Abstract
A simple and efficient strategy for the synthesis of 1-ethynylcyclohex-2-enol derivatives was developed utilizing regioselective addition of acetylides to enediones based on steric effects. Further investigation of the substrate scope of enediones indicated that all the addition reactions ocurred in good yield.
1. Introduction
Alkynylation reactions of carbonyl compounds that generate propargylic alcohols are among the most important carbon–carbon bond-forming reactions. Moreover, propargylic alcohols are important intermediates for the synthesis of natural products and pharmaceuticals [1,2,3]. In particular, 1-ethynyl-cyclohex-2-enol derivatives are very important intermediates used to prepare many biologically active natural products [4,5]. Recently, these derivatives have also been increasingly utilized in organic synthesis as precursors of stereodefined cyclic or acyclic molecules [6,7,8,9]. 1-Ethynylcyclohex-2-enol derivatives are easily obtained in a good yield from the reactions of cyclohexenone with alkali metal (Na, Li) alkynides or alkynyl Grignard reagents at low temperature [10,11,12]. In contrast, diketones need to be selectively protected with ethylene glycol before the alkynylation [13]. Liotta reported a useful method for preparing highly functionalized synthetic intermediates without the use of protecting groups based on stereoelectronic control [14], but the conditions of these reactions were harsh. As part of our continuing interest in the synthesis and bioactivities of 1-ethynylcyclohex-2-enol derivatives, a series of 1,4-enediones with high steric effects including substituted six-membered rings, tetralones, and quinolines were selected to react with a range of acetylides. A highly efficient regioselective addition of acetylides to 1,4-enediones without the use of a protecting group, and with only the aid of steric effects, was found. Herein, we describe this useful method and explore the 1,4-enedione and acetylide substrate scope.
2. Results and Discussion
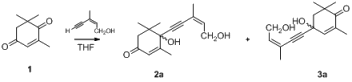
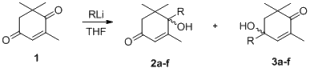
4-Ketoisophorone (1) is a versatile substrate in the synthesis of natural products and flavors, as it has two different carbonyl groups. The 1-carbonyl group, which is connected with three methyl groups, has stronger steric hindrance than the 4-carbonyl group. Initially, using 1 as a model substrate, the regioselective addition of acetylides to 1 under various experimental conditions was investigated. As shown in Table 1, the yield of the process increased from 81% to 87% when the amount of n-BuLi was raised from 2.0 to 2.1 eqv (Table 1, entry 6). Decreasing the amount of THF from 30 mL to 15 mL resulted in an increase in the yield of the addition product from 82% to 87% (Table 1, entry 6). These experiments demonstrate that the best results could be obtained with 2.1 eqv of n-BuLi in 15 mL THF (Table 1, entry 6).

Table 1.
Optimization of the reaction conditions a.
| Entry | n-BuLi (eqv) | Solvent (mL) | Time (h) b | Yield (3a) c (%) |
|---|---|---|---|---|
| 1 | 2.0 | 30 | 2 | 81 |
| 2 | 2.0 | 30 | 3 | 82 |
| 3 | 2.0 | 30 | 4 | 80 |
| 4 | 2.1 | 30 | 2 | 82 |
| 5 | 2.1 | 15 | 2 | 87 |
| 6 | 2.1 | 15 | 3 | 86 |
| 7 | 2.5 | 15 | 2 | 87 |
a All reactions were performed on a 5-mmol scale in 4-ketoisophorone and (Z)-3-methylpent-2-en-4-yn-1-ol, stirred at −78 °C for 2 h and r.t. for another 2 h. b Time for reaction at r.t. c Yields for isolated products after column chromatography.
The regioselectivity of these acetylide additions can be determined using a variety of techniques, the simplest of which is proton NMR. Since the products of these additions are enones, it is quite a straightforward matter to determine if the material in question possesses an α-enone proton (high field) or a β-enone proton (low field) [14]. In 2a, because of the dual effect of the 7′-methyl electron-donating effect and the α,β-unsaturated ketone conjugate effect, the α-H tends toward the 1H-NMR low field. In 3a, the 7′-methyl electron-donating effect does not completely overlap with the α,β-unsaturated ketone conjugate effect. Thus, in the 1H-NMR, the β-H is skewed towards the high field. In 1H-NMR, 2a [15] showed signals of all protons in a higher field than 3a—i.e., the α-enone proton of 2a at 5.76 compared to the β-enone proton of 3a at 5.85—proving the presence of steric effects (Table 2). In tetralone, in order to confirm the products’ structure, we protected the 4-carbonyl group of 4b to give 7 [16], and after addition of acetylide and deprotection, 5c was obtained (Scheme 1). NMR showed the presence of a significant difference (see Supplementary Material).
Table 2.
1H-NMR of 2a and 3a.
| 2a a | 3a b | |
|---|---|---|
| =CH | 5.83 | 5.92 |
| =CH | 5.76 | 5.85 |
| CH2OH | 4.17 | 4.25 |
| CH2 | 2.37 | 2.46 |
| vinyl CH3 | 2.05 | 2.14 |
| vinyl CH3 | 1.79 | 1.87 |
| CH3 | 1.12 | 1.23 |
| CH3 | 1.03 | 1.11 |
| CH2OH | 3.3 | 3.49 |
a 360 MHz, using CDCl3 as the solvent. b 300 MHz, using CDCl3 as the solvent.
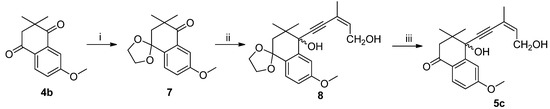
Scheme 1.
Synthesis of 5c.
Scheme 1.
Synthesis of 5c.
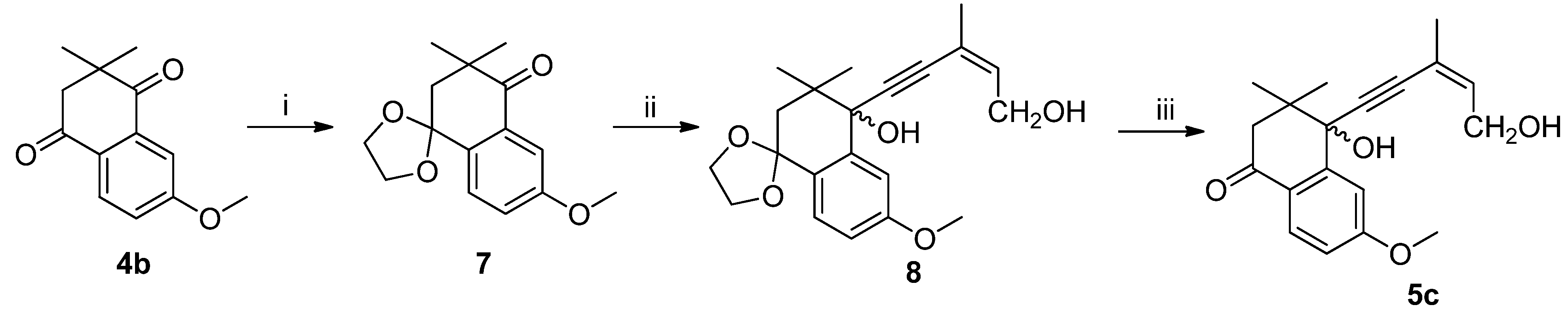
Conditions and reagents: i. p-TSA, CH(OCH3)3, HOCH2CH2OH, ether; ii. n-BuLi, (z)-3-methylpent-2-en-4-yn-1-ol, THF, −78 °C; iii. p-TSA, acetone.
With the optimal conditions identified, the scope of the regioselective additions of acetylides to enediones based on steric effects was investigated (Table 3 and Table 4). The results of these processes, summarized in Table 3 and Table 4, show that these processes take place efficiently to generate adducts in good to excellent yields. With additions of acetylides to 4-ketoisophorone, the propionates, t-butylacetylene, and phenylethyne, gave excellent yields (Table 3, entries 3–6), but alkynols gave lower yields (Table 3, entry 1). The best yield was obtained when ethyl propiolate was used as a substrate (Table 3, entry 3).
Table 3.
Regioselective additions of acetylides to 4-ketoisophorone.
| Entry | Substrate | RLi | 2 (%) | 3 (%) a |
|---|---|---|---|---|
| 1 | 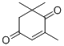 | 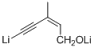 | — | 3a: 87 |
| 2 | |  | — | 3b: 91 |
| 3 | |  | — | 3c: 95 |
| 4 | | 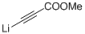 | — | 3d: 92 |
| 5 | | 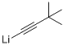 | — | 3e: 93 |
| 6 | | 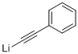 | — | 3f: 94 |
a Yields for isolated products after column chromatography.
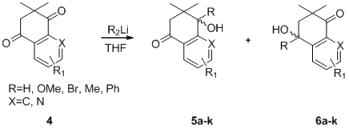
Further investigations demonstrated that substituent effects influence the yields of tertiary propargylic alcohol formation in the regioselective additions of acetylides to dimethyl dihydronaphthalene-1,4-diones. The data in Table 4 show that dimethyl dihydronaphthalene-1,4-diones with electron-withdrawing or -donating substituents at the meta and para positions of the benzene rings worked well (Table 4, entries 3–8). It is worth mentioning that dimethyl dihydroquinoline-5,8-diones also worked well, readily affording the corresponding products with the best yield and regioselectivity (Table 4, entries 9–11). Similarly, a significant yield was obtained when ethyl propiolate was used as a substrate (Table 4, entries 2 and 6).
Table 4.
Regioselective additions of acetylides to dimethyl dihydronaphthalene-1,4-diones.
| Entry | R1, X [17,18] | R2Li | 5 (%) | 6 (%) a |
|---|---|---|---|---|
| 1 | 4a: R1 = H, X = C | | — | 6a: 87 |
| 2 | 4a: R1 = H, X = C | | — | 6b: 94 |
| 3 | 4b: R1 = 7-Ome, X = C | | — | 6c: 86 |
| 4 | 4c: R1 = 6-Ome, X = C | | — | 6d: 84 |
| 5 | 4c: R1 = 6-Ome, X = C | | — | 6e: 88 |
| 6 | 4c: R1 = 6-Ome, X = C | | — | 6f: 94 |
| 7 | 4c: R1 = 6-Ome, X = C | | — | 6g: 91 |
| 8 | 4d: R1 = 7-Br, X = C | | — | 6h: 89 |
| 9 | 4e: R1 = 2-Me, X = N | | — | 6i: 94 |
| 10 | 4f: R1 = 2-Ph, X = N | | — | 6j: 96 |
| 11 | 4g: R1 = H, X = N | | — | 6k:95 |
a Yields for isolated products after column chromatography.
3. Experimental
The 1H and 13C-NMR spectra were recorded on a Bruker Avance DPX300 with tetramethylsilane as an internal standard, all NMR spectra were obtained using CDCl3 as the solvent unless otherwise noted. GC-Mass was carried out on a 6890N GC-Agilent 5973N. Melting points were measured on an Cole-Parmer melting point apparatus and are not corrected. Commercially available compounds were used in this work without further purification. The solvent tetrahydrofuran (THF) was dried by distillation from sodium and benzophenone. Substrates 4 were synthesized by our lab, while the others were commercially available. Column chromatography was performed using silica gel (200–300 mesh). TLC was performed on GF254 silica gel plates (Qingdao Haiyang Co., Ltd, Qingdao, China). NMR spectra were recorded on a Bruker Avance DPX300 spectrometer (300 MHz for 1H and 75 MHz for 13C) with tetramethylsilane as the internal standard.
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-2,6,6-trimethylcyclohex-2-enone (3a): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 3a (1.08 g, 87%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C): δ 5.94–5.91 (m, 1H), 5.90–5.84 (m, 1H), 4.41 (s, 1H), 4.26–4.24 (d, J = 6.7, 2H), 3.49 (s, 1H), 2.53–2.39 (m, 2H), 2.14 (s, 3H), 1.87 (s, 3H), 1.23 (s, 3H), 1.11 (s, 3H). 13C-NMR (CDCl3, 25 °C) : δ 199.0, 136.7, 125.5, 119.8, 92.7, 85.1, 74.4, 60.6, 41.7, 25.0, 22.7, 19.7.
4-Hydroxy-2,6,6-trimethyl-4-((trimethylsilyl)ethynyl)cyclohex-2-enone (3b): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of ethynyltrimethylsilane (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 5:1) to afford 3b (1.14 g, 91%) as a white solid, mp: 76–78 °C. 1H-NMR (CDCl3, 25 °C) δ 5.77 (d, J = 1.1 Hz, 1H), 2.71 (s, 1H), 2.38 (q, J1 = 16.4 Hz, J2 = 37.2 Hz, 2H), 2.04 (d, J = 1.3 Hz, 3H), 1.12 (s, 3H), 1.06 (s, 3H), 0.10 (s, 9H). 13C-NMR (CDCl3, 25 °C) δ 198.4, 126.0, 103.9, 92.2, 74.6, 58.2, 49.0, 41.5, 24.9, 19.5, 18.2, -0.3.MS:273.1 (M + Na+).
Ethyl 3-(1′-hydroxy-3′,5′,5′-trimethyl-4′-oxocyclohex-2′-en-1′-yl)propiolate (3c): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of ethylpropiolate (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 10:1) to afford 3c (1.19 g, 95%) as a white solid, 72–73 °C. 1H-NMR (CDCl3, 25 °C) δ 5.91 (d, J = 1.3 Hz, 1H), 4.25 (q, J1 = 7.1 Hz, J2 = 14.2 Hz, 2H), 3.52 (s, 1H), 2.47 (t, J = 12.0 Hz, 2H), 2.14 (d, J = 1.4 Hz, 3H), 1.32 (t, J = 7.2 Hz, 3H), 1.24 (s, 3H), 1.12 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ. 197.7, 158.2, 152.8, 126.8, 85.0, 78.4, 74.4, 62.3, 48.7, 42.0, 24.9, 21.7, 19.6, 13.8. MS:273.0 (M + Na+).
Methyl 3-(1′-hydroxy-3′,5′,5′-trimethyl-4′-oxocyclohex-2′-en-1′-yl)propiolate (3d): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of methyl propiolate (0.42 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 9:1) to afford 3d (1.08 g, 92%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C) δ 5.92 (d, J = 1.3 Hz, 1H), 4.22 (s, 1H), 3.79 (s, 3H), 2.49 (q, J = 6.6 Hz, 2H), 2.15 (d, J = 1.3 Hz, 3H), 1.25 (s, 3H), 1.13 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 198.0, 153.2, 126.6, 85.7, 77.4, 74.2, 52.8, 41.9, 24.8, 19.5. MS:258.9 (M + Na+).
4-(3′,3′-Dimethylbut-1′-yn-1′-yl)-4-hydroxy-2,6,6-trimethylcyclohex-2-enone (3e): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of 3,3-dimethyl-1-butyne (0.41 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether /EtOAc, 10:1) to afford 3e (1.09 g, 93%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C) δ 5.81 (d, J = 1.0 Hz, 1H), 3.0 (s, 1H), 2.43 (d, J = 8.5 Hz, 2H), 2.11 (d, J = 1.2 Hz, 3H), 1.22 (s, 9H), 1.17 (s, 3H), 1.09 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 198.6, 125.4, 95.9, 74.0, 41.5, 30.6, 27.4, 24.9, 19.6. MS:257.0 (M + Na+).
4-Hydroxy-2,6,6-trimethyl-4-(phenylethynyl)cyclohex-2-enone (3f): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of phenylacetylene (0.51 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 10:1) to afford 3f (1.19 g, 94%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C) δ 7.42–7.38 (m, 2H), 7.32–7.26 (m, 3H), 5.89 (d, J = 1.3 Hz, 1H), 3.88 (s, 1H), 2.50 (q, J1 = 19.2 Hz, J2 = 23.2 Hz, 2H), 2.16 (d, J = 1.4 Hz, 3H), 1.22 (s, 3H), 1.14 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 198.7, 131.5, 128.6, 128.1, 125.6, 121.8, 87.7, 86.7, 76.5, 74.4, 49.1, 41.8, 24.9, 21.7, 19.6. MS:277.0 (M + Na+).
(Z)-4-hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-6-methoxy-3,3-dimethyl-3,4-dihydronaphthalen-1(2H)-one (5c): To a stirred solution of 4b (2.0 g, 9.2 mmol), ethylene glycol (1.14 g, 18.4 mmol), methylorthoformate (1.46 g, 13.8 mmol) and p-TSA (0.15g, 0.87 mmol) in 150 mL diethyl ether under an atmosphere of argon at r.t for 16h. The reaction was quenched by slow addition of aqueous NaHCO3 solution (5 mL) and extracted with diethyl ether (3 × 50 mL). The organic phase was washed with water (2 × 50 mL) and dried over anhydrous Na2SO4 and filtered, filtrate concentrated under reduced pressure. The residue was subjected to silica gel chromatography using PE and EtOAc (6:1) as eluant to afford ketal 7 as a yellow oil (2.05 g, yield 85%). 7: 1H-NMR (CDCl3, 25 °C) δ 7.49–7.46 (m, 2H), 7.14 (dd, J1 = 2.8 Hz, J2 = 8.5 Hz, 1H), 4.22–4.08 (m, 4H), 3.84 (s, 3H), 2.20 (s, 2H), 1.30 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 201.9, 160.2, 135.2, 132.3, 127.3, 121.5, 109.2, 105.0, 64.9, 55.3, 45.3, 42.4, 26.6.
n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 7 (1.31 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 8 (1.54 g, 86%) as a yellow oil. 8: 1H-NMR (300 MHz, CDCl3, 25 °C) δ 7.39 (d, J = 7.5 Hz, 1H), 7.31 (d, J = 2.6 Hz, 1H), 6.89 (dd, J1 = 2.6 Hz, J2 = 8.6 Hz, 1H), 5.85 (t, J = 1.4 Hz, 1H), 4.22 (d, J = 6.8 Hz, 2H), 4.17-4.05 (m, 4H), 3.81 (s, 3H), 3.08 (s, 1H), 2.22 (d, J = 14.2 Hz, 1H), 2.03 (d, J = 14.2 Hz, 1H), 1.87 (d, J = 0.9 Hz, 3H), 1.15 (s, 3H), 1.12 (s, 3H). 13C-NMR (75 MHz, CDCl3, 25 °C) δ 160.1, 141.4, 136.1, 128.7, 127.6, 120.3, 120.3, 114.9, 111.0, 105.8, 95.2, 85.0, 75.1, 64.6, 60.9, 55.2, 42.9, 39.2, 25.2, 23.5, 22.9.
To a stirred solution of 8 (1.50 g, 4.2 mmol) in acetone (30 mL), p-TSA (60 mg) was added, and the mixture was stirred for 1 h at room temperature. A saturated NaHCO3 solution (5 mL) was added, and it was extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (3 × 20 mL), dried over anhydrous Na2SO4 and concentrated to a residue, which was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 5c (1.17 g, 89%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 7.99 (d, J = 8.6 Hz, 1H), 7.39 (d, J = 2.5 Hz, 1H), 6.92 (dd, J1 = 2.5 Hz, J2 = 8.7 Hz, 1H), 5.94-5.89 (m, 1H), 4.29 (d, J = 6.7 Hz, 2H), 3.90 (s, 3H), 2.86-2.63 (m, 3H), 1.90 (s, 3H), 1.17 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 195.7, 164.4, 136.6, 130.8, 129.5, 128.8, 123.6, 120.0, 114.2, 94.2, 85.9, 74.8, 65.5, 61.2, 55.5, 41.5, 30.5, 24.9, 22.9.
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6a): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4a (0.94 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at –78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 6a (1.24 g, 87%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.02 (d, J = 9.2 Hz, 1H), 7.90 (d, J = 8.9 Hz, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 5.94–5.89 (m, 1H), 4.28 (d, J = 6.6 Hz, 2H), 2.94 (d, J = 17.4 Hz, 2H), 2.64 (d, J = 17.4 Hz, 2H), 1.91 (d, J = 1.1 Hz, 3H), 1.77 (s, 2H), 1.21 (s, 3H), 1.17 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 197.1, 144.0, 136.5, 134.3, 130.0, 128.6, 127.0, 126.9, 120.0, 94.3, 85.9, 74.7, 61.25, 48.5, 41.4, 25.0, 23.1, 22.9.
Ethyl 3-(1′-hydroxy-3′,3′-dimethyl-4′-oxo-1′,2′,3′,4′-tetrahydronaphthalen-1′-yl)propiolate 6b: LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of ethylpropiolate (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4a (0.94 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 10:1) to afford 6b (1.34 g, 94%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 8.00 (dd, J1 = 1.0 Hz, J2 = 7.7 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.62 (dt, J1 = 1.3 Hz, J2 = 7.5 Hz, 1H), 4.22 (q, J1 = 7.1 Hz, J2 = 14.2 Hz, 2H), 3.97 (s, 1H), 2.90 (d, J = 17.3 Hz, 1H), 2.63 (d, J = 17.3 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H), 1.22 (s, 3H), 1.18 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.7, 153.0, 142.2, 134.4, 129.8, 129.0, 127.3, 126.9, 86.4, 79.0, 74.1, 62.2, 41.4, 24.6, 13.8. MS:309.0 (M+Na+).
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-7-methoxy-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6c): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at –78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4b (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 6c (1.35 g, 86%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 7.96 (d, J = 8.7 Hz, 1H), 7.39 (d, J = 2.4 Hz, 1H), 6.89 (dd, J1 = 2.4 Hz, J2 = 8.7 Hz, 1H), 5.91–5.87 (m, 1H), 4.26 (d, J = 6.4 Hz, 2H), 3.87 (s, 3H), 2.78-2.61 (m, 3H), 1.87 (s, 3H), 1.15 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 196.2, 164.3, 146.8, 136.4, 129.3, 123.4, 119.9, 114.0, 111.8, 94.1, 85.6, 74.4, 60.8, 55.4, 48.2, 41.4, 30.7, 24.8, 22.8.
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-6-methoxy-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6d): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at –78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether /EtOAc, 2:1) to afford 6d (1.32 g, 84%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 7.81 (d, J = 8.6 Hz, 1H), 7.46 (d, J = 2.8 Hz, 1H), 7.13 (dd, J1 = 2.8 Hz, J2 = 8.6 Hz, 1H), 5.90–5.85 (m, 1H), 4.27 (d, J = 6.7 Hz, 2H), 3.83 (s, 3H), 2.88 (d, J = 16.8 Hz, 1H), 2.58 (d, J = 16.8 Hz, 1H), 1.88 (d, J = 0.78 Hz, 3H), 1.20 (s, 3H), 1.13 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 197.4, 159.5, 136.7, 136.2, 131.0, 128.9, 121.5, 120.0, 109.3, 94.4, 85.4, 74.1, 60.9, 55.4, 48.2, 41.5, 24.9, 23.2, 22.9.
4-Hydroxy-6-methoxy-2,2-dimethyl-4-((trimethylsilyl)ethynyl)-3,4-dihydronaphthalen-1(2H)-one (6e): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of ethynyltrimethylsilane (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 6:1) to afford 6e (1.39 g, 88%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 7.84 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 2.7 Hz, 1H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.6 Hz, 1H), 3.85 (s, 3H), 2.92 (d, J = 16.6 Hz, 1H), 2.56 (d, J = 16.6 Hz, 1H), 2.53 (s, 1H), 1.22 (s, 3H), 1.13 (s, 3H), 0.20 (s, 9H). 13C-NMR (CDCl3, 25 °C) δ 197.1, 159.7, 131.2, 129.0, 121.5, 109.3, 105.4, 74.2, 55.5, 41.3, 24.8, −0.2. MS: 339.0 (M+Na+).
Ethyl 3-(1′-hydroxy-7′-methoxy-3′,3′-dimethyl-4′-oxo-1′,2′,3′,4′-tetrahydronaphthalen-1′-yl)propiolate (6f): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of ethylpropiolate (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 8:1) to afford 6f (1.48 g, 94%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 7.79 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 2.8 Hz, 1H), 7.15 (dd, J1 = 2.8 Hz, J2 = 8.6 Hz, 1H), 4.23 (q, J1 = 7.1 Hz, J2 = 14.2 Hz, 2H), 3.85 (s, 3H), 3.50 (d, J = 5.7 Hz, 1H), 2.92 (d, J = 17.1 Hz, 1H), 2.64 (d, J = 17.1 Hz, 1H), 1.31 (t, J = 7.1 Hz, 3H), 1.25 (s, 3H), 1.17 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.5, 160.0, 153.1, 134.7, 131.2, 129.0, 121.6, 109.7, 86.5, 78.9, 73.9, 62.2, 55.5, 41.6, 24.7, 23.3, 13.8. MS:339.0 (M+Na+).
Methyl 3-(1′-hydroxy-7′-methoxy-3′,3′-dimethyl-4′-oxo-1′,2′,3′,4′-tetrahydronaphthalen-1′-yl)propiolate (6g): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of methyl propiolate (0.42 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 9:1) to afford 6g (1.37 g, 91%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 7.78 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 2.8 Hz, 1H), 7.16 (dd, J1 = 2.8 Hz, J2 = 8.6 Hz, 1H), 3.86 (s, 3H), 3.79 (s, 3H), 3.03 (s, 1H), 2.93 (d, J = 17.1 Hz, 1H), 2.60 (d, J = 17.1 Hz, 1H), 1.23 (s, 3H), 1.16 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.3, 160.2, 153.5, 134.5, 131.2, 128.9, 121.7, 109.8, 86.9, 78.7, 74.1, 55.6, 52.8, 48.0, 41.6, 24.7, 23.1. MS:325.0 (M+Na+).
(Z)-7-Bromo-4-hydroxy-4-(5′-hydroxy-3-methylpent-3′-en-1′-yn-1′-yl)-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6h): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4d (1.33 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 6h (1.61 g, 89%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.06 (d, J = 1.8 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.55 (dd, J1 = 1.9 Hz, J2 = 8.3 Hz, 1H), 5.93–5.89 (m, 1H), 4.26 (d, J = 6.7 Hz, 2H), 4.03 (s, 1H), 2.81 (d, J = 24.1 Hz, 1H), 2.64 (d, J = 24.1 Hz, 1H), 2.59 (s, 1H), 1.88 (s, 3H), 1.16 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 196.6, 146.1, 136.6, 131.7, 130.4, 129.4, 128.7, 128.5, 120.1, 93.6, 86.3, 74.0, 60.8, 48.5, 41.5, 29.5, 24.8, 22.8, 22.7.
(Z)-5-Hydroxy-5-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-2,7,7-trimethyl-6,7-dihydroquinolin-8(5H)-one (6i): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4e (1.01 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 1:1) to afford 6i (1.40 g, 94%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.17 (d, J = 7.9 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 5.85–5.83 (m, 1H), 5.34 (s, 1H), 4.16 (d, J = 6.5 Hz, 2H), 2.99 (d, J = 17.5 Hz, 1H), 2.59 (d, J = 17.5 Hz, 1H), 2.65 (s, 3H), 1.79 (s, 3H), 1.39 (s, 3H), 0.96 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 195.8, 163.5, 160.8, 136.8, 135.4, 123.4, 122.5, 119.4, 94.3, 85.2, 73.6, 60.8, 50.2, 40.7, 24.6, 24.6, 22.6, 20.9.
(Z)-5-Hydroxy-5-(5′-hydroxy-3-methylpent-3′-en-1′-yn-1′-yl)-7,7-dimethyl-2-phenyl-6,7-dihydroquinolin-8(5H)-one (6j): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4f (1.32 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 1:1) to afford 6j (1.73 g, 96%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.35 (d, J = 8.1 Hz, 1H), 8.11 (dd, J1 = 2.7 Hz, J2 = 7.9 Hz, 2H), 7.87 (d, J = 8.2 Hz, 1H), 7.55–7.51 (m, 3H), 5.84–5.79 (m, 1H), 5.33 (s, 1H), 4.16 (s, 2H), 3.07 (d, J = 17.5 Hz, 1H), 2.62 (d, J = 17.5 Hz, 1H), 1.80 (d, J = 1.1 Hz, 3H), 1.45 (s, 3H), 0.99 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 195.7, 161.3, 160.5, 137.3, 136.6, 136.3, 130.6, 129.0, 127.5, 123.4, 120.2, 119.8, 94.6, 85.3, 73.8, 61.2, 50.6, 40.9, 24.8, 22.8, 21.0.
(Z)-5-Hydroxy-5-(5′-hydroxy-3-methylpent-3′-en-1′-yn-1′-yl)-7,7-dimethyl-6,7-dihydroquinolin-8(5H)-one (6k): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4g (0.94 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 1:2) to afford 6c (1.35 g, 95%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.77 (dd, J1 = 1.7 Hz, J2 = 4.8 Hz, 1H), 8.30 (dd, J1 = 1.7 Hz, J2 = 7.8 Hz, 1H), 7.45 (dd, J1 = 4.8 Hz, J2 = 7.8 Hz, 1H), 5.87–5.82 (m, 1H), 5.48 (s, 1H), 4.14 (d, J = 6.5 Hz, 2H), 3.00 (d, J = 17.4 Hz, 1H), 2.68 (d, J = 17.4 Hz, 1H), 3.02 (s, 1H), 1.78 (d, J = 1.2 Hz, 3H), 1.40 (d, J = 6.8 Hz, 3H), 1.01 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.0, 161.4, 153.1, 136.9, 135.2, 124.9, 123.6, 119.3, 93.9, 85.7, 73.9, 60.6, 50.0, 40.7, 24.6, 22.6, 21.1.
4. Conclusions
We have developed a novel regioselective addition of acetylides to enediones that affords the corresponding 1-ethynylcyclohex-2-enol derivatives with excellent regioselectivity and high yield. This synthetic strategy can be applied to the manufacture of pharmaceuticals, fine chemicals, and natural products. Applications of this strategy in natural products are currently being carried out in our laboratory and will be reported in due course.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/18/9/10776/s1.
Acknowledgments
We thank the National Natural Science Foundation of China (No. 20972184) and the National Hi-Tech Research and Development Program of China (No. 2011AA10A206) for their financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pu, L. Asymmetric alkynylzinc additions to aldehydes and ketones. Tetrahedron 2003, 59, 9873–9886. [Google Scholar] [CrossRef]
- Trost, B.M.; Weiss, A.H. The enantioselective addition of alkyne nucleophiles to carbonyl groups. Adv. Synth. Catal. 2009, 351, 963–983. [Google Scholar] [CrossRef]
- Ding, C.H.; Hou, X.L. Catalytic asymmetric propargylation. Chem. Rev. 2011, 111, 1914–1937. [Google Scholar] [CrossRef]
- Mayer, H.J.; Rigassi, N.; Schwietter, U. Synthesis of abscisic acid. Helv. Chim. Acta 1976, 59, 1424–1427. [Google Scholar] [CrossRef]
- Khodabocus, A. Preparation of functionalised bicycles by means of vinyl ketene-cyclohexadienone electrocyclisations: An entry into terpenoid precursors. ARKIVOC 2002, i, 25–34. [Google Scholar] [CrossRef]
- McCubbin, J.A.; Voth, S.; Krokhin, O.V. Catalytic asymmetric propargylation. J. Org. Chem. 2011, 76, 8537–8542. [Google Scholar] [CrossRef]
- Vatele, J.M. Lewis acid-catalyzed oxidative rearrangement of tertiary allylic alcohols mediated by TEMPO. Tetrahedron 2010, 66, 904–912. [Google Scholar] [CrossRef]
- Uyanik, M.; Fukatsu, R.; Ishihara, K. IBS-catalyzed oxidative rearrangement of tertiary allylic alcohols to enones with oxone. Org. Lett. 2009, 11, 3470–3473. [Google Scholar] [CrossRef]
- McCubbin, J.A.; Hosseini, H.; Krokhin, O.V. Boronic acid catalyzed friedel—Crafts reactions of allylic alcohols with electron-rich arenes and heteroarenes. J. Org. Chem. 2010, 75, 959–962. [Google Scholar] [CrossRef]
- Brandsma, L. Preparative Acetylenic Chemistry; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Fisch, A.; Coisne, J.M.; Figeys, H.P. An improved procedure for preparing and utilizing alkali metal acetylides. Synthesis 1982, 3, 211. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Huynh, V.; Moreaw, J.J.E. Uses of SiN bonds in organic synthesis. A direct synthesis of functional protected propargylic primary amines. Tetrahedron Lett. 1974, 25, 1887–1890. [Google Scholar] [CrossRef]
- Guerra-Navarro, N.A.; Palacios-Grijalva, L.N.; Angeles-Beltrán, D.; Negrón-Silva, G.E.; Lomas-Romero, L.; González-Zamora, E.; Gaviño-Ramírez, R.; Navarrete-Bolaños, J. Synthesis of new pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione (PCU) cyanosilylated derivatives using sulphated zirconia and hydrotalcite as catalysts in microwave-assisted reactions under solvent free conditions. Molecules 2011, 16, 6561–6576. [Google Scholar] [CrossRef]
- Liotta, D.; Saindane, M.; Jamison, L.; Grossman, J.; Phillips, P. Acetylide additions to enediones. regioselectivity based on stereoelectronic ontrol. J. Org. Chem. 1985, 50, 3241–3243. [Google Scholar] [CrossRef]
- Abrams, S.R.; Gusta, L.V.; Reaney, M.J.T.; Ewan, B.E. Use of Compounds to Enhance Synchrony of Germination and Emergence in Plants. U.S. Patent 5,518,995, 21 May 1996. [Google Scholar]
- Takahashi, S.; Oritani, T.; Yamashita, K. Synthesis of (+)-methyl phaseate and its isomer from (−)-β-pinene. Agric. Biol. Chem. 1989, 53, 2711–2718. [Google Scholar] [CrossRef]
- Nyangulu, J.M.; Nelson, K.M.; Rose, P.A.; Gai, Y.; Loewen, M.; Lougheed, B.; Quail, J.W.; Culter, A.J.; Abrams, S.R. Synthesis and biological activity of tetralone abscisic acid analogues. Org. Biomol. Chem. 2006, 4, 1400–1412. [Google Scholar] [CrossRef]
- Han, X.Q.; Zhou, Z.Y.; Wan, C.; Xiao, Y.M.; Qin, Z.H. Co(acac)2-catalyzed allylic and benzylic oxidation by tert-butyl hydroperoxide. Synthesis 2013, 45, 615–620. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).