B4C structures at different temperatures. It is evident that at temperatures below 100 K, all elastic constants maintain an almost constant value, attributed to the minimal vibration of atoms at those temperatures. All elastic constants except show a decreasing trend with increasing temperature. exhibits almost no change with temperature, demonstrating the anisotropic behavior of boron carbide. It was observed that all volume-dependent excluding exhibit decreasing trends with the volume changes (shown in Figure 3). In contrast, has an increasing trend at volumes lower than the equilibrium volume. After reaching the equilibrium volume, the rate of increase diminishes significantly, resulting in an almost constant value at higher volumes. Since, in the temperature-dependent results, the zero temperature corresponds to the ground state of the material, the effects of structural shrinking at temperatures below zero Kelvin are ignored. Thus, the observed trends in align closely with those of at volumes higher than the equilibrium volume.
3.4. Temperature-Dependent Thermal Expansion Coefficient and Bulk Modulus
The temperature-dependent thermal expansion coefficient (TEC) of boron carbide is also predicted from the quasi-harmonic approach. As a result of strong covalent bonds between B and C atoms in boron carbide, its thermal expansion coefficient is measured to be much lower compared to other materials such as metals and polymers (e.g., zinc: [77], aluminum: [77], and polyethylene: [78] at room temperature). Figure 6a shows the anticipated temperature-dependent TEC of B4C boron carbide alongside existing literature data [26,79,80,81,82]. Most of the available data are from experiments performed at room temperature (298 K). For example, Tsagareishvili [80], Hollenberg [82], and Telle [81] measured the TEC for boron carbide at room temperature as , , and respectively. The calculated TEC in this study at room temperature is predicted to be , a value consistent with the established range of previously recorded data. At elevated temperatures, the extent of the established data is significantly reduced, and the available data are often derived from experiments conducted on the polycrystalline structures of boron carbide, typically exhibiting defects [26]. The presence of defects such as vacancies, impurities, and point defects can distort the regular arrangement of atoms and lattice symmetry, contributing to a different TEC [83]. Moreover, the grain boundaries and various crystallographic orientations in polycrystalline structures result in an overall expansion that may be lower than that of a perfect single crystal [83]. Kuliiev et al. [26] predicted that the TEC of B4C within the temperature range 298–1273 K follows an almost linear trend with an average of . Thevenot [79] used the following equation to determine the TEC of boron carbide: in °C). However, in the two previous temperature-dependent studies, the TEC at room temperature was lower than the established range, resulting in an overall lower TEC at elevated temperatures compared to the case of the perfect single crystal B4C studied here.
Additionally, a comparison between the TEC of advanced ceramics, including boron carbide (B4C), alumina (Al2O3) [34], and silicon carbide (SiC) [84] along with the room temperature experimental results [85,86] of them are illustrated in Figure 6b to capture the trends in TEC for these materials. It is evident that these materials exhibit a similar pattern, characterized by a very low to negligible increase in TEC at temperatures below 100 K, followed by a sharp rise until approximately 500–700 K, and then a gradual increase until it approaches a nearly constant value. This trend is attributed to the transition from internal energy dominance to vibrational entropy dominance, indicating significant changes in the material’s behavior as temperature increases. At low temperatures (<100 K), the atomic vibrations and motions are minimal leading to less expansion when subjected to a temperature change [47,50]. As the temperature rises to an intermediate range, the vibrational motion of atoms becomes more prominent resulting in a more rapid expansion. As the temperature increases, reaching an elevated range (e.g., near melting point), the material may undergo phase transitions that affect its thermal expansion behavior [47]. At these higher temperatures, the TEC still increases, but the rate of increase tends to be more gradual. The material may approach a nearly constant value as it reaches a state where further increases in temperature have a diminishing impact on the expansion behavior.
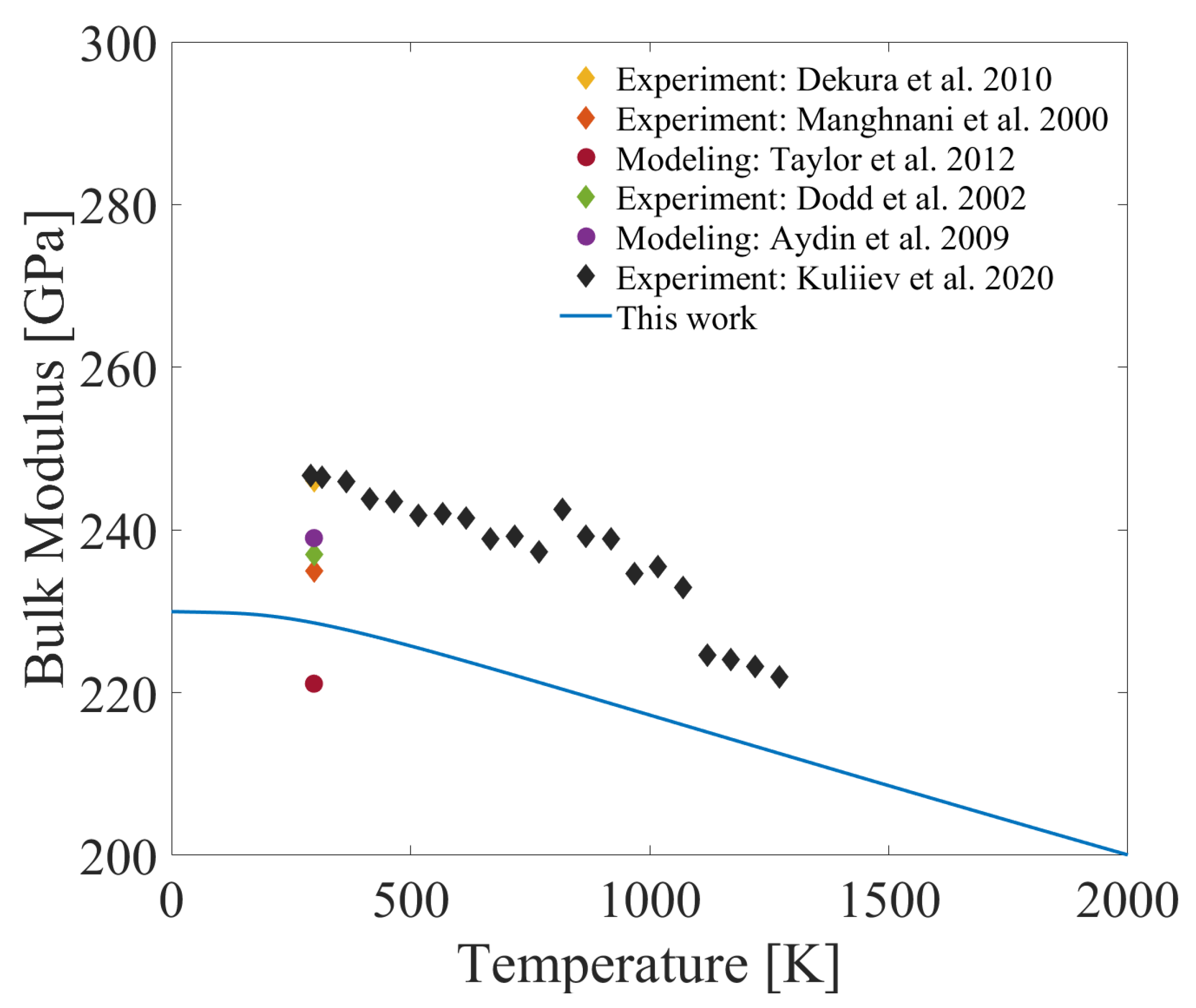
As the final result, the temperature-dependent bulk modulus of boron carbide is presented in Figure 7. Similar to the TEC results, most of the available data are from experiments and modeling performed at room temperature rather than elevated temperatures. Previously performed experiments at room temperature resulted in the following data: 246 GPa by Dekura et al. [72], 235 GPa by Manghnani et al. [29], and 237 GPa by Dodd et al. [27]. Moreover, the bulk modulus was predicted as 221 GPa by Taylor et al. [28] and 298 GPa by Aydin et al. [32]. The first-principles calculation produced in this present study with the phonon model resulted in 228 GPa at room temperature, which is in alignment with the data from the literature. In the same experiment that Kuliiev et al. [26] obtained TEC, they also provided the bulk modulus at higher temperatures. The results from our study follow the same decreasing trend of their work and the highest error value is equal to 9%. This error, along with the variations in trends, can be attributed to differences in the studied structure. The experimental studies use polycrystals with defects, whereas our investigation focuses on a perfect single crystal structure. Similar to the elastic constants, the bulk modulus exhibits negligible changes at temperatures below 100 K.
3.5. Limitations
A potential limitation of this work is the use of the quasi-harmonic approximation, which incorporates the volume dependence of phonon frequencies as a component of anharmonic effects. In the quasi-harmonic approximation, phonon–phonon coupling is neglected, leading to limitations, particularly near the melting temperature where this coupling becomes significant [47]. Another potential limitation of this study was the exclusive consideration of the B4C configuration of boron carbide. The small difference between the atomic number of 5B and 6C makes the precise composition and distribution of carbon atoms difficult to measure [79]. In addition, this chemical similarity induces substitutional disorder, causing changes in the local atomic arrangement and carbon concentration in boron carbide. The substitution of the central carbon in the CCC chain with the boron in the polar site of the icosahedra results in a more thermodynamically stable structure with a lower energy with a configuration of C11CpCBC [87,88,89]. These variations in boron carbide configurations and chain arrangements lead to different mechanical behavior and stabilities in structures [28,90]. Exploring various stoichiometries in boron carbide provides opportunities for future investigations to further refine our comprehension of this material in extreme environments.
4. Conclusions
In conclusion, this study investigates the temperature-dependent elastic and thermal properties of rhombohedral B4C boron carbide, revealing its intricate mechanical response and thermal behavior. Using a quasi-harmonic approach involving static DFT simulations and first-principles phonon calculations, the exploration of elastic constants, thermal properties (Helmholtz free energy, entropy, and heat capacity), and thermal expansion coefficient (TEC) versus temperature provided valuable insights into the material’s stability and performance at elevated temperatures. The comprehensive validation of our first-principles calculations against structural parameters and E-V fitting with mBM EOS establishes the reliability of our findings. Moreover, the comparison of TEC with other advanced ceramics demonstrates similar trends emphasizing the influence of atomic structure on material properties. Given the unexplored behavior of single crystal boron carbide at elevated temperatures, the obtained TEC ( K−1) and bulk modulus (228 GPa) from the quasi-harmonic approach were compared with the room temperature results from literature experiments [27,29,72,80,81,82] and models [28,31,32]. This comparison showed that the predicted results aligned closely with literature data (TEC = 5.73–9.50 K−1, B = 221–246 GPa). This work presents the variation in elastic stiffness constants, average thermal expansion coefficient, and bulk modulus of single crystal B4C across a broad temperature range from 0 to 2000 K. Overall, this work advances our fundamental understanding of boron carbide at elevated temperatures and provides crucial information for optimizing its design across various extreme environment applications, including satellite and aerospace components [6,12], as well as radiation shielding [10,91].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ceramics7010015/s1 Figure S1: Temperature-dependent thermodynamic properties of B4C for different volumes. (a) Helmholtz free energy, (b) entropy, and (c) heat capacity at constant volume.
Author Contributions
Conceptualization, S.S., W.S. and J.D.H.; methodology, S.S.; software, S.S.; validation, S.S., W.S. and J.D.H.; formal analysis, S.S.; investigation, S.S.; resources, W.S. and J.D.H.; writing—original draft preparation, S.S.; writing—review and editing, W.S. and J.D.H.; visualization, S.S.; supervision, W.S. and J.D.H.; project administration, W.S. and J.D.H.; funding acquisition, J.D.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by the Natural Sciences and Engineering Research Council of Canada Discovery Grant (Grant #2023-04457), and contributions from the George Ford Chair in Materials Engineering to JDH.
Data Availability Statement
Data will be made available on request.
Acknowledgments
This research was enabled in part by support provided by the Digital Research Alliance of Canada (www.alliancecan.ca (accessed on 13 December 2023)) via Cedar cluster.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DFT | Density Functional Theory |
| TEC | Thermal Expansion Coefficient |
| MD | Molecular Dynamics |
| EOS | Equation of State |
| BM | Birch–Murnaghan |
| mBM | Modified Birch–Murnaghan |
| DOS | Density of States |
| VASP | Vienna Ab Initio Simulation Package |
| GGA | Generalized Gradient Approximation |
| SCF | Self-Consistent Field |
References
- Roy, T.; Subramanian, C.; Suri, A. Pressureless sintering of boron carbide. Ceram. Int. 2006, 32, 227–233. [Google Scholar] [CrossRef]
- Swab, J.J.; Meredith, C.S.; Casem, D.T.; Gamble, W.R. Static and dynamic compression strength of hot-pressed boron carbide using a dumbbell-shaped specimen. J. Mater. Sci. 2017, 52, 10073–10084. [Google Scholar] [CrossRef]
- Vargas-Gonzalez, L.; Speyer, R.F.; Campbell, J. Flexural strength, fracture toughness, and hardness of silicon carbide and boron carbide armor ceramics. Int. J. Appl. Ceram. Technol. 2010, 7, 643–651. [Google Scholar] [CrossRef]
- Werheit, H.; Leithe-Jasper, A.; Tanaka, T.; Rotter, H.; Schwetz, K. Some properties of single-crystal boron carbide. J. Solid State Chem. 2004, 177, 575–579. [Google Scholar] [CrossRef]
- Matkovich, V.I.; Samsonov, G.V.; Hagenmuller, P. Boron and Refractory Borides; Springer: Berlin/Heidelberg, Germany, 1977. [Google Scholar]
- Varshney, V. Molecular dynamics methodologies for predicting thermal transport in aerospace polymers and their composites. In Hybrid Atomic-Scale Interface Design for Materials Functionality; Elsevier: Amsterdam, The Netherlands, 2021; pp. 19–34. [Google Scholar]
- El Messiry, M. Protective Armor Engineering Design; Apple Academic Press: Palm Bay, FL, USA, 2019. [Google Scholar]
- Turatti, A.M.; Pereira, A.S. Wear resistant boron carbide compacts produced by pressureless sintering. Ceram. Int. 2017, 43, 7970–7977. [Google Scholar] [CrossRef]
- Domnich, V.; Reynaud, S.; Haber, R.A.; Chhowalla, M. Boron carbide: Structure, properties, and stability under stress. J. Am. Ceram. Soc. 2011, 94, 3605–3628. [Google Scholar] [CrossRef]
- Sarıyer, D.; Küçer, R.; Küçer, N. Neutron shielding properties of concretes containing boron carbide and ferro–boron. Procedia-Soc. Behav. Sci. 2015, 195, 1752–1756. [Google Scholar] [CrossRef]
- Chevalier, J.; Gremillard, L. Ceramics for medical applications: A picture for the next 20 years. J. Eur. Ceram. Soc. 2009, 29, 1245–1255. [Google Scholar] [CrossRef]
- Levchenko, I.; Bazaka, K.; Belmonte, T.; Keidar, M.; Xu, S. Advanced Materials for Next-Generation Spacecraft. Adv. Mater. 2018, 30, 1802201. [Google Scholar] [CrossRef]
- Goldman, N. Computational Approaches for Chemistry under Extreme Conditions; Springer: Berlin/Heidelberg, Germany, 2019; Volume 28. [Google Scholar]
- Mishin, Y. Machine-learning interatomic potentials for materials science. Acta Mater. 2021, 214, 116980. [Google Scholar] [CrossRef]
- Suri, A.; Subramanian, C.; Sonber, J.; Murthy, T.C. Synthesis and consolidation of boron carbide: A review. Int. Mater. Rev. 2010, 55, 4–40. [Google Scholar] [CrossRef]
- Konovalikhin, S.; Ponomarev, V. Carbon in boron carbide: The crystal structure of B11.4C3.6. Russ. J. Inorg. Chem. 2009, 54, 197–203. [Google Scholar] [CrossRef]
- Morawiec, A. Review of deterministic methods of calculation of polycrystal elastic constants. Textures Microstruct. 1994, 22, 139–167. [Google Scholar] [CrossRef]
- Fahrenholtz, W.G.; Hilmas, G.E. Ultra-high temperature ceramics: Materials for extreme environments. Scr. Mater. 2017, 129, 94–99. [Google Scholar] [CrossRef]
- Saha, D.; Ghara, D.K.; Pal, M. Nanoporous γ-alumina based novel sensor to detect trace moisture in high temperature and high pressure environment. Sens. Actuators B Chem. 2016, 222, 1043–1049. [Google Scholar] [CrossRef]
- Medvedovski, E. Influence of corrosion and mechanical loads on advanced ceramic components. Ceram. Int. 2013, 39, 2723–2741. [Google Scholar] [CrossRef]
- Chang, J.; Chen, Z.; Hogan, J.D. Molecular Dynamics Simulations Correlating Mechanical Property Changes of Alumina with Atomic Voids under Triaxial Tension Loading. Modelling 2023, 4, 211–223. [Google Scholar] [CrossRef]
- Yang, A.; Romanyk, D.; Hogan, J.D. High-velocity impact study of an advanced ceramic using finite element model coupling with a machine learning approach. Ceram. Int. 2023, 49, 10481–10498. [Google Scholar] [CrossRef]
- Parsazadeh, M.; Fisher, G.; McDonald, A.; Hogan, J.D. Computational modelling of the effect of microstructure on the abrasive wear resistance of tungsten-carbide nickel composite coatings under sub-critical cyclic impact loading. Ceram. Int. 2022, 48, 14338–14348. [Google Scholar] [CrossRef]
- Manafi Farid, H.; McDonald, A.; Hogan, J.D. Impact Deposition Behavior of Al/B4C Cold-Sprayed Composite Coatings: Understanding the Role of Porosity on Particle Retention. Materials 2023, 16, 2525. [Google Scholar] [CrossRef]
- Gogotsi, G.; Groushevsky, Y.L.; Dashevskaya, O.; Gogotsi, Y.G.; Lavrenko, V. Complex investigation of hot-pressed boron carbide. J. Less Common Met. 1986, 117, 225–230. [Google Scholar] [CrossRef]
- Kuliiev, R.; Orlovskaya, N.; Hyer, H.; Sohn, Y.; Lugovy, M.; Ha, D.; Radovic, M.; Castle, E.G.; Reece, M.J.; Vallachira Warriam Sasikumar, P.; et al. Spark Plasma Sintered B4C—Structural, Thermal, Electrical and Mechanical Properties. Materials 2020, 13, 1612. [Google Scholar] [CrossRef]
- Dodd, S.; Saunders, G.; James, B. Temperature and pressure dependences of the elastic properties of ceramic boron carbide (B4C). J. Mater. Sci. 2002, 37, 2731–2736. [Google Scholar] [CrossRef]
- Taylor, D.E.; McCauley, J.W.; Wright, T. The effects of stoichiometry on the mechanical properties of icosahedral boron carbide under loading. J. Phys. Condens. Matter 2012, 24, 505402. [Google Scholar] [CrossRef] [PubMed]
- Manghnani, M.H.; Wang, Y.; Li, F.; Zinin, P.; Rafaniello, W. Elastic and vibrational properties of B4C to 21 GPa. Sci. Technol. High Press. 2000, 2, 25–30. [Google Scholar]
- Gieske, J.; Aselage, T.; Emin, D. Elastic properties of boron carbides. In AIP Conference Proceedings; American Institute of Physics: College Park, MD, USA, 1991; Volume 231, pp. 376–379. [Google Scholar]
- Lee, S.; Bylander, D.; Kleinman, L. Elastic moduli of B12 and its compounds. Phys. Rev. B 1992, 45, 3245. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Simsek, M. Hypothetically superhard boron carbide structures with a B11C icosahedron and three-atom chain. Phys. Status Solidi (B) 2009, 246, 62–70. [Google Scholar] [CrossRef]
- Farbaniec, L.; Hogan, J.; McCauley, J.; Ramesh, K. Anisotropy of mechanical properties in a hot-pressed boron carbide. Int. J. Appl. Ceram. Technol. 2016, 13, 1008–1016. [Google Scholar] [CrossRef]
- Shang, S.L.; Zhang, H.; Wang, Y.; Liu, Z.K. Temperature-dependent elastic stiffness constants of α-and θ-Al2O3 from first-principles calculations. J. Phys. Condens. Matter 2010, 22, 375403. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.L.; Wang, Y.; Kim, D.; Liu, Z.K. First-principles thermodynamics from phonon and Debye model: Application to Ni and Ni3Al. Comput. Mater. Sci. 2010, 47, 1040–1048. [Google Scholar] [CrossRef]
- Tang, B.; He, Y.; Goddard III, W.A.; An, Q. First principles predicting enhanced ductility of boride carbide through magnesium microalloying. J. Am. Ceram. Soc. 2019, 102, 5514–5523. [Google Scholar] [CrossRef]
- Shen, Y.; Reddy, K.M.; Li, J.; Chen, M.; An, Q. Atomistic origin of shear induced quasi-plastic deformation in boron carbide. Acta Mater. 2023, 249, 118828. [Google Scholar] [CrossRef]
- Kocer, E.; Ko, T.W.; Behler, J. Neural Network Potentials: A Concise Overview of Methods. Annu. Rev. Phys. Chem. 2022, 73. [Google Scholar] [CrossRef]
- Zuo, Y.; Chen, C.; Li, X.; Deng, Z.; Chen, Y.; Behler, J.; Csányi, G.; Shapeev, A.V.; Thompson, A.P.; Wood, M.A.; et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 2020, 124, 731–745. [Google Scholar] [CrossRef]
- Shang, S.; Wang, Y.; Liu, Z.K. First-principles calculations of phonon and thermodynamic properties in the boron-alkaline earth metal binary systems: B-Ca, B-Sr, and B-Ba. Phys. Rev. B 2007, 75, 024302. [Google Scholar] [CrossRef]
- Rou, S.; Chandran, K.R. First principles calculation of single-crystal elastic constants of titanium tetraboride (Ti3B4) and experimental validation. J. Am. Ceram. Soc. 2018, 101, 4308–4320. [Google Scholar] [CrossRef]
- Nikolussi, M.; Shang, S.; Gressmann, T.; Leineweber, A.; Mittemeijer, E.; Wang, Y.; Liu, Z.K. Extreme elastic anisotropy of cementite, Fe3C: First-principles calculations and experimental evidence. Scr. Mater. 2008, 59, 814–817. [Google Scholar] [CrossRef]
- Cheng, C.; Reddy, K.M.; Hirata, A.; Fujita, T.; Chen, M. Structure and mechanical properties of boron-rich boron carbides. J. Eur. Ceram. Soc. 2017, 37, 4514–4523. [Google Scholar] [CrossRef]
- DeVries, M.; Subhash, G.; Awasthi, A. Shocked ceramics melt: An atomistic analysis of thermodynamic behavior of boron carbide. Phys. Rev. B 2020, 101, 144107. [Google Scholar] [CrossRef]
- Pillai, H.G.; Madam, A.K.; Chandra, S.; Cheruvalath, V.M. Semiconducting B13C2 system: Structure search and DFT-based analysis. Mater. Res. Express 2019, 6, 046544. [Google Scholar] [CrossRef]
- Li, L.; Weidner, D.J.; Brodholt, J.; Alfe, D.; Price, G.D.; Caracas, R.; Wentzcovitch, R. Elasticity of CaSiO3 perovskite at high pressure and high temperature. Phys. Earth Planet. Inter. 2006, 155, 249–259. [Google Scholar] [CrossRef]
- Ritz, E.T.; Li, S.J.; Benedek, N.A. Thermal expansion in insulating solids from first principles. J. Appl. Phys. 2019, 126, 171102. [Google Scholar] [CrossRef]
- Eschrig, H. The Fundamentals of Density Functional Theory; Springer: Berlin/Heidelberg, Germany, 1996; Volume 32. [Google Scholar]
- McGaughey, A.J.; Jain, A.; Kim, H.Y.; Fu, B. Phonon properties and thermal conductivity from first principles, lattice dynamics, and the Boltzmann transport equation. J. Appl. Phys. 2019, 125, 011101. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1996. [Google Scholar]
- Fultz, B. Vibrational thermodynamics of materials. Prog. Mater. Sci. 2010, 55, 247–352. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.K.; Chen, L.Q. Thermodynamic properties of Al, Ni, NiAl, and Ni3Al from first-principles calculations. Acta Mater. 2004, 52, 2665–2671. [Google Scholar] [CrossRef]
- Wang, D.Y.; Yan, Q.; Wang, B.; Wang, Y.X.; Yang, J.; Yang, G. Predicted boron-carbide compounds: A first-principles study. J. Chem. Phys. 2014, 140, 224704. [Google Scholar] [CrossRef] [PubMed]
- Birch, F. Finite strain isotherm and velocities for single-crystal and polycrystalline NaCl at high pressures and 300 K. J. Geophys. Res. Solid Earth 1978, 83, 1257–1268. [Google Scholar] [CrossRef]
- Teter, D.; Gibbs, G.V.; Boisen, M.B., Jr.; Allan, D.; Teter, M. First-principles study of several hypothetical silica framework structures. Phys. Rev. B 1995, 52, 8064. [Google Scholar] [CrossRef]
- Togo, A. First-principles phonon calculations with phonopy and phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223. [Google Scholar] [CrossRef]
- Heywang, W.; Zaininger, K. Silicon: The semiconductor material. In Silicon: Evolution and Future of a Technology; Springer: Berlin/Heidelberg, Germany, 2004; pp. 25–42. [Google Scholar]
- Nekrashevich, S.; Gritsenko, V. Electronic structure of silicon dioxide (a review). Phys. Solid State 2014, 56, 207–222. [Google Scholar] [CrossRef]
- Hushur, A.; Manghnani, M.H.; Werheit, H.; Dera, P.; Williams, Q. High-pressure phase transition makes B4. 3C boron carbide a wide-gap semiconductor. J. Phys. Condens. Matter 2016, 28, 045403. [Google Scholar] [CrossRef] [PubMed]
- Brown-Shaklee, H.J.; Neuman, E.W.; Fahrenholtz, W.G.; Hilmas, G.E. Optical characterization of boron carbide powders synthesized with varying B-to-C ratios. J. Am. Ceram. Soc. 2023, 106, 1932–1944. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Pulay, P. Convergence acceleration of iterative sequences. The case of SCF iteration. Chem. Phys. Lett. 1980, 73, 393–398. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Kwei, G.H.; Morosin, B. Structures of the boron-rich boron carbides from neutron powder diffraction: Implications for the nature of the inter-icosahedral chains. J. Phys. Chem. 1996, 100, 8031–8039. [Google Scholar] [CrossRef]
- McClellan, K.; Chu, F.; Roper, J.; Shindo, I. Room temperature single crystal elastic constants of boron carbide. J. Mater. Sci. 2001, 36, 3403–3407. [Google Scholar] [CrossRef]
- Dekura, H.; Shirai, K.; Yanase, A. Metallicity of boron carbides at high pressure. In Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2010; Volume 215, p. 012117. [Google Scholar]
- Bylander, D.; Kleinman, L.; Lee, S. Self-consistent calculations of the energy bands and bonding properties of B12C3. Phys. Rev. B 1990, 42, 1394. [Google Scholar] [CrossRef] [PubMed]
- Project, T.M. Materials Data on B4C (mp-696746) by Materials Project (v2023.11.1). Available online: https://next-gen.materialsproject.org/materials/mp-696746 (accessed on 3 December 2023).
- An, Q.; Goddard III, W.A. Atomistic origin of brittle failure of boron carbide from large-scale reactive dynamics simulations: Suggestions toward improved ductility. Phys. Rev. Lett. 2015, 115, 105501. [Google Scholar] [CrossRef] [PubMed]
- Pavlovskii, M.S.; Andryushin, N. Calculating the Lattice Dynamics in the RFe3(BO3)4 Crystals in the Quasi-Harmonic Approximation. Phys. Solid State 2019, 61, 2019–2025. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Li, D. Assessment of thermal expansion coefficient for pure metals. Calphad 2013, 43, 7–17. [Google Scholar] [CrossRef]
- Shen, M.; Hansen, W.N.; Romo, P.C. Thermal expansion of the polyethylene unit cell. J. Chem. Phys. 1969, 51, 425–430. [Google Scholar] [CrossRef]
- Thevenot, F. Boron carbide—a comprehensive review. J. Eur. Ceram. Soc. 1990, 6, 205–225. [Google Scholar] [CrossRef]
- Tsagareishvili, G.; Nakashidze, T.; Jobava, J.S.; Lomidze, G.; Khulelidze, D.; Tsagareishvili, D.S.; Tsagareishvili, O. Thermal expansion of boron and boron carbide. J. Less Common Met. 1986, 117, 159–161. [Google Scholar] [CrossRef]
- Telle, R. Boride–eine neue Hartstoffgeneration? Chem. Unserer Zeit 1988, 22, 93–99. [Google Scholar] [CrossRef]
- Hollenberg, G. Thermally induced stresses and fractures in boron carbide pellets Bull. Am. Ceram. Soc. 1980, 59, 538–548. [Google Scholar]
- Wang, K.; Reeber, R. Thermal defects and thermal expansion of ionic crystals at high temperatures. Phys. Status Solidi (A) 1994, 146, 621–627. [Google Scholar] [CrossRef]
- Talwar, D.; Sherbondy, J.C. Thermal expansion coefficient of 3C–SiC. Appl. Phys. Lett. 1995, 67, 3301–3303. [Google Scholar] [CrossRef]
- White, G.K.; Roberts, R.B. Thermal expansion of reference materials: Tungsten and α-Al2O3. High Temp.-High Press. 1983, 15, 321–328. [Google Scholar]
- Li, Z.; Bradt, R. Thermal expansion and elastic anisotropies of SiC as related to polytype structure. In Proceedings of the Silicon Carbide”87, Columbus, OH, USA, 2–5 August 1989. [Google Scholar]
- Shirai, K.; Sakuma, K.; Uemura, N. Theoretical study of the structure of boron carbide B13C2. Phys. Rev. B 2014, 90, 064109. [Google Scholar] [CrossRef]
- Ektarawong, A.; Simak, S.I.; Alling, B. Structural models of increasing complexity for icosahedral boron carbide with compositions throughout the single-phase region from first principles. Phys. Rev. B 2018, 97, 174104. [Google Scholar] [CrossRef]
- Rasim, K.; Ramlau, R.; Leithe-Jasper, A.; Mori, T.; Burkhardt, U.; Borrmann, H.; Schnelle, W.; Carbogno, C.; Scheffler, M.; Grin, Y. Local atomic arrangements and band structure of boron carbide. Angew. Chem. 2018, 130, 6238–6243. [Google Scholar] [CrossRef]
- Jay, A.; Hardouin Duparc, O.; Sjakste, J.; Vast, N. Theoretical phase diagram of boron carbide from ambient to high pressure and temperature. J. Appl. Phys. 2019, 125, 185902. [Google Scholar] [CrossRef]
- Vignesh, S.; JT, W.J.; Nagaveena, S.; Sharma, K.; Khan, A. Boron carbide dispersed epoxy composites for gamma radiation shielding applications. Vacuum 2022, 205, 111474. [Google Scholar]
Figure 2.
DFT energy versus volume of the cell for boron carbide and the fitted mBM EOS.

Figure 3.
Calculated independent elastic constants (a) and and (b) , , , and for single crystal boron carbide using DFT and their trend with cell volume change alongside with literature data for equilibrium volume [28].
Figure 3.
Calculated independent elastic constants (a) and and (b) , , , and for single crystal boron carbide using DFT and their trend with cell volume change alongside with literature data for equilibrium volume [28].

Figure 4.
(a) Free energy vs. volume of B4C at different temperatures, and the equilibrium volume for each temperature shown by the red curve. (b) Volume vs temperature of B4C calculated from phonon calculations.
Figure 4.
(a) Free energy vs. volume of B4C at different temperatures, and the equilibrium volume for each temperature shown by the red curve. (b) Volume vs temperature of B4C calculated from phonon calculations.

Figure 5.
Temperature-dependent 6 independent elastic constants of B4C (a) and and (b) , , , and .

Figure 6.
Temperature-dependent thermal expansion coefficient of (a) B4C alongside the literature data [26,79,80,81,82] and (b) comparison between TEC of advanced ceramics alongside their room temperature modeling [34,84] and experimental data [85,86].

Figure 7.
Temperature-dependent bulk modulus of B4C alongside the bulk modulus obtained from other experiments [26,27,29,72] and modeling works [28,32].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of the relaxed lattice parameters, cell volume, and total energy of B4C in the rhombohedral system.
Table 1.
Comparison of the relaxed lattice parameters, cell volume, and total energy of B4C in the rhombohedral system.
| (Å) | Cell Volume (Å3) | Energy per Atom (eV/atom) | ||
|---|---|---|---|---|
| Calculation | ||||
| Present work | 5.185 | 65.89 | 111.12 | −7.239 |
| Taylor et al. (2012) [28] | 5.190 | 66.01 | 112.09 | |
| Materials Project [74] | 5.180 | 65.95 | 110.65 | |
| Dekura et al. (2010) [72] | 5.110 | 66.00 | −7.231 | |
| Bylander et al. (1990) [73] | 5.182 | 65.61 | ||
| Experiment | ||||
| Kwei et al. (1996) [70] | 5.155 | 65.67 | ||
| McClellan et al. (2001) [71] | 5.190 | 65.18 |
Table 2.
Comparison of the elastic constants of B4C in the rhombohedral system.
| (GPa) | ||||||
|---|---|---|---|---|---|---|
| Present work | 465 | 125 | 103 | 36 | 498 | 175 |
| Taylor et al. (2012) [28] | 486 | 188 | 64 | 14 | 518 | 133 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sheikhi, S.; Stroberg, W.; Hogan, J.D. Temperature-Dependent Elastic Properties of B4C from First-Principles Calculations and Phonon Modeling. Ceramics 2024, 7, 235-249. https://doi.org/10.3390/ceramics7010015
AMA Style
Sheikhi S, Stroberg W, Hogan JD. Temperature-Dependent Elastic Properties of B4C from First-Principles Calculations and Phonon Modeling. Ceramics. 2024; 7(1):235-249. https://doi.org/10.3390/ceramics7010015
Chicago/Turabian StyleSheikhi, Sara, Wylie Stroberg, and James D. Hogan. 2024. "Temperature-Dependent Elastic Properties of B4C from First-Principles Calculations and Phonon Modeling" Ceramics 7, no. 1: 235-249. https://doi.org/10.3390/ceramics7010015